The Role of Autophagy for the Regeneration of the Aging Liver


Abstract
1. Introduction
1.1. Aging Increases the Incidence of Malignancies
1.2. Aging Increases the Risk of Liver Resection Due to Impaired Liver Regeneration
1.3. Aging Impairs Autophagy
1.4. Impaired Regeneration of the Aged Liver is Related to Impaired Autophagy
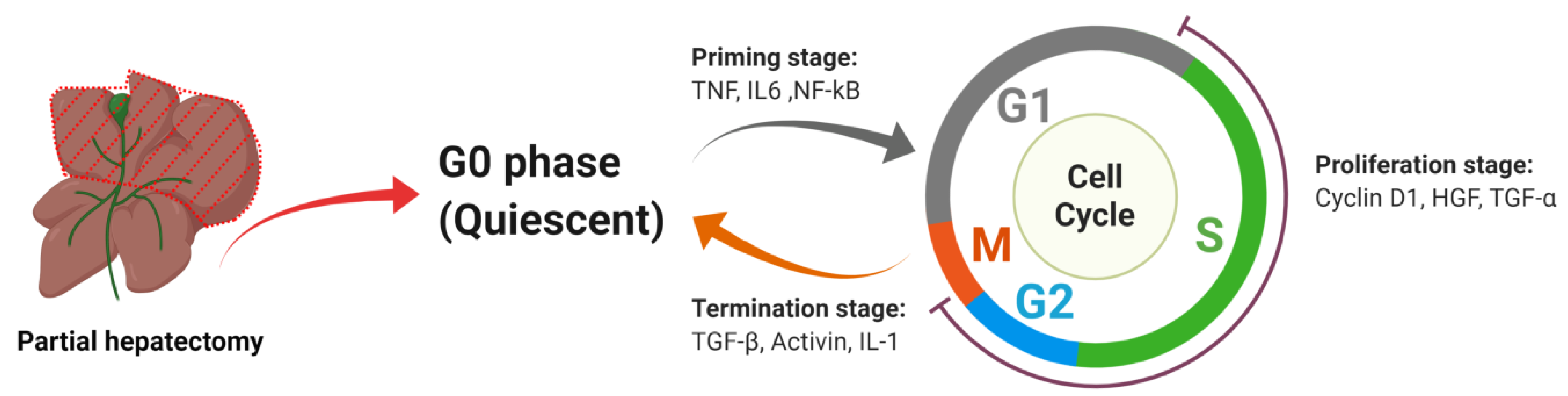
2. Liver Regeneration
2.1. The Powerful Regenerative Capacity of the Liver Is the Pathophysiological Basis for Successful Partial Hepatectomy
2.2. Under Most Circumstances Regeneration of the Liver Is Achieved by the Division of the Remaining Mature Hepatocytes
2.2.1. Priming Stage
TNF
IL6
NF-kB
2.2.2. Proliferation Stage
Cyclin D1
HGF
TGF-α
2.2.3. Termination Stage
TGF-β
Activin
IL-1α/β
2.3. The Effects of Aging on Liver Morphology, Metabolism and Regeneration
2.4. Liver Progenitor Cells may Enhance Liver Regeneration under Certain Circumstances
2.5. The Effects of Aging on Liver Progenitor Cells
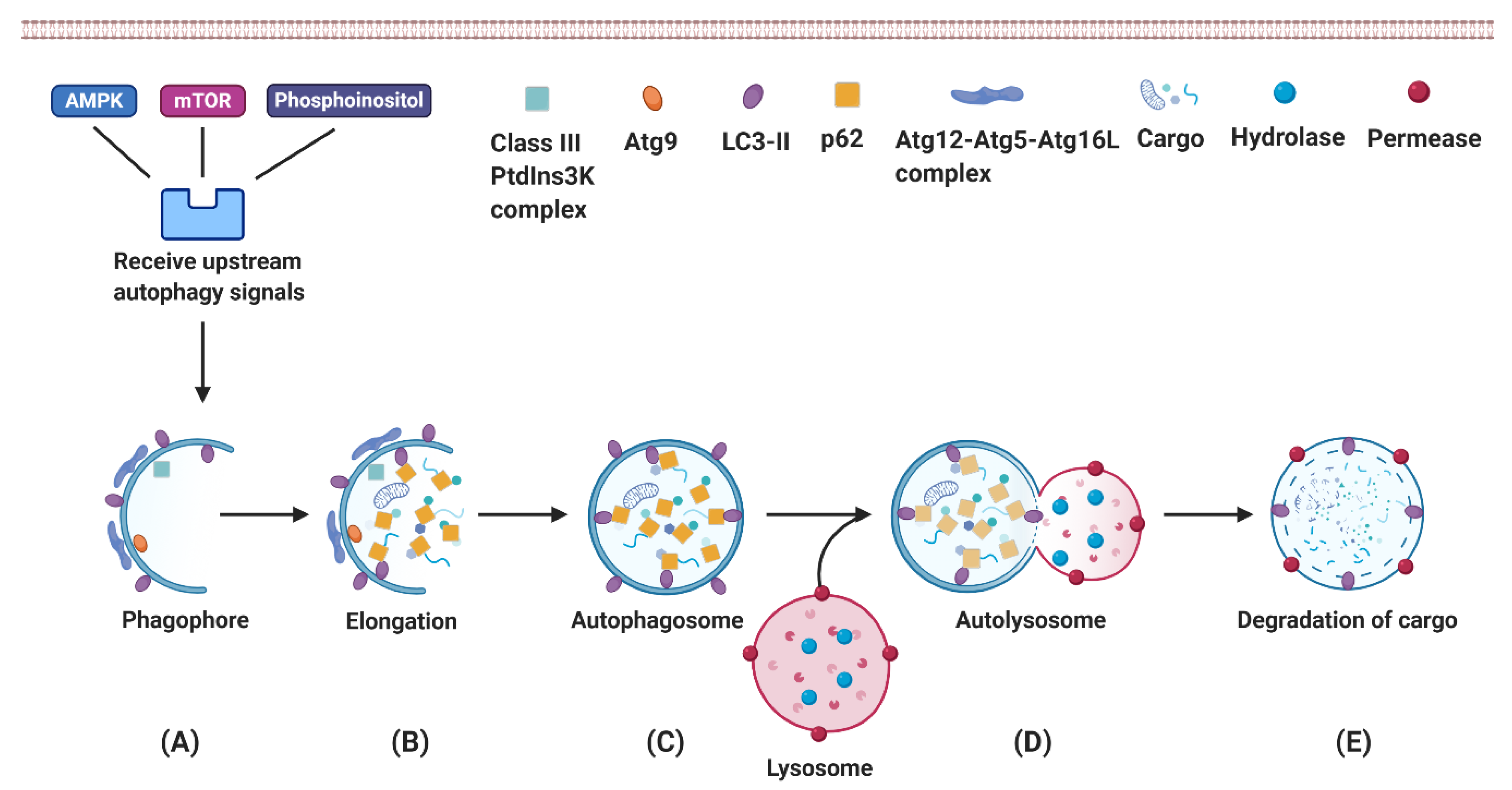
3. Autophagy
3.1. Autophagy Is an Essential Mechanism for Eukaryotes to Recycle Intracellular Components
3.2. Autophagy Plays an Essential Role in Hepatic Physiological Processes
3.3. Autophagy Provides Energy as Needed for Liver Regeneration
3.4. Age-Related Alterations Impair Autophagy Activity
3.4.1. Age-Related Decline of AMPK Activation Impairs Autophagosome Formation
3.4.2. Age-Related Lipofuscin Accumulation in Lysosomes Reduces the Efficiency of the Degradation Process
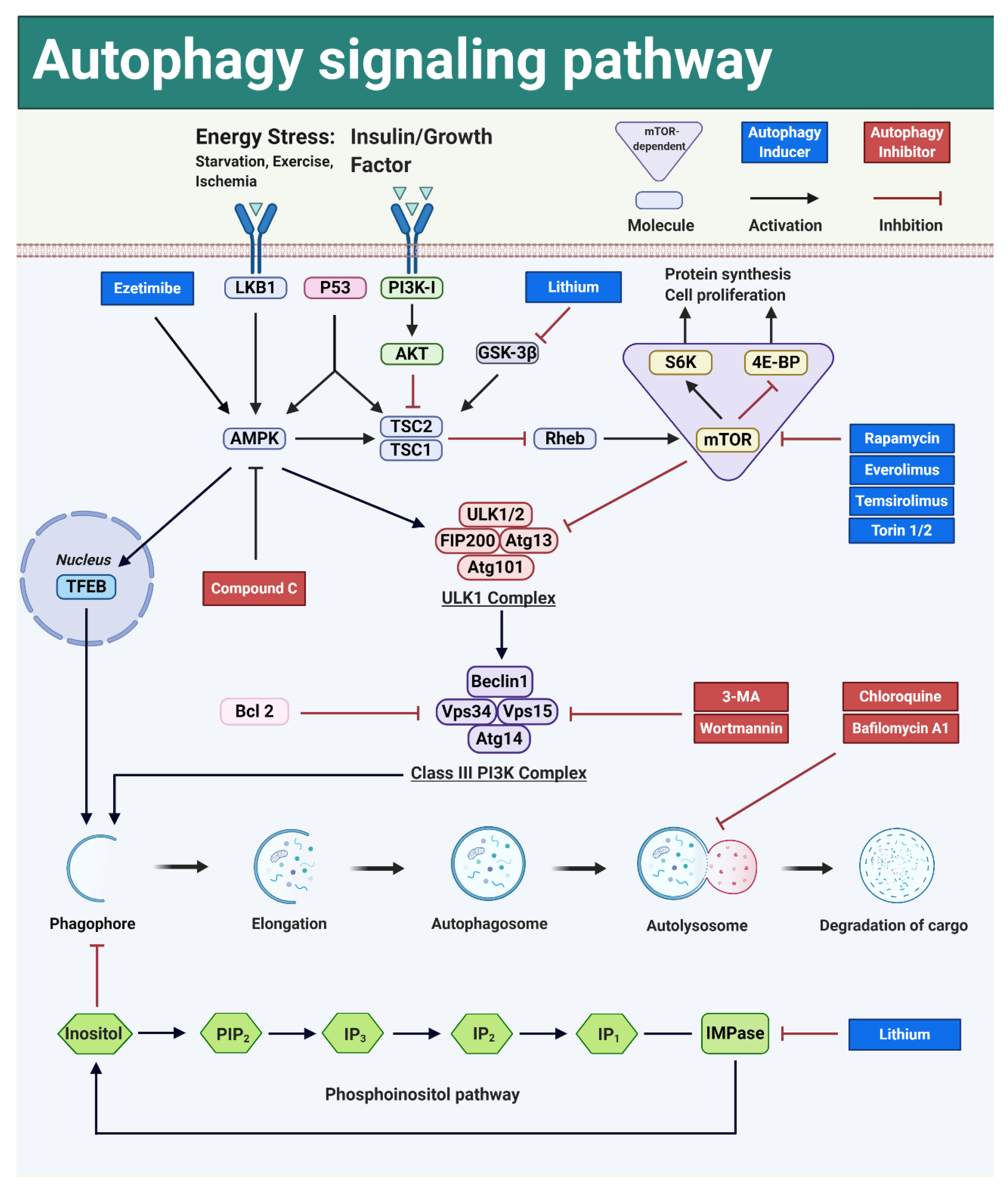
3.5. Autophagy Can Be Modulated by Interfering with Key Signal Transduction Pathways
3.5.1. Autophagy Can Be Activated by Interfering with the mTOR-Dependent Signal Transduction Pathways
PI3K-AKT-mTOR
LKB1-AMPK-mTOR
p53-AMPK-mTOR
mTOR-Dependent Autophagy Induction and Liver Regeneration
3.5.2. mTOR-Independent Autophagy Pathways Are Not Interrelated with Cell Proliferation Pathways
AMPK-ULK1
AMPK-TFEB
Phosphoinositol Pathway
mTOR-Independent Autophagy Inducers and Liver Regeneration
