Structural Mechanisms of Store-Operated and Mitochondrial Calcium Regulation: Initiation Points for Drug Discovery

 , , , , and
, , , , and 
Abstract
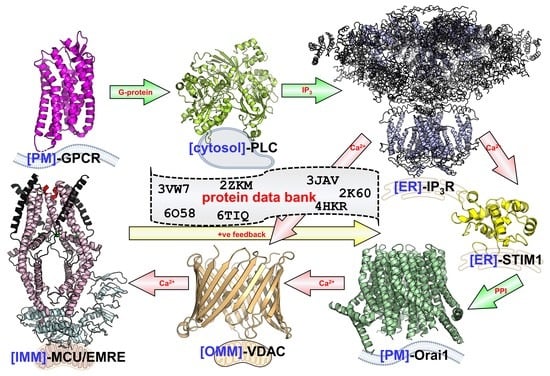
1. Cytosolic and Stored Calcium
1.1. Mobilization of S/ER Calcium Stores
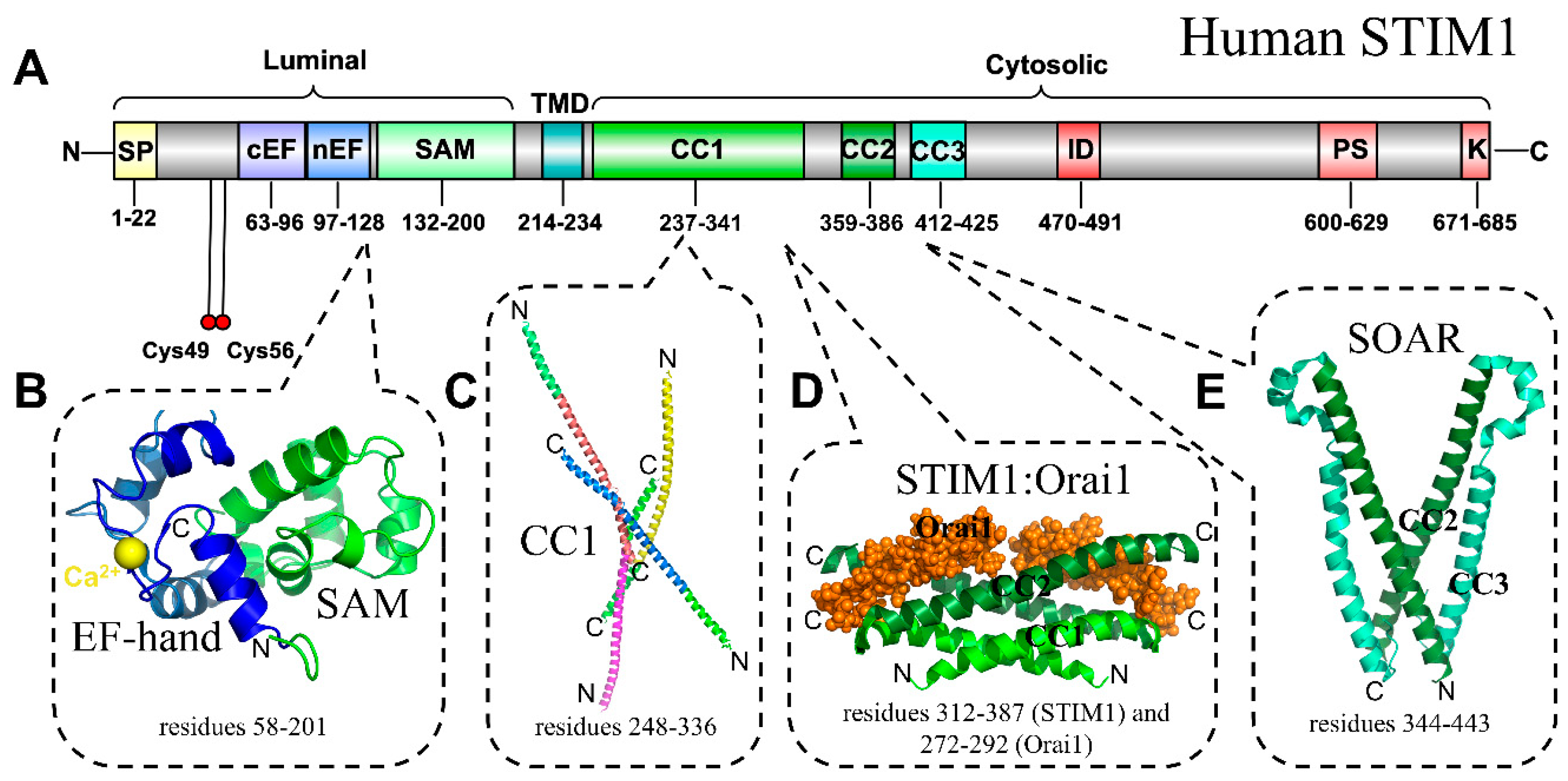
1.2. Store-Operated Calcium Entry


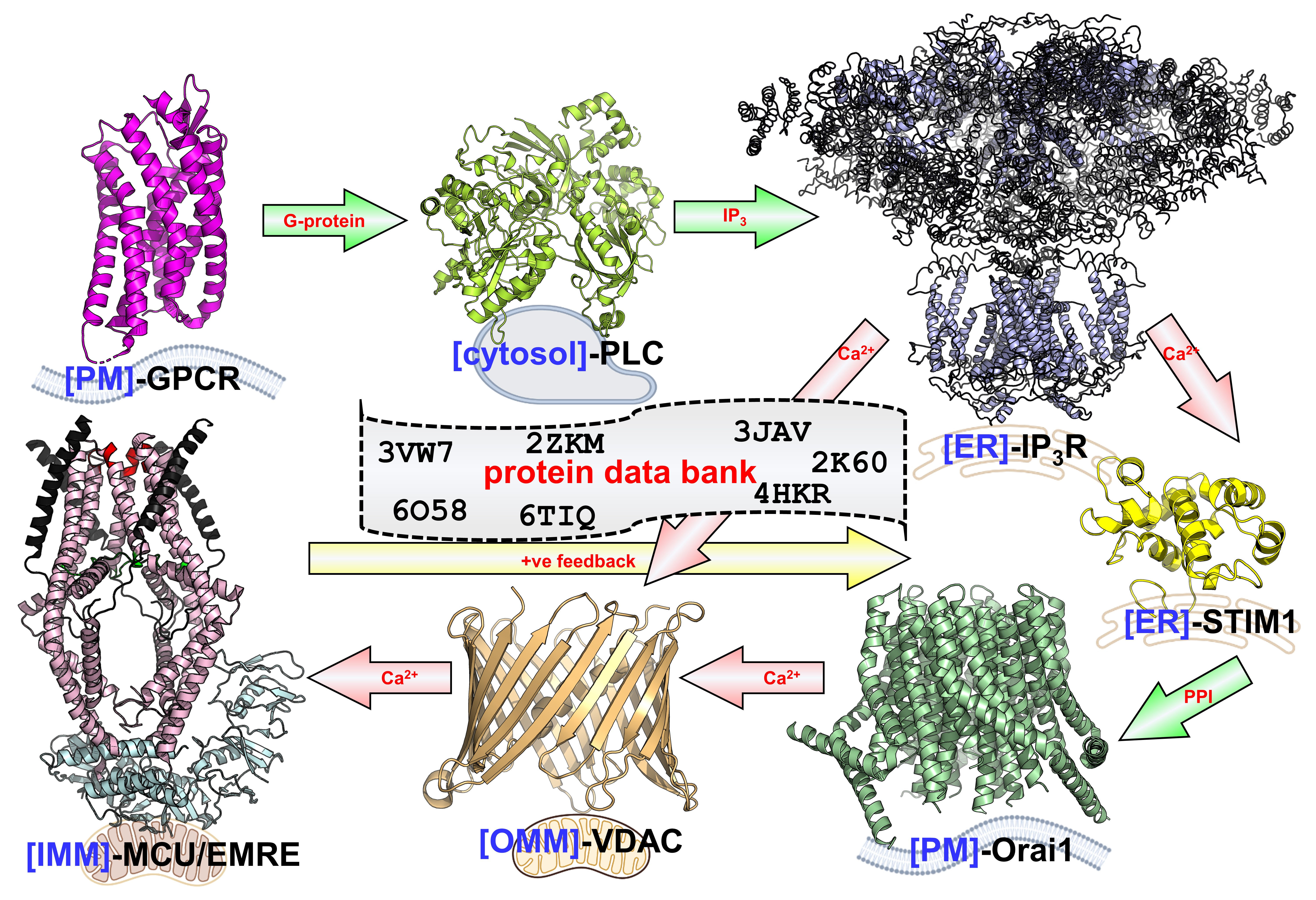
1.3. STIM1, Orai1 and Disease
1.4. Mitochondrial Calcium Uptake and SOCE Crosstalk
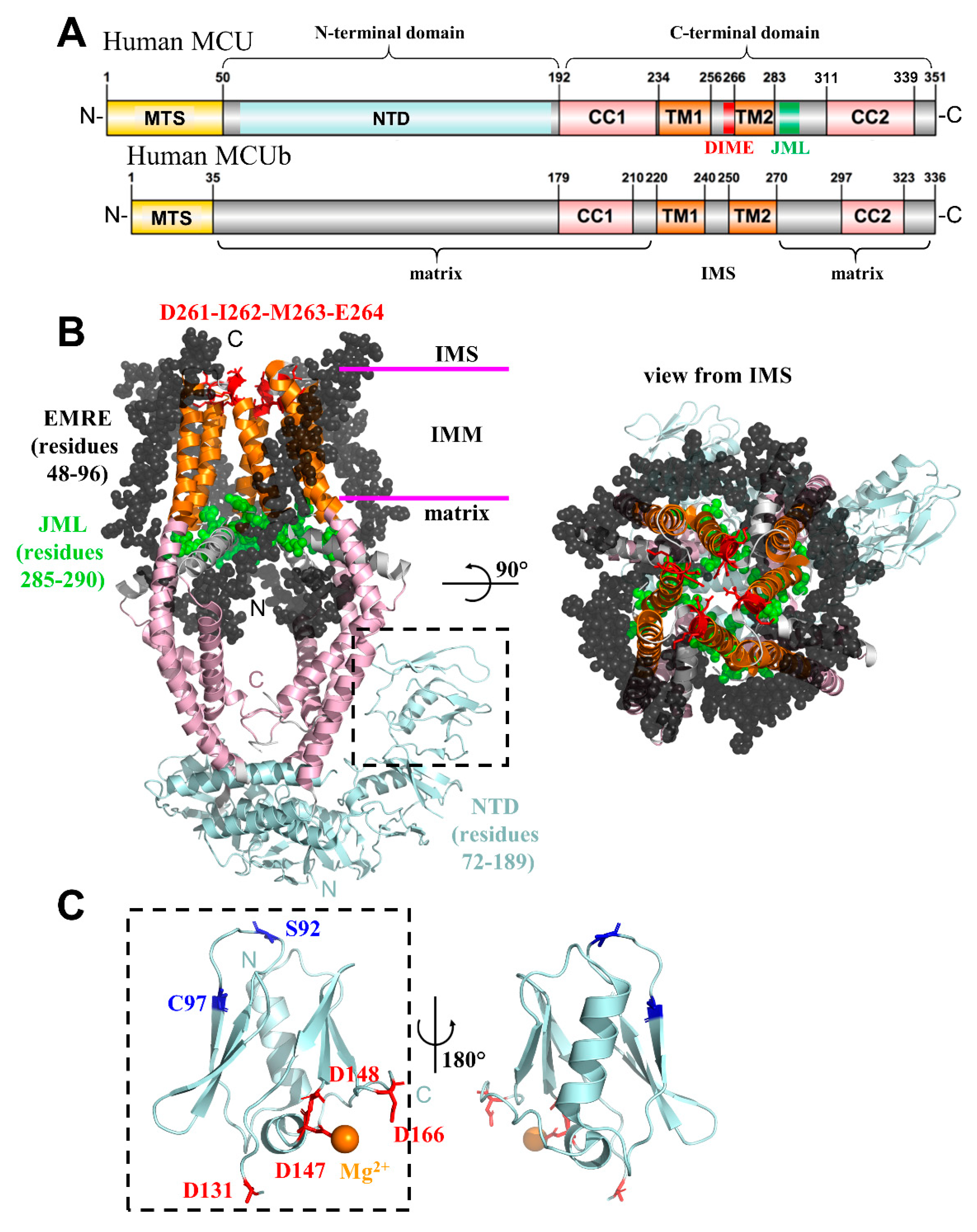
2. Mitochondrial Calcium Uniporter (MCU)
2.1. Essential Mitochondrial Calcium Uniporter Regulator (EMRE)
2.2. Mitochondrial Calcium Uptake (MICU) Proteins
2.3. Mitochondrial Calcium Uniporter Dominant Negative Beta (MCUb) Subunit
2.4. Mitochondrial Calcium Uniporter Regulator-1 (MCUR1)
2.5. MCU and Disease
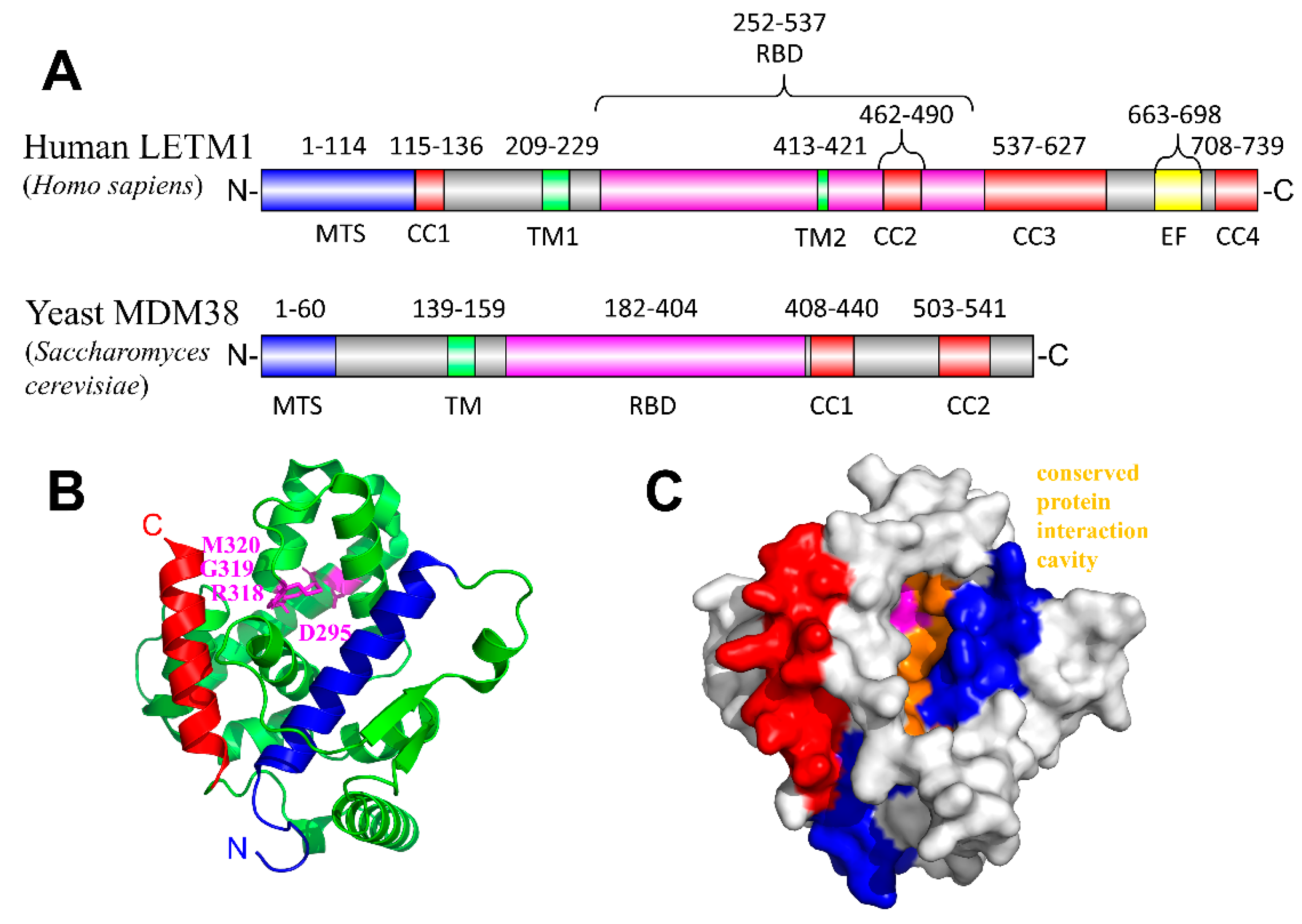
3. Leucine Zipper EF-Hand Containing Transmembrane Protein-1 (LETM1)
LETM1 and Disease
4. Generating New Therapeutics and Diagnostics from Protein Structures
4.1. Store-Operated and Mitochondrial Ca2+ Entry Proteins as Drug Targets
4.2. Initiating Protein Structure-based Drug Discovery
4.3. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef] [PubMed]
- Rimessi, A.; Giorgi, C.; Pinton, P.; Rizzuto, R. The versatility of mitochondrial calcium signals: From stimulation of cell metabolism to induction of cell death. Biochim. Biophys. Acta 2008, 1777, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Deluca, H.F.; Engstrom, G.W. Calcium uptake by rat kidney mitochondria. Proc. Natl. Acad. Sci. USA 1961, 47, 1744–1750. [Google Scholar] [CrossRef]
- Amberger, A.; Weiss, H.; Haller, T.; Kock, G.; Hermann, M.; Widschwendter, M.; Margreiter, R. A subpopulation of mitochondria prevents cytosolic calcium overload in endothelial cells after cold ischemia/reperfusion. Transplantation 2001, 71, 1821–1827. [Google Scholar] [CrossRef]
- Ly, L.D.; Ly, D.D.; Nguyen, N.T.; Kim, J.H.; Yoo, H.; Chung, J.; Lee, M.S.; Cha, S.K.; Park, K.S. Mitochondrial Ca(2+) Uptake Relieves Palmitate-Induced Cytosolic Ca(2+) Overload in MIN6 Cells. Mol. Cells 2020, 43, 66–75. [Google Scholar]
- Yi, M.; Weaver, D.; Hajnoczky, G. Control of mitochondrial motility and distribution by the calcium signal: A homeostatic circuit. J. Cell Biol. 2004, 167, 661–672. [Google Scholar] [CrossRef]
- Peng, T.I.; Jou, M.J. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 2010, 1201, 183–188. [Google Scholar] [CrossRef]
- Patron, M.; Raffaello, A.; Granatiero, V.; Tosatto, A.; Merli, G.; De Stefani, D.; Wright, L.; Pallafacchina, G.; Terrin, A.; Mammucari, C.; et al. The mitochondrial calcium uniporter (MCU): Molecular identity and physiological roles. J. Biol. Chem. 2013, 288, 10750–10758. [Google Scholar] [CrossRef]
- Cardenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgo, J.; Muller, M.; Vais, H.; Cheung, K.H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef]
- Jouaville, L.S.; Pinton, P.; Bastianutto, C.; Rutter, G.A.; Rizzuto, R. Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA 1999, 96, 13807–13812. [Google Scholar] [CrossRef] [PubMed]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Zhao, L.; Clapham, D.E. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 2009, 326, 144–147. [Google Scholar] [CrossRef]
- Lin, Q.T.; Stathopulos, P.B. Molecular Mechanisms of Leucine Zipper EF-Hand Containing Transmembrane Protein-1 Function in Health and Disease. Int. J. Mol. Sci. 2019, 20, 286. [Google Scholar] [CrossRef]
- Feske, S. Calcium signalling in lymphocyte activation and disease. Nat. Rev. Immunol. 2007, 7, 690–702. [Google Scholar] [CrossRef]
- Feske, S.; Skolnik, E.Y.; Prakriya, M. Ion channels and transporters in lymphocyte function and immunity. Nat. Rev. Immunol. 2012, 12, 532–547. [Google Scholar] [CrossRef]
- Luik, R.M.; Wang, B.; Prakriya, M.; Wu, M.M.; Lewis, R.S. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature 2008, 454, 538–542. [Google Scholar] [CrossRef]
- Montero, M.; Barrero, M.J.; Torrecilla, F.; Lobaton, C.D.; Moreno, A.; Alvarez, J. Stimulation by thimerosal of histamine-induced Ca(2+) release in intact HeLa cells seen with aequorin targeted to the endoplasmic reticulum. Cell Calcium 2001, 30, 181–190. [Google Scholar] [CrossRef]
- Suzuki, J.; Kanemaru, K.; Ishii, K.; Ohkura, M.; Okubo, Y.; Iino, M. Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat. Commun. 2014, 5, 4153. [Google Scholar] [CrossRef]
- Yu, R.; Hinkle, P.M. Rapid turnover of calcium in the endoplasmic reticulum during signaling. Studies with cameleon calcium indicators. J. Biol. Chem. 2000, 275, 23648–23653. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Stathopulos, P.B.; Seo, M.D.; Enomoto, M.; Amador, F.J.; Ishiyama, N.; Ikura, M. Themes and variations in ER/SR calcium release channels: Structure and function. Physiology (Bethesda) 2012, 27, 331–342. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Amador, F.J.; Stathopulos, P.B.; Enomoto, M.; Ikura, M. Ryanodine receptor calcium release channels: Lessons from structure-function studies. FEBS J. 2013, 280, 5456–5470. [Google Scholar] [CrossRef]
- Fedorenko, O.A.; Popugaeva, E.; Enomoto, M.; Stathopulos, P.B.; Ikura, M.; Bezprozvanny, I. Intracellular calcium channels: Inositol-1,4,5-trisphosphate receptors. Eur. J. Pharmacol. 2014, 739, 39–48. [Google Scholar] [CrossRef]
- Stathopulos, P.B.; Ikura, M. Store operated calcium entry: From concept to structural mechanisms. Cell Calcium 2017, 63, 3–7. [Google Scholar] [CrossRef]
- Novello, M.J.; Zhu, J.; Feng, Q.; Ikura, M.; Stathopulos, P.B. Structural elements of stromal interaction molecule function. Cell Calcium 2018, 73, 88–94. [Google Scholar] [CrossRef]
- Putney, J.W., Jr. A model for receptor-regulated calcium entry. Cell Calcium 1986, 7, 1–12. [Google Scholar] [CrossRef]
- Smyth, J.T.; Dehaven, W.I.; Jones, B.F.; Mercer, J.C.; Trebak, M.; Vazquez, G.; Putney, J.W., Jr. Emerging perspectives in store-operated Ca2+ entry: Roles of Orai, Stim and TRP. Biochim. Biophys. Acta 2006, 1763, 1147–1160. [Google Scholar] [CrossRef]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E., Jr.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Vig, M.; Beck, A.; Billingsley, J.M.; Lis, A.; Parvez, S.; Peinelt, C.; Koomoa, D.L.; Soboloff, J.; Gill, D.L.; Fleig, A.; et al. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr. Biol. 2006, 16, 2073–2079. [Google Scholar] [CrossRef]
- Vig, M.; Peinelt, C.; Beck, A.; Koomoa, D.L.; Rabah, D.; Koblan-Huberson, M.; Kraft, S.; Turner, H.; Fleig, A.; Penner, R.; et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 2006, 312, 1220–1223. [Google Scholar] [CrossRef]
- Yeromin, A.V.; Zhang, S.L.; Jiang, W.; Yu, Y.; Safrina, O.; Cahalan, M.D. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature 2006, 443, 226–229. [Google Scholar] [CrossRef]
- Hohendanner, F.; McCulloch, A.D.; Blatter, L.A.; Michailova, A.P. Calcium and IP3 dynamics in cardiac myocytes: Experimental and computational perspectives and approaches. Front. Pharmacol. 2014, 5, 35. [Google Scholar] [CrossRef]
- Hilger, D.; Masureel, M.; Kobilka, B.K. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 2018, 25, 4–12. [Google Scholar] [CrossRef]
- Kadamur, G.; Ross, E.M. Mammalian phospholipase C. Annu. Rev. Physiol. 2013, 75, 127–154. [Google Scholar] [CrossRef]
- Smrcka, A.V.; Fisher, I. G-protein betagamma subunits as multi-functional scaffolds and transducers in G-protein-coupled receptor signaling. Cell Mol. Life Sci. 2019, 76, 4447–4459. [Google Scholar] [CrossRef]
- Nieman, M.T. Protease-activated receptors in hemostasis. Blood 2016, 128, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Scarlata, S. The role of phospholipase Cbeta on the plasma membrane and in the cytosol: How modular domains enable novel functions. Adv. Biol. Regul. 2019, 73, 100636. [Google Scholar] [CrossRef] [PubMed]