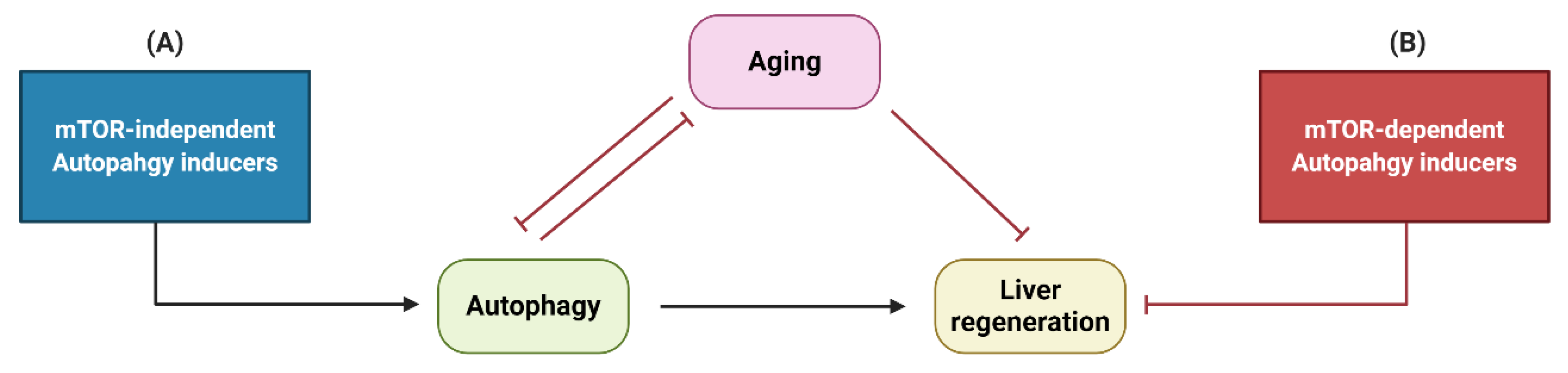
4. Interactions between Autophagy, Liver Regeneration and Aging
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPK | AMP-activated protein kinase |
| AMP | Adenosine monophosphate |
| ASPP2 | Apoptosis-stimulating protein two of p53 |
| ATP | Adenosine triphosphate |
| Atg5 | Autophagy related 5 protein |
| Atg7 | Autophagy related 7 protein |
| Atg13 | Autophagy related 13 protein |
| Bcl2 | B-cell CLL/lymphoma 2 |
| BrdU | 5-Bromo-2′-Deoxyuridine |
| CDE | Choline-Deficient, Ethionine-supplemented |
| CDK | Cyclin-dependent kinase |
| CXCL7 | Chemokine (C-X-C motif) ligand 7 |
| C/EBP | CCAAT enhancer binding protein |
| DAPK-1 | Death-associated protein kinase 1 |
| DNA | Deoxyribonucleic Acid |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| ER | Endoplasmic reticulum |
| ERK1/2 | Extracellular signal-regulated kinase 1/2 |
| FFAs | Free fatty acids |
| GSK-3β | Glycogen synthase kinase 3 beta |
| HCs | Hepatocytes |
| HEK | Human embryonic kidney |
| HGF | Hepatocyte growth factor |
| HSC | Hepatic stellate cells |
| HSCs | Hepatic stem cells |
| IL-1 | Interleukin 1 |
| IL-6 | Interleukin 6 |
| IL-6R | Interleukin 6 receptor |
| IMPase | Inositol monophosphatase |
| Ins | Inositol |
| IPMK | Inositol Polyphosphate Multikinase |
| IRI | Ischemia-reperfusion injury |
| KD | Knockdown |
| KO | Knockout |
| LBWR | Liver to body weight ratio |
| LC3 | Microtubule-associated protein 1A/1B-light chain 3 |
| LKB1 | Liver kinase B1 |
| LPCs | Liver progenitor cells |
| LPS | Lipopolysaccharide |
| LSCs | Liver stem cells |
| LWRR | Liver weight recovery rate |
| MAPKs | Mitogen-activated protein kinases |
| MEFs | Mouse Embryonic Fibroblasts |
| mTOR | mammalian Target of Rapamycin |
| mTORC1 | mammalian Target of Rapamycin complex 1 |
| mTORC2 | mammalian Target of Rapamycin complex 2 |
| NADH | Nicotinamide adenine dinucleotide |
| NAFLD | Non-alcoholic fatty liver disease |
| NF-kB | Nuclear factor kappa B |
| NPC1L1 | Niemann-Pick C1-Like 1 |
| OCs | Oval cells |
| PCs | Progenitor cells |
| PCNA | Proliferating cell nuclear antigen |
| PDK1 | Phosphoinositide dependent kinase 1 |
| PH | Partial hepatectomy |
| PIP3 | Phosphatidylinositol-3,4,5-triphosphate |
| PI3K | Phosphatidylinositol 3-kinase |
| PVL | Portal vein ligation |
| Rb | Retinoblastoma |
| Rheb | Ras homolog enriched in brain |
| ROS | Reactive oxygen species |
| SD | Sprague Dawley |
| S6K1 | p70S6 kinase 1 |
| SIRT1 | Silent information regulator 1 |
| Ski | Sloan-Kettering Institute |
| SnoN | Ski-related novel protein N |
| STAT-3 | Signal transducer and activator of transcription 3 |
| TFEB | Transcription Factor EB |
| TGF-α | Transforming growth factor alpha |
| TGF-β | Transforming growth factor beta |
| TNF-α | Tumor necrosis factor alpha |
| TNFR-1 | Tumor necrosis factor receptor 1 |
| TSC1/2 | Tuberous sclerosis complex 1/2 |
| ULK1 | Unc-51 like autophagy activating kinase 1 |
| 3-MA | 3-methyladenine |
| 4E-BP | eIF4E binding protein |
References
- Roser, M. Life Expectancy. 2019. Available online: https://ourworldindata.org/life-expect (accessed on 27 February 2019).
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Anantharaju, A.; Feller, A.; Chedid, A. Aging Liver. Gerontology 2002, 48, 343–353. [Google Scholar] [CrossRef]
- Wynne, H.A.; Cope, L.H.; Mutch, E.; Rawlins, M.D.; Woodhouse, K.W.; James, O.F. The effect of age upon liver volume and apparent liver blood flow in healthy man. Hepatology 1989, 9, 297–301. [Google Scholar] [CrossRef]
- Liu, A.; Guo, E.; Yang, J.; Yang, Y.; Liu, S.; Jiang, X.; Hu, Q.; Dirsch, O.; Dahmen, U.; Zhang, C.; et al. Young plasma reverses age-dependent alterations in hepatic function through the restoration of autophagy. Aging Cell 2018, 17, e12708. [Google Scholar] [CrossRef]
- Pibiri, M. Liver regeneration in aged mice: New insights. Aging 2018, 10, 1801–1824. [Google Scholar] [CrossRef]