- Konieczny, V.; Tovey, S.C.; Mataragka, S.; Prole, D.L.; Taylor, C.W. Cyclic AMP Recruits a Discrete Intracellular Ca(2+) Store by Unmasking Hypersensitive IP3 Receptors. Cell. Rep. 2017, 18, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Meena, A.; Tovey, S.C.; Taylor, C.W. Sustained signalling by PTH modulates IP3 accumulation and IP3 receptors through cyclic AMP junctions. J. Cell Sci. 2015, 128, 408–420. [Google Scholar] [CrossRef]
- Tovey, S.C.; Taylor, C.W. Cyclic AMP directs inositol (1,4,5)-trisphosphate-evoked Ca2+ signalling to different intracellular Ca2+ stores. J. Cell Sci. 2013, 126, 2305–2313. [Google Scholar] [CrossRef]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Trenker, R.; Jura, N. Receptor tyrosine kinase activation: From the ligand perspective. Curr. Opin. Cell Biol. 2020, 63, 174–185. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, Y.; Zhang, S.; Haneef, K.; Liu, W. Structural and immunogenomic insights into B-cell receptor activation. J. Genet. Genom. 2020, 47, 27–35. [Google Scholar] [CrossRef]
- Treanor, B. B-cell receptor: From resting state to activate. Immunology 2012, 136, 21–27. [Google Scholar] [CrossRef]
- Kim, Y.J.; Sekiya, F.; Poulin, B.; Bae, Y.S.; Rhee, S.G. Mechanism of B-cell receptor-induced phosphorylation and activation of phospholipase C-gamma2. Mol. Cell Biol. 2004, 24, 9986–9999. [Google Scholar] [CrossRef]
- Mahtani, T.; Treanor, B. Beyond the CRAC: Diversification of ion signaling in B cells. Immunol. Rev. 2019, 291, 104–122. [Google Scholar] [CrossRef]
- Schamel, W.W.; Alarcon, B.; Minguet, S. The TCR is an allosterically regulated macromolecular machinery changing its conformation while working. Immunol. Rev. 2019, 291, 8–25. [Google Scholar] [CrossRef]
- Xu, X.; Li, H.; Xu, C. Structural understanding of T cell receptor triggering. Cell Mol. Immunol. 2020, 17, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Olivera, A.; Beaven, M.A.; Metcalfe, D.D. Mast cells signal their importance in health and disease. J. Allergy Clin. Immunol. 2018, 142, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Bournazos, S.; Ravetch, J.V. Fcgamma receptor pathways during active and passive immunization. Immunol. Rev. 2015, 268, 88–103. [Google Scholar] [CrossRef] [PubMed]
- Rougier, J.S.; Abriel, H. Cardiac voltage-gated calcium channel macromolecular complexes. Biochim. Biophys. Acta 2016, 1863, 1806–1812. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yan, N.; Yan, Z. Structure-Function Relationship of the Voltage-Gated Calcium Channel Cav1.1 Complex. Adv. Exp. Med. Biol. 2017, 981, 23–39. [Google Scholar]
- Pallien, T.; Klussmann, E. New aspects in cardiac L-type Ca2+ channel regulation. Biochem. Soc. Trans. 2020, 48, 39–49. [Google Scholar] [CrossRef]
- Eisner, D.A.; Caldwell, J.L.; Kistamas, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef]
- Thul, R. Translating intracellular calcium signaling into models. Cold Spring Harb. Protoc. 2014, 2014, 463–471. [Google Scholar] [CrossRef]
- Guse, A.H.; da Silva, C.P.; Berg, I.; Skapenko, A.L.; Weber, K.; Heyer, P.; Hohenegger, M.; Ashamu, G.A.; Schulze-Koops, H.; Potter, B.V.; et al. Regulation of calcium signalling in T lymphocytes by the second messenger cyclic ADP-ribose. Nature 1999, 398, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Kiselyov, K.; Shin, D.M.; Shcheynikov, N.; Kurosaki, T.; Muallem, S. Regulation of Ca2+-release-activated Ca2+ current (Icrac) by ryanodine receptors in inositol 1,4,5-trisphosphate-receptor-deficient DT40 cells. Biochem. J. 2001, 360, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Schwarzmann, N.; Kunerth, S.; Weber, K.; Mayr, G.W.; Guse, A.H. Knock-down of the type 3 ryanodine receptor impairs sustained Ca2+ signaling via the T cell receptor/CD3 complex. J. Biol. Chem. 2002, 277, 50636–50642. [Google Scholar] [CrossRef] [PubMed]
- Smrcka, A.V. Regulation of phosphatidylinositol-specific phospholipase C at the nuclear envelope in cardiac myocytes. J. Cardiovasc. Pharmacol. 2015, 65, 203–210. [Google Scholar] [CrossRef]
- Siltari, A.; Korpela, R.; Vapaatalo, H. Bradykinin -induced vasodilatation: Role of age, ACE1-inhibitory peptide, mas- and bradykinin receptors. Peptides 2016, 85, 46–55. [Google Scholar] [CrossRef]
- Thangam, E.B.; Jemima, E.A.; Singh, H.; Baig, M.S.; Khan, M.; Mathias, C.B.; Church, M.K.; Saluja, R. The Role of Histamine and Histamine Receptors in Mast Cell-Mediated Allergy and Inflammation: The Hunt for New Therapeutic Targets. Front. Immunol. 2018, 9, 1873. [Google Scholar] [CrossRef]
- Galvin, C.D.; Hardiman, O.; Nolan, C.M. IGF-1 receptor mediates differentiation of primary cultures of mouse skeletal myoblasts. Mol. Cell. Endocrinol. 2003, 200, 19–29. [Google Scholar] [CrossRef]
- Molhoek, K.R.; Shada, A.L.; Smolkin, M.; Chowbina, S.; Papin, J.; Brautigan, D.L.; Slingluff, C.L., Jr. Comprehensive analysis of receptor tyrosine kinase activation in human melanomas reveals autocrine signaling through IGF-1R. Melanoma Res. 2011, 21, 274–284. [Google Scholar] [CrossRef]
- Goodnow, C.C.; Sprent, J.; Fazekas de St Groth, B.; Vinuesa, C.G. Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature 2005, 435, 590–597. [Google Scholar] [CrossRef]
- Cai, X. Molecular evolution and functional divergence of the Ca(2+) sensor protein in store-operated Ca(2+) entry: Stromal interaction molecule. PLoS ONE 2007, 2, e609. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, M.; Nishikawa, T.; Back, S.I.; Ishiyama, N.; Zheng, L.; Stathopulos, P.B.; Ikura, M. Coordination of a Single Calcium Ion in the EF-hand Maintains the Off State of the Stromal Interaction Molecule Luminal Domain. J. Mol. Biol. 2020, 432, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Stathopulos, P.B.; Li, G.Y.; Plevin, M.J.; Ames, J.B.; Ikura, M. Stored Ca2+ depletion-induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF-SAM region: An initiation mechanism for capacitive Ca2+ entry. J. Biol. Chem. 2006, 281, 35855–35862. [Google Scholar] [CrossRef] [PubMed]
- Muik, M.; Fahrner, M.; Schindl, R.; Stathopulos, P.; Frischauf, I.; Derler, I.; Plenk, P.; Lackner, B.; Groschner, K.; Ikura, M.; et al. STIM1 couples to ORAI1 via an intramolecular transition into an extended conformation. EMBO J. 2011, 30, 1678–1689. [Google Scholar] [CrossRef] [PubMed]
- Stathopulos, P.B.; Zheng, L.; Li, G.Y.; Plevin, M.J.; Ikura, M. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell 2008, 135, 110–122. [Google Scholar] [CrossRef]
- Zhou, Y.; Srinivasan, P.; Razavi, S.; Seymour, S.; Meraner, P.; Gudlur, A.; Stathopulos, P.B.; Ikura, M.; Rao, A.; Hogan, P.G. Initial activation of STIM1, the regulator of store-operated calcium entry. Nat. Struct. Mol. Biol. 2013, 20, 973–981. [Google Scholar] [CrossRef]
- Baba, Y.; Hayashi, K.; Fujii, Y.; Mizushima, A.; Watarai, H.; Wakamori, M.; Numaga, T.; Mori, Y.; Iino, M.; Hikida, M.; et al. Coupling of STIM1 to store-operated Ca2+ entry through its constitutive and inducible movement in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2006, 103, 16704–16709. [Google Scholar] [CrossRef]
- Liou, J.; Fivaz, M.; Inoue, T.; Meyer, T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc. Natl. Acad. Sci. USA 2007, 104, 9301–9306. [Google Scholar] [CrossRef] [PubMed]
- Luik, R.M.; Wu, M.M.; Buchanan, J.; Lewis, R.S. The elementary unit of store-operated Ca2+ entry: Local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J. Cell Biol. 2006, 174, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.M.; Buchanan, J.; Luik, R.M.; Lewis, R.S. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J. Cell Biol. 2006, 174, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Muik, M.; Frischauf, I.; Derler, I.; Fahrner, M.; Bergsmann, J.; Eder, P.; Schindl, R.; Hesch, C.; Polzinger, B.; Fritsch, R.; et al. Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J. Biol. Chem. 2008, 283, 8014–8022. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.P.; Zeng, W.; Dorwart, M.R.; Choi, Y.J.; Worley, P.F.; Muallem, S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 2009, 11, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Lange, I.; Feske, S. A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem. Biophys. Res. Commun. 2009, 385, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Hoth, M.; Penner, R. Calcium release-activated calcium current in rat mast cells. J. Physiol. 1993, 465, 359–386. [Google Scholar] [CrossRef] [PubMed]
- Zweifach, A.; Lewis, R.S. Rapid inactivation of depletion-activated calcium current (ICRAC) due to local calcium feedback. J. Gen. Physiol. 1995, 105, 209–226. [Google Scholar] [CrossRef]
- Derler, I.; Fahrner, M.; Muik, M.; Lackner, B.; Schindl, R.; Groschner, K.; Romanin, C. A Ca2(+)release-activated Ca2(+) (CRAC) modulatory domain (CMD) within STIM1 mediates fast Ca2(+)-dependent inactivation of ORAI1 channels. J. Biol. Chem. 2009, 284, 24933–24938. [Google Scholar] [CrossRef]
- Mullins, F.M.; Lewis, R.S. The inactivation domain of STIM1 is functionally coupled with the Orai1 pore to enable Ca2+-dependent inactivation. J. Gen. Physiol. 2016, 147, 153–164. [Google Scholar] [CrossRef]
- Mullins, F.M.; Yen, M.; Lewis, R.S. Orai1 pore residues control CRAC channel inactivation independently of calmodulin. J. Gen. Physiol. 2016, 147, 137–152. [Google Scholar] [CrossRef]
- Mullins, F.M.; Park, C.Y.; Dolmetsch, R.E.; Lewis, R.S. STIM1 and calmodulin interact with Orai1 to induce Ca2+-dependent inactivation of CRAC channels. Proc. Natl. Acad. Sci. USA 2009, 106, 15495–15500. [Google Scholar] [CrossRef]
- Choi, Y.J.; Zhao, Y.; Bhattacharya, M.; Stathopulos, P.B. Structural perturbations induced by Asn131 and Asn171 glycosylation converge within the EFSAM core and enhance stromal interaction molecule-1 mediated store operated calcium entry. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Gui, L.; Zhu, J.; Lu, X.; Sims, S.M.; Lu, W.Y.; Stathopulos, P.B.; Feng, Q. S-Nitrosylation of STIM1 by Neuronal Nitric Oxide Synthase Inhibits Store-Operated Ca(2+) Entry. J. Mol. Biol. 2018, 430, 1773–1785. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Lu, X.; Feng, Q.; Stathopulos, P.B. A charge-sensing region in the stromal interaction molecule 1 luminal domain confers stabilization-mediated inhibition of SOCE in response to S-nitrosylation. J. Biol. Chem. 2018, 293, 8900–8911. [Google Scholar] [CrossRef] [PubMed]
- Zhu-Mauldin, X.; Marsh, S.A.; Zou, L.; Marchase, R.B.; Chatham, J.C. Modification of STIM1 by O-linked N-acetylglucosamine (O-GlcNAc) attenuates store-operated calcium entry in neonatal cardiomyocytes. J. Biol. Chem. 2012, 287, 39094–39106. [Google Scholar] [CrossRef]
- Korzeniowski, M.K.; Popovic, M.A.; Szentpetery, Z.; Varnai, P.; Stojilkovic, S.S.; Balla, T. Dependence of STIM1/Orai1-mediated calcium entry on plasma membrane phosphoinositides. J. Biol. Chem. 2009, 284, 21027–21035. [Google Scholar] [CrossRef]
- Lopez, E.; Jardin, I.; Berna-Erro, A.; Bermejo, N.; Salido, G.M.; Sage, S.O.; Rosado, J.A.; Redondo, P.C. STIM1 tyrosine-phosphorylation is required for STIM1-Orai1 association in human platelets. Cell Signal 2012, 24, 1315–1322. [Google Scholar] [CrossRef]
- Manji, S.S.; Parker, N.J.; Williams, R.T.; van Stekelenburg, L.; Pearson, R.B.; Dziadek, M.; Smith, P.J. STIM1: A novel phosphoprotein located at the cell surface. Biochim. Biophys. Acta 2000, 1481, 147–155. [Google Scholar] [CrossRef]
- Pozo-Guisado, E.; Campbell, D.G.; Deak, M.; Alvarez-Barrientos, A.; Morrice, N.A.; Alvarez, I.S.; Alessi, D.R.; Martin-Romero, F.J. Phosphorylation of STIM1 at ERK1/2 target sites modulates store-operated calcium entry. J. Cell Sci. 2010, 123, 3084–3093. [Google Scholar] [CrossRef]
- Smyth, J.T.; Beg, A.M.; Wu, S.; Putney, J.W., Jr.; Rusan, N.M. Phosphoregulation of STIM1 leads to exclusion of the endoplasmic reticulum from the mitotic spindle. Curr. Biol. 2012, 22, 1487–1493. [Google Scholar] [CrossRef]
- Smyth, J.T.; Petranka, J.G.; Boyles, R.R.; DeHaven, W.I.; Fukushima, M.; Johnson, K.L.; Williams, J.G.; Putney, J.W., Jr. Phosphorylation of STIM1 underlies suppression of store-operated calcium entry during mitosis. Nat. Cell Biol. 2009, 11, 1465–1472. [Google Scholar] [CrossRef]
- Thompson, J.L.; Lai-Zhao, Y.; Stathopulos, P.B.; Grossfield, A.; Shuttleworth, T.J. Phosphorylation-mediated structural changes within the SOAR domain of stromal interaction molecule 1 enable specific activation of distinct Orai channels. J. Biol. Chem. 2018, 293, 3145–3155. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.T.; Manji, S.S.; Parker, N.J.; Hancock, M.S.; Van Stekelenburg, L.; Eid, J.P.; Senior, P.V.; Kazenwadel, J.S.; Shandala, T.; Saint, R.; et al. Identification and characterization of the STIM (stromal interaction molecule) gene family: Coding for a novel class of transmembrane proteins. Biochem. J. 2001, 357, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Yazbeck, P.; Tauseef, M.; Kruse, K.; Amin, M.R.; Sheikh, R.; Feske, S.; Komarova, Y.; Mehta, D. STIM1 Phosphorylation at Y361 Recruits Orai1 to STIM1 Puncta and Induces Ca(2+) Entry. Sci. Rep. 2017, 7, 42758. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, B.J.; Irrinki, K.M.; Mallilankaraman, K.; Lien, Y.C.; Wang, Y.; Bhanumathy, C.D.; Subbiah, R.; Ritchie, M.F.; Soboloff, J.; Baba, Y.; et al. S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J. Cell Biol. 2010, 190, 391–405. [Google Scholar] [CrossRef]
- Lupas, A.; Van Dyke, M.; Stock, J. Predicting coiled coils from protein sequences. Science 1991, 252, 1162–1164. [Google Scholar] [CrossRef]
- Cui, B.; Yang, X.; Li, S.; Lin, Z.; Wang, Z.; Dong, C.; Shen, Y. The inhibitory helix controls the intramolecular conformational switching of the C-terminus of STIM1. PLoS ONE 2013, 8, e74735. [Google Scholar] [CrossRef]
- Stathopulos, P.B.; Schindl, R.; Fahrner, M.; Zheng, L.; Gasmi-Seabrook, G.M.; Muik, M.; Romanin, C.; Ikura, M. STIM1/Orai1 coiled-coil interplay in the regulation of store-operated calcium entry. Nat. Commun. 2013, 4, 2963. [Google Scholar] [CrossRef]
- Yang, X.; Jin, H.; Cai, X.; Li, S.; Shen, Y. Structural and mechanistic insights into the activation of Stromal interaction molecule 1 (STIM1). Proc. Natl. Acad. Sci. USA 2012, 109, 5657–5662. [Google Scholar] [CrossRef]
- Muik, M.; Fahrner, M.; Derler, I.; Schindl, R.; Bergsmann, J.; Frischauf, I.; Groschner, K.; Romanin, C. A Cytosolic Homomerization and a Modulatory Domain within STIM1 C Terminus Determine Coupling to ORAI1 Channels. J. Biol. Chem. 2009, 284, 8421–8426. [Google Scholar] [CrossRef]
- Covington, E.D.; Wu, M.M.; Lewis, R.S. Essential role for the CRAC activation domain in store-dependent oligomerization of STIM1. Mol. Biol. Cell 2010, 21, 1897–1907. [Google Scholar] [CrossRef]
- Berridge, M.J.; Irvine, R.F. Inositol trisphosphate, a novel second messenger in cellular signal transduction. Nature 1984, 312, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Streb, H.; Irvine, R.F.; Berridge, M.J.; Schulz, I. Release of Ca2+ from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate. Nature 1983, 306, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Stathopulos, P.B.; Schindl, R.; Li, G.Y.; Romanin, C.; Ikura, M. Auto-inhibitory role of the EF-SAM domain of STIM proteins in store-operated calcium entry. Proc. Natl. Acad. Sci. USA 2011, 108, 1337–1342. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Wei, M.; He, L.; Liu, C.; Wu, B.; Zhang, S.L.; Jing, J.; Liang, X.; Senes, A.; Tan, P.; et al. Inside-out Ca(2+) signalling prompted by STIM1 conformational switch. Nat. Commun. 2015, 6, 7826. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, M.; Muik, M.; Schindl, R.; Butorac, C.; Stathopulos, P.; Zheng, L.; Jardin, I.; Ikura, M.; Romanin, C. A coiled-coil clamp controls both conformation and clustering of stromal interaction molecule 1 (STIM1). J. Biol. Chem. 2014, 289, 33231–33244. [Google Scholar] [CrossRef] [PubMed]