- Zhu, C.Z.; Ikemoto, T.; Utsunomiya, T.; Yamada, S.; Morine, Y.; Imura, S.; Arakawa, Y.; Takasu, C.; Ishikawa, D.; Shimada, M. Senescence-related genes possibly responsible for poor liver regeneration after hepatectomy in elderly patients. J. Gastroenterol. Hepatol. 2014, 29, 1102–1108. [Google Scholar] [CrossRef]
- Zhao, J.W.; Xu, H.Y.; Li, Y.; Gong, L.L.; Zheng, G.; Wang, X.F.; Luan, W.J.; Li, S.L.; Ma, F.X.; Ni, L.H.; et al. NAFLD Induction Delays Postoperative Liver Regeneration of ALPPS in Rats. Dig. Dis. Sci. 2019, 64, 456–468. [Google Scholar] [CrossRef]
- Horiguchi, N.; Ishac, E.J.N.; Gao, B. Liver regeneration is suppressed in alcoholic cirrhosis: Correlation with decreased STAT3 activation. Alcohol 2007, 41, 271–280. [Google Scholar] [CrossRef]
- Sheedfar, F.; Di Biase, S.; Koonen, D.; Vinciguerra, M. Liver diseases and aging: Friends or foes? Aging Cell 2013, 12, 950–954. [Google Scholar] [CrossRef]
- Chun, Y.; Kim, J. Autophagy: An Essential Degradation Program for Cellular Homeostasis and Life. Cells 2018, 7, 278. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Tacke, F. Relevance of Autophagy in Parenchymal and Non-Parenchymal Liver Cells for Health and Disease. Cells 2019, 8, 16. [Google Scholar] [CrossRef]
- Toshima, T.; Shirabe, K.; Fukuhara, T.; Ikegami, T.; Yoshizumi, T.; Soejima, Y.; Ikeda, T.; Okano, S.; Maehara, Y. Suppression of Autophagy During Liver Regeneration Impairs Energy Charge and Hepatocyte Senescence in Mice. Hepatology 2014, 60, 290–300. [Google Scholar] [CrossRef]
- Liu, A.; Yang, J.; Hu, Q.; Dirsch, O.; Dahmen, U.; Zhang, C.; Gewirtz, D.A.; Fang, H.; Sun, J. Young plasma attenuates age-dependent liver ischemia reperfusion injury. FASEB J. 2019, 33, 3063–3073. [Google Scholar] [CrossRef]
- Escobar, K.A.; Cole, N.H.; Mermier, C.M.; VanDusseldorp, T.A. Autophagy and aging: Maintaining the proteome through exercise and caloric restriction. Aging Cell 2019, 18, e12876. [Google Scholar] [CrossRef]
- Bi, J.; Yang, L.; Wang, T.; Zhang, J.; Li, T.; Ren, Y.; Wu, R. Irisin Improves Autophagy of Aged Hepatocytes via Increasing Telomerase Activity in Liver Injury. Oxidative Med. Cell. Longev. 2020, 2020, 6946037. [Google Scholar] [CrossRef]