- Calloway, N.; Owens, T.; Corwith, K.; Rodgers, W.; Holowka, D.; Baird, B. Stimulated association of STIM1 and Orai1 is regulated by the balance of PtdIns(4,5)P(2) between distinct membrane pools. J. Cell Sci. 2011, 124, 2602–2610. [Google Scholar] [CrossRef]
- Walsh, C.M.; Chvanov, M.; Haynes, L.P.; Petersen, O.H.; Tepikin, A.V.; Burgoyne, R.D. Role of phosphoinositides in STIM1 dynamics and store-operated calcium entry. Biochem. J. 2009, 425, 159–168. [Google Scholar] [CrossRef]
- Huang, G.N.; Zeng, W.; Kim, J.Y.; Yuan, J.P.; Han, L.; Muallem, S.; Worley, P.F. STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels. Nat. Cell Biol. 2006, 8, 1003–1010. [Google Scholar] [CrossRef]
- McNally, B.A.; Somasundaram, A.; Jairaman, A.; Yamashita, M.; Prakriya, M. The C- and N-terminal STIM1 binding sites on Orai1 are required for both trapping and gating CRAC channels. J. Physiol. 2013, 591, 2833–2850. [Google Scholar] [CrossRef]
- Hou, X.; Pedi, L.; Diver, M.M.; Long, S.B. Crystal structure of the calcium release-activated calcium channel Orai. Science 2012, 338, 1308–1313. [Google Scholar] [CrossRef]
- Picard, C.; McCarl, C.A.; Papolos, A.; Khalil, S.; Luthy, K.; Hivroz, C.; LeDeist, F.; Rieux-Laucat, F.; Rechavi, G.; Rao, A.; et al. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N. Engl. J. Med. 2009, 360, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Byun, M.; Abhyankar, A.; Lelarge, V.; Plancoulaine, S.; Palanduz, A.; Telhan, L.; Boisson, B.; Picard, C.; Dewell, S.; Zhao, C.; et al. Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J. Exp. Med. 2010, 207, 2307–2312. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, S.; Rensing-Ehl, A.; Speckmann, C.; Bengsch, B.; Schmitt-Graeff, A.; Bondzio, I.; Maul-Pavicic, A.; Bass, T.; Vraetz, T.; Strahm, B.; et al. Antiviral and regulatory T cell immunity in a patient with stromal interaction molecule 1 deficiency. J. Immunol. 2012, 188, 1523–1533. [Google Scholar] [CrossRef] [PubMed]
- Schaballie, H.; Rodriguez, R.; Martin, E.; Moens, L.; Frans, G.; Lenoir, C.; Dutre, J.; Canioni, D.; Bossuyt, X.; Fischer, A.; et al. A novel hypomorphic mutation in STIM1 results in a late-onset immunodeficiency. J. Allergy Clin. Immunol. 2015, 136, 816–819 e4. [Google Scholar] [CrossRef]
- Maus, M.; Jairaman, A.; Stathopulos, P.B.; Muik, M.; Fahrner, M.; Weidinger, C.; Benson, M.; Fuchs, S.; Ehl, S.; Romanin, C.; et al. Missense mutation in immunodeficient patients shows the multifunctional roles of coiled-coil domain 3 (CC3) in STIM1 activation. Proc. Natl. Acad. Sci. USA 2015, 112, 6206–6211. [Google Scholar] [CrossRef]
- McCarl, C.A.; Picard, C.; Khalil, S.; Kawasaki, T.; Rother, J.; Papolos, A.; Kutok, J.; Hivroz, C.; Ledeist, F.; Plogmann, K.; et al. ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. J. Allergy Clin. Immunol. 2009, 124, 1311–1318 e7. [Google Scholar] [CrossRef]
- Chou, J.; Badran, Y.R.; Yee, C.S.K.; Bainter, W.; Ohsumi, T.K.; Al-Hammadi, S.; Pai, S.Y.; Feske, S.; Geha, R.S. A novel mutation in ORAI1 presenting with combined immunodeficiency and residual T-cell function. J. Allergy Clin. Immunol. 2015, 136, 479–482 e1. [Google Scholar] [CrossRef]
- Feske, S. CRAC channels and disease-From human CRAC channelopathies and animal models to novel drugs. Cell Calcium 2019, 80, 112–116. [Google Scholar] [CrossRef]
- Lacruz, R.S.; Feske, S. Diseases caused by mutations in ORAI1 and STIM1. Ann. N. Y. Acad. Sci. 2015, 1356, 45–79. [Google Scholar] [CrossRef]
- Misceo, D.; Holmgren, A.; Louch, W.E.; Holme, P.A.; Mizobuchi, M.; Morales, R.J.; De Paula, A.M.; Stray-Pedersen, A.; Lyle, R.; Dalhus, B.; et al. A dominant STIM1 mutation causes Stormorken syndrome. Hum. Mutat. 2014, 35, 556–564. [Google Scholar] [CrossRef]
- Bohm, J.; Chevessier, F.; Maues De Paula, A.; Koch, C.; Attarian, S.; Feger, C.; Hantai, D.; Laforet, P.; Ghorab, K.; Vallat, J.M.; et al. Constitutive activation of the calcium sensor STIM1 causes tubular-aggregate myopathy. Am. J. Hum. Genet. 2013, 92, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Bohm, J.; Chevessier, F.; Koch, C.; Peche, G.A.; Mora, M.; Morandi, L.; Pasanisi, B.; Moroni, I.; Tasca, G.; Fattori, F.; et al. Clinical, histological and genetic characterisation of patients with tubular aggregate myopathy caused by mutations in STIM1. J. Med. Genet. 2014, 51, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.C.; Rossius, M.; Zitzelsberger, M.; Vorgerd, M.; Muller-Felber, W.; Ertl-Wagner, B.; Zhang, Y.; Brinkmeier, H.; Senderek, J.; Schoser, B. 50 years to diagnosis: Autosomal dominant tubular aggregate myopathy caused by a novel STIM1 mutation. Neuromuscul. Disord. 2015, 25, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Nesin, V.; Wiley, G.; Kousi, M.; Ong, E.C.; Lehmann, T.; Nicholl, D.J.; Suri, M.; Shahrizaila, N.; Katsanis, N.; Gaffney, P.M.; et al. Activating mutations in STIM1 and ORAI1 cause overlapping syndromes of tubular myopathy and congenital miosis. Proc. Natl. Acad. Sci. USA 2014, 111, 4197–4202. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Noguchi, S.; Hara, Y.; Hayashi, Y.K.; Motomura, K.; Miyatake, S.; Murakami, N.; Tanaka, S.; Yamashita, S.; Kizu, R.; et al. Dominant mutations in ORAI1 cause tubular aggregate myopathy with hypocalcemia via constitutive activation of store-operated Ca(2)(+) channels. Hum. Mol. Genet. 2015, 24, 637–648. [Google Scholar] [CrossRef]
- Bohm, J.; Laporte, J. Gain-of-function mutations in STIM1 and ORAI1 causing tubular aggregate myopathy and Stormorken syndrome. Cell Calcium 2018, 76, 1–9. [Google Scholar] [CrossRef]
- Hoth, M.; Fanger, C.M.; Lewis, R.S. Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. J. Cell Biol. 1997, 137, 633–648. [Google Scholar] [CrossRef]
- Hoth, M.; Button, D.C.; Lewis, R.S. Mitochondrial control of calcium-channel gating: A mechanism for sustained signaling and transcriptional activation in T lymphocytes. Proc. Natl. Acad. Sci. USA 2000, 97, 10607–10612. [Google Scholar] [CrossRef]
- Quintana, A.; Schwarz, E.C.; Schwindling, C.; Lipp, P.; Kaestner, L.; Hoth, M. Sustained activity of calcium release-activated calcium channels requires translocation of mitochondria to the plasma membrane. J. Biol. Chem. 2006, 281, 40302–40309. [Google Scholar] [CrossRef]
- Naghdi, S.; Waldeck-Weiermair, M.; Fertschai, I.; Poteser, M.; Graier, W.F.; Malli, R. Mitochondrial Ca2+ uptake and not mitochondrial motility is required for STIM1-Orai1-dependent store-operated Ca2+ entry. J. Cell Sci. 2010, 123, 2553–2564. [Google Scholar] [CrossRef]
- Gilabert, J.A.; Parekh, A.B. Respiring mitochondria determine the pattern of activation and inactivation of the store-operated Ca(2+) current I(CRAC). EMBO J. 2000, 19, 6401–6407. [Google Scholar] [CrossRef] [PubMed]
- Samanta, K.; Douglas, S.; Parekh, A.B. Mitochondrial calcium uniporter MCU supports cytoplasmic Ca2+ oscillations, store-operated Ca2+ entry and Ca2+-dependent gene expression in response to receptor stimulation. PLoS ONE 2014, 9, e101188. [Google Scholar] [CrossRef] [PubMed]
- Kostic, M.; Sekler, I. Functional properties and mode of regulation of the mitochondrial Na(+)/Ca(2+) exchanger, NCLX. Semin Cell Dev. Biol. 2019, 94, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Ben-Kasus Nissim, T.; Zhang, X.; Elazar, A.; Roy, S.; Stolwijk, J.A.; Zhou, Y.; Motiani, R.K.; Gueguinou, M.; Hempel, N.; Hershfinkel, M.; et al. Mitochondria control store-operated Ca(2+) entry through Na(+) and redox signals. EMBO J. 2017, 36, 797–815. [Google Scholar] [CrossRef] [PubMed]
- Villalobos, C.; Gutierrez, L.G.; Hernandez-Morales, M.; Del Bosque, D.; Nunez, L. Mitochondrial control of store-operated Ca(2+) channels in cancer: Pharmacological implications. Pharmacol. Res. 2018, 135, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Pinton, P. The mitochondrial calcium uniporter complex: Molecular components, structure and physiopathological implications. J. Physiol. 2014, 592, 829–839. [Google Scholar] [CrossRef]
- Giacomello, M.; Drago, I.; Pizzo, P.; Pozzan, T. Mitochondrial Ca2+ as a key regulator of cell life and death. Cell Death Differ. 2007, 14, 1267–1274. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; De, S. Mitochondrial VDAC, the Na(+)/Ca(2+) Exchanger, and the Ca(2+) Uniporter in Ca(2+) Dynamics and Signaling. Adv. Exp. Med. Biol. 2017, 981, 323–347. [Google Scholar]
- Mallilankaraman, K.; Doonan, P.; Cardenas, C.; Chandramoorthy, H.C.; Muller, M.; Miller, R.; Hoffman, N.E.; Gandhirajan, R.K.; Molgo, J.; Birnbaum, M.J.; et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 2012, 151, 630–644. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pozzan, T. Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef]
- Baradaran, R.; Wang, C.; Siliciano, A.F.; Long, S.B. Cryo-EM structures of fungal and metazoan mitochondrial calcium uniporters. Nature 2018, 559, 580–584. [Google Scholar] [CrossRef]
- Fan, C.; Fan, M.; Orlando, B.J.; Fastman, N.M.; Zhang, J.; Xu, Y.; Chambers, M.G.; Xu, X.; Perry, K.; Liao, M.; et al. X-ray and cryo-EM structures of the mitochondrial calcium uniporter. Nature 2018, 559, 575–579. [Google Scholar] [CrossRef]
- Nguyen, N.X.; Armache, J.P.; Lee, C.; Yang, Y.; Zeng, W.; Mootha, V.K.; Cheng, Y.; Bai, X.C.; Jiang, Y. Cryo-EM structure of a fungal mitochondrial calcium uniporter. Nature 2018, 559, 570–574. [Google Scholar] [CrossRef]
- Wang, Y.; Nguyen, N.X.; She, J.; Zeng, W.; Yang, Y.; Bai, X.C.; Jiang, Y. Structural Mechanism of EMRE-Dependent Gating of the Human Mitochondrial Calcium Uniporter. Cell 2019, 177, 1252–1261 e13. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Wu, M.; Yin, Y.; Herzik, M.A., Jr.; Lander, G.C.; Lee, S.Y. Cryo-EM structure of a mitochondrial calcium uniporter. Science 2018, 361, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Perocchi, F.; Gohil, V.M.; Girgis, H.S.; Bao, X.R.; McCombs, J.E.; Palmer, A.E.; Mootha, V.K. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature 2010, 467, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Plovanich, M.; Bogorad, R.L.; Sancak, Y.; Kamer, K.J.; Strittmatter, L.; Li, A.A.; Girgis, H.S.; Kuchimanchi, S.; De Groot, J.; Speciner, L.; et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE 2013, 8, e55785. [Google Scholar] [CrossRef]
- Sancak, Y.; Markhard, A.L.; Kitami, T.; Kovacs-Bogdan, E.; Kamer, K.J.; Udeshi, N.D.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 2013, 342, 1379–1382. [Google Scholar] [CrossRef]
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabo, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376. [Google Scholar] [CrossRef]
- Mallilankaraman, K.; Cardenas, C.; Doonan, P.J.; Chandramoorthy, H.C.; Irrinki, K.M.; Golenar, T.; Csordas, G.; Madireddi, P.; Yang, J.; Muller, M.; et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 2012, 14, 1336–1343. [Google Scholar] [CrossRef]
- Martell, J.D.; Deerinck, T.J.; Sancak, Y.; Poulos, T.L.; Mootha, V.K.; Sosinsky, G.E.; Ellisman, M.H.; Ting, A.Y. Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat. Biotechnol. 2012, 30, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Oxenoid, K.; Dong, Y.; Cao, C.; Cui, T.; Sancak, Y.; Markhard, A.L.; Grabarek, Z.; Kong, L.; Liu, Z.; Ouyang, B.; et al. Architecture of the mitochondrial calcium uniporter. Nature 2016, 533, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Min, C.K.; Kim, T.G.; Song, H.K.; Lim, Y.; Kim, D.; Shin, K.; Kang, M.; Kang, J.Y.; Youn, H.S.; et al. Structure and function of the N-terminal domain of the human mitochondrial calcium uniporter. EMBO Rep. 2015, 16, 1318–1333. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Shanmughapriya, S.; Mok, M.C.Y.; Dong, Z.; Tomar, D.; Carvalho, E.; Rajan, S.; Junop, M.S.; Madesh, M.; Stathopulos, P.B. Structural Insights into Mitochondrial Calcium Uniporter Regulation by Divalent Cations. Cell Chem. Biol. 2016, 23, 1157–1169. [Google Scholar] [CrossRef]
- Dong, Z.; Shanmughapriya, S.; Tomar, D.; Siddiqui, N.; Lynch, S.; Nemani, N.; Breves, S.L.; Zhang, X.; Tripathi, A.; Palaniappan, P.; et al. Mitochondrial Ca(2+) Uniporter Is a Mitochondrial Luminal Redox Sensor that Augments MCU Channel Activity. Mol. Cell 2017, 65, 1014–1028 e7. [Google Scholar] [CrossRef]
- Vais, H.; Mallilankaraman, K.; Mak, D.D.; Hoff, H.; Payne, R.; Tanis, J.E.; Foskett, J.K. EMRE Is a Matrix Ca(2+) Sensor that Governs Gatekeeping of the Mitochondrial Ca(2+) Uniporter. Cell Rep. 2016, 14, 403–410. [Google Scholar] [CrossRef]
- Tsai, M.F.; Phillips, C.B.; Ranaghan, M.; Tsai, C.W.; Wu, Y.; Willliams, C.; Miller, C. Dual functions of a small regulatory subunit in the mitochondrial calcium uniporter complex. Elife 2016, 5, e15545. [Google Scholar] [CrossRef]
- Hung, V.; Zou, P.; Rhee, H.W.; Udeshi, N.D.; Cracan, V.; Svinkina, T.; Carr, S.A.; Mootha, V.K.; Ting, A.Y. Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol. Cell 2014, 55, 332–341. [Google Scholar] [CrossRef]
- Csordas, G.; Golenar, T.; Seifert, E.L.; Kamer, K.J.; Sancak, Y.; Perocchi, F.; Moffat, C.; Weaver, D.; de la Fuente Perez, S.; Bogorad, R.; et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2)(+) uniporter. Cell Metab. 2013, 17, 976–987. [Google Scholar] [CrossRef]
- Patron, M.; Checchetto, V.; Raffaello, A.; Teardo, E.; Vecellio Reane, D.; Mantoan, M.; Granatiero, V.; Szabo, I.; De Stefani, D.; Rizzuto, R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 2014, 53, 726–737. [Google Scholar] [CrossRef]
- Patron, M.; Granatiero, V.; Espino, J.; Rizzuto, R.; De Stefani, D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 2019, 26, 179–195. [Google Scholar] [CrossRef] [PubMed]
- Kamer, K.J.; Grabarek, Z.; Mootha, V.K. High-affinity cooperative Ca(2+) binding by MICU1-MICU2 serves as an on-off switch for the uniporter. EMBO Rep. 2017, 18, 1397–1411. [Google Scholar] [CrossRef] [PubMed]
- Kamer, K.J.; Jiang, W.; Kaushik, V.K.; Mootha, V.K.; Grabarek, Z. Crystal structure of MICU2 and comparison with MICU1 reveal insights into the uniporter gating mechanism. Proc. Natl. Acad. Sci. USA 2019, 116, 3546–3555. [Google Scholar] [CrossRef] [PubMed]
- Kamer, K.J.; Mootha, V.K. MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter. EMBO Rep. 2014, 15, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Wang, M.; Wang, J.; Nie, Z.; Wu, G.; Yang, X.; Shen, Y. Dimerization of MICU Proteins Controls Ca(2+) Influx through the Mitochondrial Ca(2+) Uniporter. Cell Rep. 2019, 26, 1203–1212 e4. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, Y.; Park, T.; Kang, J.Y.; Mun, S.A.; Jin, M.; Yang, J.; Eom, S.H. Structure of the MICU1-MICU2 heterodimer provides insights into the gatekeeping threshold shift. IUCrJ 2020, 7, 355–365. [Google Scholar] [CrossRef]
- Wang, L.; Yang, X.; Li, S.; Wang, Z.; Liu, Y.; Feng, J.; Zhu, Y.; Shen, Y. Structural and mechanistic insights into MICU1 regulation of mitochondrial calcium uptake. EMBO J. 2014, 33, 594–604. [Google Scholar] [CrossRef]
- Wu, W.; Shen, Q.; Lei, Z.; Qiu, Z.; Li, D.; Pei, H.; Zheng, J.; Jia, Z. The crystal structure of MICU2 provides insight into Ca(2+) binding and MICU1-MICU2 heterodimer formation. EMBO Rep. 2019, 20, e47488. [Google Scholar] [CrossRef]
- Zhuo, W.; Zhou, H.; Guo, R.; Yi, J.; Sui, Y.; Zhang, L.; Zeng, W.; Wang, P.; Yang, M. Structure of intact human MCU supercomplex with the auxiliary MICU subunits. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lambert, J.P.; Luongo, T.S.; Tomar, D.; Jadiya, P.; Gao, E.; Zhang, X.; Lucchese, A.M.; Kolmetzky, D.W.; Shah, N.S.; Elrod, J.W. MCUB Regulates the Molecular Composition of the Mitochondrial Calcium Uniporter Channel to Limit Mitochondrial Calcium Overload During Stress. Circulation 2019, 140, 1720–1733. [Google Scholar] [CrossRef]