- Terman, A.; Brunk, U.T. Autophagy in cardiac myocyte homeostasis, aging, and pathology. Cardiovasc. Res. 2005, 68, 355–365. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Bergamini, E.; Brunk, U.T.; Dröge, W.; Ffrench, M.; Terman, A. Autophagy and Aging: The Importance of Maintaining “Clean” Cells. Autophagy 2005, 1, 131–140. [Google Scholar] [CrossRef]
- Brunk, U.T.; Terman, A. The mitochondrial-lysosomal axis theory of aging—Accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur. J. Biochem. 2002, 269, 1996–2002. [Google Scholar] [CrossRef]
- Reznick, R.M.; Zong, H.; Li, J.; Morino, K.; Moore, I.K.; Yu, H.J.; Liu, Z.-X.; Dong, J.; Mustard, K.J.; Hawley, S.A.; et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007, 5, 151–156. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Age-related changes in AMPK activation: Role for AMPK phosphatases and inhibitory phosphorylation by upstream signaling pathways. Ageing Res. Rev. 2016, 28, 15–26. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 2012, 11, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Alexandrino, H.; Rolo, A.; Tralhão, J.G.; Castro e Sousa, F.; Palmeira, C. Mitochondria in liver regeneration: energy metabolism and posthepatectomy liver dysfunction. In Mitochondrial Biology and Experimental Therapeutics; Oliveira, P.J., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 127–152. [Google Scholar]
- Enkhbold, C.; Morine, Y.; Utsunomiya, T.; Imura, S.; Ikemoto, T.; Arakawa, Y.; Saito, Y.; Yamada, S.; Ishikawa, D.; Shimada, M. Dysfunction of liver regeneration in aged liver after partial hepatectomy. J. Gastroenterol. Hepatol. 2015, 30, 1217–1224. [Google Scholar] [CrossRef]
- Shi, H.; Zhang, Y.; Ji, J.; Xu, P.; Shi, H.; Yue, X.; Ren, F.; Chen, Y.; Duan, Z.; Chen, D. Deficiency of apoptosis-stimulating protein two of p53 promotes liver regeneration in mice by activating mammalian target of rapamycin. Sci. Rep. 2018, 8, 17927. [Google Scholar] [CrossRef]