- Tomar, D.; Dong, Z.; Shanmughapriya, S.; Koch, D.A.; Thomas, T.; Hoffman, N.E.; Timbalia, S.A.; Goldman, S.J.; Breves, S.L.; Corbally, D.P.; et al. MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics. Cell Rep. 2016, 15, 1673–1685. [Google Scholar] [CrossRef] [PubMed]
- Vais, H.; Tanis, J.E.; Muller, M.; Payne, R.; Mallilankaraman, K.; Foskett, J.K. MCUR1, CCDC90A, Is a Regulator of the Mitochondrial Calcium Uniporter. Cell Metab. 2015, 22, 533–535. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Sileikyte, J.; Forte, M. The Mitochondrial Permeability Transition in Mitochondrial Disorders. Oxid Med. Cell Longev. 2019, 2019, 3403075. [Google Scholar] [CrossRef]
- Kwong, J.Q.; Molkentin, J.D. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab. 2015, 21, 206–214. [Google Scholar] [CrossRef]
- Zoratti, M.; Szabo, I. The mitochondrial permeability transition. Biochim. Biophys. Acta 1995, 1241, 139–176. [Google Scholar] [CrossRef]
- Bhosale, G.; Sharpe, J.A.; Koh, A.; Kouli, A.; Szabadkai, G.; Duchen, M.R. Pathological consequences of MICU1 mutations on mitochondrial calcium signalling and bioenergetics. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1009–1017. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Richardson, A.P. The mitochondrial permeability transition: A current perspective on its identity and role in ischaemia/reperfusion injury. J. Mol. Cell Cardiol. 2015, 78, 129–141. [Google Scholar] [CrossRef]
- Liao, Y.; Dong, Y.; Cheng, J. The Function of the Mitochondrial Calcium Uniporter in Neurodegenerative Disorders. Int. J. Mol. Sci. 2017, 18, 248. [Google Scholar] [CrossRef]
- Tarasov, A.I.; Semplici, F.; Ravier, M.A.; Bellomo, E.A.; Pullen, T.J.; Gilon, P.; Sekler, I.; Rizzuto, R.; Rutter, G.A. The mitochondrial Ca2+ uniporter MCU is essential for glucose-induced ATP increases in pancreatic beta-cells. PLoS ONE 2012, 7, e39722. [Google Scholar] [CrossRef]
- Vultur, A.; Gibhardt, C.S.; Stanisz, H.; Bogeski, I. The role of the mitochondrial calcium uniporter (MCU) complex in cancer. Pflugers Arch. 2018, 470, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Debattisti, V.; Horn, A.; Singh, R.; Seifert, E.L.; Hogarth, M.W.; Mazala, D.A.; Huang, K.T.; Horvath, R.; Jaiswal, J.K.; Hajnoczky, G. Dysregulation of Mitochondrial Ca(2+) Uptake and Sarcolemma Repair Underlie Muscle Weakness and Wasting in Patients and Mice Lacking MICU1. Cell Rep. 2019, 29, 1274–1286. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.V.; Szabadkai, G.; Sharpe, J.A.; Parry, D.A.; Torelli, S.; Childs, A.M.; Kriek, M.; Phadke, R.; Johnson, C.A.; Roberts, N.Y.; et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat. Genet. 2014, 46, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Smith, D.; Kamer, K.J.; Griffin, H.; Childs, A.M.; Pysden, K.; Titov, D.; Duff, J.; Pyle, A.; Taylor, R.W.; Yu-Wai-Man, P.; et al. Homozygous deletion in MICU1 presenting with fatigue and lethargy in childhood. Neurol. Genet. 2016, 2, e59. [Google Scholar] [CrossRef] [PubMed]
- Musa, S.; Eyaid, W.; Kamer, K.; Ali, R.; Al-Mureikhi, M.; Shahbeck, N.; Al Mesaifri, F.; Makhseed, N.; Mohamed, Z.; AlShehhi, W.A.; et al. A Middle Eastern Founder Mutation Expands the Genotypic and Phenotypic Spectrum of Mitochondrial MICU1 Deficiency: A Report of 13 Patients. JIMD Rep. 2019, 43, 79–83. [Google Scholar]
- Shamseldin, H.E.; Alasmari, A.; Salih, M.A.; Samman, M.M.; Mian, S.A.; Alshidi, T.; Ibrahim, N.; Hashem, M.; Faqeih, E.; Al-Mohanna, F.; et al. A null mutation in MICU2 causes abnormal mitochondrial calcium homeostasis and a severe neurodevelopmental disorder. Brain 2017, 140, 2806–2813. [Google Scholar] [CrossRef]
- Gordienko, D.V.; Greenwood, I.A.; Bolton, T.B. Direct visualization of sarcoplasmic reticulum regions discharging Ca(2+)sparks in vascular myocytes. Cell Calcium 2001, 29, 13–28. [Google Scholar] [CrossRef]
- Hajnoczky, G.; Hager, R.; Thomas, A.P. Mitochondria suppress local feedback activation of inositol 1,4, 5-trisphosphate receptors by Ca2+. J. Biol. Chem. 1999, 274, 14157–14162. [Google Scholar] [CrossRef]
- Marchant, J.S.; Ramos, V.; Parker, I. Structural and functional relationships between Ca2+ puffs and mitochondria in Xenopus oocytes. Am. J. Physiol. Cell Physiol. 2002, 282, C1374–C1386. [Google Scholar] [CrossRef]
- Pacher, P.; Thomas, A.P.; Hajnoczky, G. Ca2+ marks: Miniature calcium signals in single mitochondria driven by ryanodine receptors. Proc. Natl. Acad. Sci. USA 2002, 99, 2380–2385. [Google Scholar] [CrossRef]
- Antony, A.N.; Paillard, M.; Moffat, C.; Juskeviciute, E.; Correnti, J.; Bolon, B.; Rubin, E.; Csordas, G.; Seifert, E.L.; Hoek, J.B.; et al. MICU1 regulation of mitochondrial Ca(2+) uptake dictates survival and tissue regeneration. Nat. Commun. 2016, 7, 10955. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.; Nowikovsky, K. LETM1: Essential for Mitochondrial Biology and Cation Homeostasis? Trends Biochem. Sci. 2019, 44, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Doonan, P.J.; Chandramoorthy, H.C.; Hoffman, N.E.; Zhang, X.; Cardenas, C.; Shanmughapriya, S.; Rajan, S.; Vallem, S.; Chen, X.; Foskett, J.K.; et al. LETM1-dependent mitochondrial Ca2+ flux modulates cellular bioenergetics and proliferation. FASEB J. 2014, 28, 4936–4949. [Google Scholar] [CrossRef]
- Jiang, D.; Zhao, L.; Clish, C.B.; Clapham, D.E. Letm1, the mitochondrial Ca2+/H+ antiporter, is essential for normal glucose metabolism and alters brain function in Wolf-Hirschhorn syndrome. Proc. Natl. Acad. Sci. USA 2013, 110, E2249–E2254. [Google Scholar] [CrossRef] [PubMed]
- Nowikovsky, K.; Reipert, S.; Devenish, R.J.; Schweyen, R.J. Mdm38 protein depletion causes loss of mitochondrial K+/H+ exchange activity, osmotic swelling and mitophagy. Cell Death Differ. 2007, 14, 1647–1656. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Fu, Z.; Ji, Y.; Guan, X.; Guo, S.; Ding, Z.; Yang, X.; Cong, Y.; Shen, Y. Leucine zipper-EF-hand containing transmembrane protein 1 (LETM1) forms a Ca(2+)/H(+) antiporter. Sci. Rep. 2016, 6, 34174. [Google Scholar] [CrossRef] [PubMed]
- Tamai, S.; Iida, H.; Yokota, S.; Sayano, T.; Kiguchiya, S.; Ishihara, N.; Hayashi, J.; Mihara, K.; Oka, T. Characterization of the mitochondrial protein LETM1, which maintains the mitochondrial tubular shapes and interacts with the AAA-ATPase BCS1L. J. Cell Sci. 2008, 121, 2588–2600. [Google Scholar] [CrossRef]
- Tsai, M.F.; Jiang, D.; Zhao, L.; Clapham, D.; Miller, C. Functional reconstitution of the mitochondrial Ca2+/H+ antiporter Letm1. J. Gen. Physiol. 2014, 143, 67–73. [Google Scholar] [CrossRef]
- Endele, S.; Fuhry, M.; Pak, S.J.; Zabel, B.U.; Winterpacht, A. LETM1, a novel gene encoding a putative EF-hand Ca(2+)-binding protein, flanks the Wolf-Hirschhorn syndrome (WHS) critical region and is deleted in most WHS patients. Genomics 1999, 60, 218–225. [Google Scholar] [CrossRef]
- Rutherford, E.L.; Lowery, L.A. Exploring the developmental mechanisms underlying Wolf-Hirschhorn Syndrome: Evidence for defects in neural crest cell migration. Dev. Biol. 2016, 420, 1–10. [Google Scholar] [CrossRef][Green Version]
- Blomen, V.A.; Majek, P.; Jae, L.T.; Bigenzahn, J.W.; Nieuwenhuis, J.; Staring, J.; Sacco, R.; van Diemen, F.R.; Olk, N.; Stukalov, A.; et al. Gene essentiality and synthetic lethality in haploid human cells. Science 2015, 350, 1092–1096. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Birsoy, K.; Hughes, N.W.; Krupczak, K.M.; Post, Y.; Wei, J.J.; Lander, E.S.; Sabatini, D.M. Identification and characterization of essential genes in the human genome. Science 2015, 350, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Collins, T.J.; Lipp, P.; Berridge, M.J.; Bootman, M.D. Mitochondrial Ca(2+) uptake depends on the spatial and temporal profile of cytosolic Ca(2+) signals. J. Biol. Chem. 2001, 276, 26411–26420. [Google Scholar] [CrossRef] [PubMed]