- Lin, C.W.; Chen, Y.S.; Lin, C.C.; Chen, Y.J.; Lo, G.H.; Lee, P.H.; Kuo, P.L.; Dai, C.Y.; Huang, J.F.; Chung, W.L.; et al. Amiodarone as an autophagy promoter reduces liver injury and enhances liver regeneration and survival in mice after partial hepatectomy. Sci. Rep. 2015, 5, 15807. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Kodama, T.; Hikita, H.; Tanaka, S.; Shigekawa, M.; Nawa, T.; Shimizu, S.; Li, W.; Miyagi, T.; Hiramatsu, N.; et al. Carbamazepine promotes liver regeneration and survival in mice. J. Hepatol. 2013, 59, 1239–1245. [Google Scholar] [CrossRef]
- Jia, C.J.; Sun, H.; Dai, C.L. Autophagy Contributes to Liver Regeneration After Portal Vein Ligation in Rats. Med. Sci. Monit. 2019, 25, 5674–5682. [Google Scholar] [CrossRef]
- Cheng, Y.J.; Wang, B.; Zhou, H.; Dang, S.P.; Jin, M.; Shi, Y.F.; Hao, L.; Yang, Z.X.; Zhang, Y.Y. Autophagy is Required for the Maintenance of Liver Progenitor Cell Functionality. Cell Physiol. Biochem. 2015, 36, 1163–1174. [Google Scholar] [CrossRef]
- Gebhardt, R.; Baldysiak-Figiel, A.; Krugel, V.; Ueberham, E.; Gaunitz, F. Hepatocellular expression of glutamine synthetase: An indicator of morphogen actions as master regulators of zonation in adult liver. Prog. Histochem. Cyto. 2007, 41, 201–266. [Google Scholar] [CrossRef]
- Yang, L.L.; Luo, Y. Liver regeneration after partial hepatectomy. Shijie Huaren Xiaohua Zazhi 2016, 24, 67–74. [Google Scholar] [CrossRef]
- Michalopoulos, G.K.; DeFrances, M.C. Liver Regeneration. Science 1997, 276, 60–66. [Google Scholar] [CrossRef]
- Fausto, N. Liver regeneration. J. Hepatol. 2000, 32, 19–31. [Google Scholar] [CrossRef]
- Loffreda, S.; Rai, R.; Yang, S.Q.; Lin, H.Z.; Diehl, A.M. Bile ducts and portal and central veins are major producers of tumor necrosis factor alpha in regenerating rat liver. Gastroenterology 1997, 112, 2089–2098. [Google Scholar] [CrossRef]
- Li, W.; Liang, X.P.; Kellendonk, C.; Poli, V.; Taub, R. STAT3 contributes to the mitogenic response of hepatocytes during liver regeneration. J. Biol. Chem. 2002, 277, 28411–28417. [Google Scholar] [CrossRef] [PubMed]
- Taub, R. Liver regeneration: From myth to mechanism. Nat. Rev. Mol. Cell Biol. 2004, 5, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Souissi, I.; Najjar, I.; Ah-Koon, L.; Schischmanoff, P.O.; Lesage, D.; Le Coquil, S.; Roger, C.; Dusanter-Fourt, I.; Varin-Blank, N.; Cao, A.; et al. A STAT3-decoy oligonucleotide induces cell death in a human colorectal carcinoma cell line by blocking nuclear transfer of STAT3 and STAT3-bound NF-κB. BMC Cell Biol. 2011, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; McBride, K.M.; Reich, N.C. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha3. Proc. Natl. Acad. Sci. USA 2005, 102, 8150–8155. [Google Scholar] [CrossRef]
- Guégan, J.-P.; Frémin, C.; Baffet, G. The MAPK MEK1/2-ERK1/2 Pathway and Its Implication in Hepatocyte Cell Cycle Control. Int. J. Hepatol. 2012, 2012, 328372. [Google Scholar] [CrossRef] [PubMed]
- Talarmin, H.; Rescan, C.; Cariou, S.; Glaise, D.; Zanninelli, G.; Bilodeau, M.; Loyer, P.; Guguen-Guillouzo, C.; Baffet, G. The mitogen-activated protein kinase kinase/extracellular signal-regulated kinase cascade activation is a key signalling pathway involved in the regulation of G(1) phase progression in proliferating hepatocytes. Mol. Cell Biol. 1999, 19, 6003–6011. [Google Scholar] [CrossRef]
- Yamada, Y.; Kirillova, I.; Peschon, J.J.; Fausto, N. Initiation of liver growth by tumor necrosis factor: Deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 1441–1446. [Google Scholar] [CrossRef]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef]
- Yamasaki, K.; Taga, T.; Hirata, Y.; Yawata, H.; Kawanishi, Y.; Seed, B.; Taniguchi, T.; Hirano, T.; Kishimoto, T. Cloning and Expression of the Human Interleukin-6 (Bsf-2/Ifn-Beta-2) Receptor. Science 1988, 241, 825–828. [Google Scholar] [CrossRef] [PubMed]
- Taga, T.; Hibi, M.; Hirata, Y.; Yamasaki, K.; Yasukawa, K.; Matsuda, T.; Hirano, T.; Kishimoto, T. Interleukin-6 Triggers the Association of Its Receptor with a Possible Signal Transducer, Gp130. Cell 1989, 58, 573–581. [Google Scholar] [CrossRef]