- Santo-Domingo, J.; Demaurex, N. Calcium uptake mechanisms of mitochondria. Biochim. Biophys. Acta 2010, 1797, 907–912. [Google Scholar] [CrossRef]
- Santo-Domingo, J.; Demaurex, N. Perspectives on: SGP symposium on mitochondrial physiology and medicine: The renaissance of mitochondrial pH. J. Gen. Physiol. 2012, 139, 415–423. [Google Scholar] [CrossRef]
- Aral, C.; Demirkesen, S.; Bircan, R.; Yasar Sirin, D. Melatonin reverses the oxidative stress and mitochondrial dysfunction caused by LETM1 silencing. Cell Biol. Int. 2020, 44, 795–807. [Google Scholar] [CrossRef]
- Huang, E.; Qu, D.; Huang, T.; Rizzi, N.; Boonying, W.; Krolak, D.; Ciana, P.; Woulfe, J.; Klein, C.; Slack, R.S.; et al. PINK1-mediated phosphorylation of LETM1 regulates mitochondrial calcium transport and protects neurons against mitochondrial stress. Nat. Commun. 2017, 8, 1399. [Google Scholar] [CrossRef]
- Yoo, C.M.; Rhee, H.W. APEX, a Master Key To Resolve Membrane Topology in Live Cells. Biochemistry 2020, 59, 250–259. [Google Scholar] [CrossRef]
- Lupo, D.; Vollmer, C.; Deckers, M.; Mick, D.U.; Tews, I.; Sinning, I.; Rehling, P. Mdm38 is a 14-3-3-like receptor and associates with the protein synthesis machinery at the inner mitochondrial membrane. Traffic 2011, 12, 1457–1466. [Google Scholar] [CrossRef]
- Nakamura, S.; Matsui, A.; Akabane, S.; Tamura, Y.; Hatano, A.; Miyano, Y.; Omote, H.; Kajikawa, M.; Maenaka, K.; Moriyama, Y.; et al. The mitochondrial inner membrane protein LETM1 modulates cristae organization through its LETM domain. Commun. Biol. 2020, 3, 99. [Google Scholar] [CrossRef]
- Huang, B.; Zhang, J.; Zhang, X.; Huang, C.; Hu, G.; Li, S.; Xie, T.; Liu, M.; Xu, Y. Suppression of LETM1 by siRNA inhibits cell proliferation and invasion of bladder cancer cells. Oncol. Rep. 2017, 38, 2935–2940. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.; Li, Y.; Kim, S.J.; Byun, H.S.; Huang, S.M.; Hwang, S.K.; Yang, K.J.; Park, K.A.; Won, M.; Hong, J.; et al. Association of LETM1 and MRPL36 contributes to the regulation of mitochondrial ATP production and necrotic cell death. Cancer Res. 2009, 69, 3397–3404. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.; Li, Y.; Kim, S.J.; Sohn, K.C.; Yang, K.J.; Park, K.A.; Byun, H.S.; Won, M.; Hong, J.; Hur, G.M.; et al. Regulation of OPA1-mediated mitochondrial fusion by leucine zipper/EF-hand-containing transmembrane protein-1 plays a role in apoptosis. Cell Signal. 2009, 21, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Ni, W.; Cui, C.; Qi, W.; Piao, L.; Xuan, Y. Identification of LETM1 as a marker of cancer stem-like cells and predictor of poor prognosis in esophageal squamous cell carcinoma. Hum. Pathol. 2018, 81, 148–156. [Google Scholar] [CrossRef]
- Hou, X.; Burstein, S.R.; Long, S.B. Structures reveal opening of the store-operated calcium channel Orai. Elife 2018, 7, e36758. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, G.; Yu, Y.; Chen, X.; Ji, R.; Lu, J.; Li, X.; Zhang, X.; Yang, X.; Shen, Y. Molecular understanding of calcium permeation through the open Orai channel. PLoS Biol. 2019, 17, e3000096. [Google Scholar] [CrossRef]
- Yuan, Y.; Cao, C.; Wen, M.; Li, M.; Dong, Y.; Wu, L.; Wu, J.; Cui, T.; Li, D.; Chou, J.J.; et al. Structural Characterization of the N-Terminal Domain of the Dictyostelium discoideum Mitochondrial Calcium Uniporter. ACS Omega. 2020, 5, 6452–6460. [Google Scholar] [CrossRef]
- Adlakha, J.; Karamichali, I.; Sangwallek, J.; Deiss, S.; Bar, K.; Coles, M.; Hartmann, M.D.; Lupas, A.N.; Hernandez Alvarez, B. Characterization of MCU-Binding Proteins MCUR1 and CCDC90B-Representatives of a Protein Family Conserved in Prokaryotes and Eukaryotic Organelles. Structure 2019, 27, 464–475 e6. [Google Scholar] [CrossRef]
- Bosanac, I.; Alattia, J.R.; Mal, T.K.; Chan, J.; Talarico, S.; Tong, F.K.; Tong, K.I.; Yoshikawa, F.; Furuichi, T.; Iwai, M.; et al. Structure of the inositol 1,4,5-trisphosphate receptor binding core in complex with its ligand. Nature 2002, 420, 696–700. [Google Scholar] [CrossRef]
- Bosanac, I.; Yamazaki, H.; Matsu-Ura, T.; Michikawa, T.; Mikoshiba, K.; Ikura, M. Crystal structure of the ligand binding suppressor domain of type 1 inositol 1,4,5-trisphosphate receptor. Mol. Cell 2005, 17, 193–203. [Google Scholar] [CrossRef]
- Amador, F.J.; Kimlicka, L.; Stathopulos, P.B.; Gasmi-Seabrook, G.M.; Maclennan, D.H.; Van Petegem, F.; Ikura, M. Type 2 ryanodine receptor domain A contains a unique and dynamic alpha-helix that transitions to a beta-strand in a mutant linked with a heritable cardiomyopathy. J. Mol. Biol. 2013, 425, 4034–4046. [Google Scholar] [CrossRef] [PubMed]
- Amador, F.J.; Liu, S.; Ishiyama, N.; Plevin, M.J.; Wilson, A.; MacLennan, D.H.; Ikura, M. Crystal structure of type I ryanodine receptor amino-terminal beta-trefoil domain reveals a disease-associated mutation “hot spot” loop. Proc. Natl. Acad. Sci. USA 2009, 106, 11040–11044. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.D.; Velamakanni, S.; Ishiyama, N.; Stathopulos, P.B.; Rossi, A.M.; Khan, S.A.; Dale, P.; Li, C.; Ames, J.B.; Ikura, M.; et al. Structural and functional conservation of key domains in InsP3 and ryanodine receptors. Nature 2012, 483, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Tung, C.C.; Lobo, P.A.; Kimlicka, L.; Van Petegem, F. The amino-terminal disease hotspot of ryanodine receptors forms a cytoplasmic vestibule. Nature 2010, 468, 585–588. [Google Scholar] [CrossRef] [PubMed]
- des Georges, A.; Clarke, O.B.; Zalk, R.; Yuan, Q.; Condon, K.J.; Grassucci, R.A.; Hendrickson, W.A.; Marks, A.R.; Frank, J. Structural Basis for Gating and Activation of RyR1. Cell 2016, 167, 145–157 e17. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Shen, H.; Wu, J.; Guo, W.; Pan, X.; Wang, R.; Chen, S.R.; Yan, N. Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science 2016, 354, aah5324. [Google Scholar] [CrossRef]
- Fan, G.; Baker, M.L.; Wang, Z.; Baker, M.R.; Sinyagovskiy, P.A.; Chiu, W.; Ludtke, S.J.; Serysheva, I.I. Gating machinery of InsP3R channels revealed by electron cryomicroscopy. Nature 2015, 527, 336–341. [Google Scholar] [CrossRef]
- Liao, J.; Li, H.; Zeng, W.; Sauer, D.B.; Belmares, R.; Jiang, Y. Structural insight into the ion-exchange mechanism of the sodium/calcium exchanger. Science 2012, 335, 686–690. [Google Scholar] [CrossRef]
- Toyoshima, C.; Nomura, H.; Tsuda, T. Lumenal gating mechanism revealed in calcium pump crystal structures with phosphate analogues. Nature 2004, 432, 361–368. [Google Scholar] [CrossRef]
- Tsunekawa, N.; Ogawa, H.; Tsueda, J.; Akiba, T.; Toyoshima, C. Mechanism of the E2 to E1 transition in Ca(2+) pump revealed by crystal structures of gating residue mutants. Proc. Natl. Acad. Sci. USA 2018, 115, 12722–12727. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, G.; Wu, J.; Wu, Q.; Gao, S.; Yan, Z.; Lei, J.; Yan, N. Molecular Basis for Ligand Modulation of a Mammalian Voltage-Gated Ca(2+) Channel. Cell 2019, 177, 1495–1506 e12. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Gamal El-Din, T.M.; Swanson, T.M.; Pryde, D.C.; Scheuer, T.; Zheng, N.; Catterall, W.A. Structural basis for inhibition of a voltage-gated Ca(2+) channel by Ca(2+) antagonist drugs. Nature 2016, 537, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Imbrici, P.; Nicolotti, O.; Leonetti, F.; Conte, D.; Liantonio, A. Ion Channels in Drug Discovery and Safety Pharmacology. Methods Mol. Biol. 2018, 1800, 313–326. [Google Scholar] [PubMed]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Wulff, H.; Christophersen, P.; Colussi, P.; Chandy, K.G.; Yarov-Yarovoy, V. Antibodies and venom peptides: New modalities for ion channels. Nat. Rev. Drug Discov. 2019, 18, 339–357. [Google Scholar] [CrossRef]
- Faulds, D.; Goa, K.L.; Benfield, P. Cyclosporin. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in immunoregulatory disorders. Drugs 1993, 45, 953–1040. [Google Scholar] [CrossRef]
- Flores, C.; Fouquet, G.; Moura, I.C.; Maciel, T.T.; Hermine, O. Lessons to Learn From Low-Dose Cyclosporin-A: A New Approach for Unexpected Clinical Applications. Front. Immunol. 2019, 10, 588. [Google Scholar] [CrossRef]
- Garcia-Rivas Gde, J.; Carvajal, K.; Correa, F.; Zazueta, C. Ru360, a specific mitochondrial calcium uptake inhibitor, improves cardiac post-ischaemic functional recovery in rats in vivo. Br. J. Pharmacol. 2006, 149, 829–837. [Google Scholar] [CrossRef]
- Zhang, S.Z.; Gao, Q.; Cao, C.M.; Bruce, I.C.; Xia, Q. Involvement of the mitochondrial calcium uniporter in cardioprotection by ischemic preconditioning. Life Sci. 2006, 78, 738–745. [Google Scholar] [CrossRef]
- de Jesus Garcia-Rivas, G.; Guerrero-Hernandez, A.; Guerrero-Serna, G.; Rodriguez-Zavala, J.S.; Zazueta, C. Inhibition of the mitochondrial calcium uniporter by the oxo-bridged dinuclear ruthenium amine complex (Ru360) prevents from irreversible injury in postischemic rat heart. FEBS J. 2005, 272, 3477–3488. [Google Scholar] [CrossRef]
- Woods, J.J.; Nemani, N.; Shanmughapriya, S.; Kumar, A.; Zhang, M.; Nathan, S.R.; Thomas, M.; Carvalho, E.; Ramachandran, K.; Srikantan, S.; et al. A Selective and Cell-Permeable Mitochondrial Calcium Uniporter (MCU) Inhibitor Preserves Mitochondrial Bioenergetics after Hypoxia/Reoxygenation Injury. ACS Cent. Sci. 2019, 5, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Arduino, D.M.; Wettmarshausen, J.; Vais, H.; Navas-Navarro, P.; Cheng, Y.; Leimpek, A.; Ma, Z.; Delrio-Lorenzo, A.; Giordano, A.; Garcia-Perez, C.; et al. Systematic Identification of MCU Modulators by Orthogonal Interspecies Chemical Screening. Mol. Cell 2017, 67, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Kon, N.; Murakoshi, M.; Isobe, A.; Kagechika, K.; Miyoshi, N.; Nagayama, T. DS16570511 is a small-molecule inhibitor of the mitochondrial calcium uniporter. Cell Death Discov. 2017, 3, 17045. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, G.; Vallese, F.; Jourde, B.; Bergsdorf, C.; Sturlese, M.; De Mario, A.; Techer-Etienne, V.; Haasen, D.; Oberhauser, B.; Schleeger, S.; et al. A High-Throughput Screening Identifies MICU1 Targeting Compounds. Cell Rep. 2020, 30, 2321–2331 e6. [Google Scholar] [CrossRef] [PubMed]
- Goto, J.; Suzuki, A.Z.; Ozaki, S.; Matsumoto, N.; Nakamura, T.; Ebisui, E.; Fleig, A.; Penner, R.; Mikoshiba, K. Two novel 2-aminoethyl diphenylborinate (2-APB) analogues differentially activate and inhibit store-operated Ca(2+) entry via STIM proteins. Cell Calcium 2010, 47, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.D.; Srikanth, S.; Tan, Y.V.; Yee, M.K.; Jew, M.; Damoiseaux, R.; Jung, M.E.; Shimizu, S.; An, D.S.; Ribalet, B.; et al. Calcium signaling via Orai1 is essential for induction of the nuclear orphan receptor pathway to drive Th17 differentiation. J. Immunol. 2014, 192, 110–122. [Google Scholar] [CrossRef]
- Stauderman, K.A. CRAC channels as targets for drug discovery and development. Cell Calcium 2018, 74, 147–159. [Google Scholar] [CrossRef]
- Zhang, H.Z.; Xu, X.L.; Chen, H.Y.; Ali, S.; Wang, D.; Yu, J.W.; Xu, T.; Nan, F.J. Discovery and structural optimization of 1-phenyl-3-(1-phenylethyl)urea derivatives as novel inhibitors of CRAC channel. Acta Pharmacol. Sin. 2015, 36, 1137–1144. [Google Scholar] [CrossRef][Green Version]
- Sadaghiani, A.M.; Lee, S.M.; Odegaard, J.I.; Leveson-Gower, D.B.; McPherson, O.M.; Novick, P.; Kim, M.R.; Koehler, A.N.; Negrin, R.; Dolmetsch, R.E.; et al. Identification of Orai1 channel inhibitors by using minimal functional domains to screen small molecule microarrays. Chem. Biol. 2014, 21, 1278–1292. [Google Scholar] [CrossRef]
- Azimi, I.; Flanagan, J.U.; Stevenson, R.J.; Inserra, M.; Vetter, I.; Monteith, G.R.; Denny, W.A. Evaluation of known and novel inhibitors of Orai1-mediated store operated Ca(2+) entry in MDA-MB-231 breast cancer cells using a Fluorescence Imaging Plate Reader assay. Bioorg. Med. Chem. 2017, 25, 440–449. [Google Scholar] [CrossRef]
- Rahman, S.; Rahman, T. Unveiling some FDA-approved drugs as inhibitors of the store-operated Ca(2+) entry pathway. Sci. Rep. 2017, 7, 12881. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15--Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Andricopulo, A.D.; Montanari, C.A. Structure-activity relationships for the design of small-molecule inhibitors. Mini. Rev. Med. Chem. 2005, 5, 585–593. [Google Scholar] [CrossRef]
- Gruber, S.J.; Cornea, R.L.; Li, J.; Peterson, K.C.; Schaaf, T.M.; Gillispie, G.D.; Dahl, R.; Zsebo, K.M.; Robia, S.L.; Thomas, D.D. Discovery of enzyme modulators via high-throughput time-resolved FRET in living cells. J. Biomol. Screen. 2014, 19, 215–222. [Google Scholar] [CrossRef]
- Schaaf, T.M.; Peterson, K.C.; Grant, B.D.; Bawaskar, P.; Yuen, S.; Li, J.; Muretta, J.M.; Gillispie, G.D.; Thomas, D.D. High-Throughput Spectral and Lifetime-Based FRET Screening in Living Cells to Identify Small-Molecule Effectors of SERCA. SLAS Discov. 2017, 22, 262–273. [Google Scholar]
- Rebbeck, R.T.; Essawy, M.M.; Nitu, F.R.; Grant, B.D.; Gillispie, G.D.; Thomas, D.D.; Bers, D.M.; Cornea, R.L. High-Throughput Screens to Discover Small-Molecule Modulators of Ryanodine Receptor Calcium Release Channels. SLAS Discov. 2017, 22, 176–186. [Google Scholar] [CrossRef]
- Rebbeck, R.T.; Singh, D.P.; Janicek, K.A.; Bers, D.M.; Thomas, D.D.; Launikonis, B.S.; Cornea, R.L. RyR1-targeted drug discovery pipeline integrating FRET-based high-throughput screening and human myofiber dynamic Ca(2+) assays. Sci. Rep. 2020, 10, 1791. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Surface Receptor or Channel | Extracellular Signal | Immediate Effect of Receptor Activation | Downstream S/ER Ca2+ Release Channel | References |
|---|---|---|---|---|
| GPCR (e.g., PAR1) | Protein and small molecule ligands (e.g., thrombin) | PLCβ activation | IP3R | [38,39,40,41,42] |
| a Polymodal GPCR (e.g., M3R and PTHR1) | Protein and small molecule ligands (e.g., carbachol/PTH) | PLCβ and adenylyl cyclase activation | IP3R | [43,44,45] |
| RTK (e.g., IGF-1R) | Protein ligands (e.g., IGF-1) | PLCγ activation | IP3R | [46,47,48] |
| b BCR | IgM-binding antigens | PLCγ activation | IP3R | [49,50,51,52] |
| b TCR | Antigen presenting cell (MHC) | PLCγ activation | IP3R | [16,17,53,54] |
| Fc | Antigen-antibody complex | PLCγ activation | IP3R | [16,17,55,56] |
| VGIC (e.g., L-type Ca2+ channel) | Membrane depolarization | Ca2+ influx from the extracellular space | RyR | [57,58,59,60,61] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noble, M.; Lin, Q.-T.; Sirko, C.; Houpt, J.A.; Novello, M.J.; Stathopulos, P.B. Structural Mechanisms of Store-Operated and Mitochondrial Calcium Regulation: Initiation Points for Drug Discovery. Int. J. Mol. Sci. 2020, 21, 3642. https://doi.org/10.3390/ijms21103642
Noble M, Lin Q-T, Sirko C, Houpt JA, Novello MJ, Stathopulos PB. Structural Mechanisms of Store-Operated and Mitochondrial Calcium Regulation: Initiation Points for Drug Discovery. International Journal of Molecular Sciences. 2020; 21(10):3642. https://doi.org/10.3390/ijms21103642
Chicago/Turabian StyleNoble, Megan, Qi-Tong Lin, Christian Sirko, Jacob A. Houpt, Matthew J. Novello, and Peter B. Stathopulos. 2020. "Structural Mechanisms of Store-Operated and Mitochondrial Calcium Regulation: Initiation Points for Drug Discovery" International Journal of Molecular Sciences 21, no. 10: 3642. https://doi.org/10.3390/ijms21103642
APA StyleNoble, M., Lin, Q.-T., Sirko, C., Houpt, J. A., Novello, M. J., & Stathopulos, P. B. (2020). Structural Mechanisms of Store-Operated and Mitochondrial Calcium Regulation: Initiation Points for Drug Discovery. International Journal of Molecular Sciences, 21(10), 3642. https://doi.org/10.3390/ijms21103642