- Hibi, M.; Murakami, M.; Saito, M.; Hirano, T.; Taga, T.; Kishimoto, T. Molecular-Cloning and Expression of an Il-6 Signal Transducer, Gp130. Cell 1990, 63, 1149–1157. [Google Scholar] [CrossRef]
- Taga, T.; Kishimoto, T. gp130 and the interleukin-6 family of cytokines. Annu. Rev. Immunol. 1997, 15, 797–819. [Google Scholar] [CrossRef]
- Cressman, D.E.; Greenbaum, L.E.; DeAngelis, R.A.; Ciliberto, G.; Furth, E.E.; Poli, V.; Taub, R. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science 1996, 274, 1379–1383. [Google Scholar] [CrossRef]
- Streetz, K.L.; Luedde, T.; Manns, M.P.; Trautwein, C. Interleukin 6 and liver regeneration. Gut 2000, 47, 309–312. [Google Scholar] [CrossRef]
- Akira, S.; Isshiki, H.; Sugita, T.; Tanabe, O.; Kinoshita, S.; Nishio, Y.; Nakajima, T.; Hirano, T.; Kishimoto, T. A Nuclear Factor for Il-6 Expression (Nf-Il6) Is a Member of a C/Ebp Family. EMBO J. 1990, 9, 1897–1906. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Wen, Z.; Darnell, J. Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar] [CrossRef]
- Trautwein, C.; Caelles, C.; Vandergeer, P.; Hunter, T.; Karin, M.; Chojkier, M. Transactivation by Nf-Il6 Lap Is Enhanced by Phosphorylation of Its Activation Domain. Nature 1993, 364, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Cressman, D.E.; Diamond, R.H.; Taub, R. Rapid activation of the Stat3 transcription complex in liver regeneration. Hepatology 1995, 21, 1443–1449. [Google Scholar] [CrossRef]
- Trautwein, C.; Rakemann, T.; Niehof, M.; RoseJohn, S.; Manns, M.P. Acute-phase response factor, increased binding, and target gene transcription during liver regeneration. Gastroenterology 1996, 110, 1854–1862. [Google Scholar] [CrossRef] [PubMed]
- Niehof, M.; Manns, M.P.; Trautwein, C. CREB controls LAP/C/EBP beta transcription. Mol. Cell Biol. 1997, 17, 3600–3613. [Google Scholar] [CrossRef] [PubMed]
- Tewari, M.; Dobrzanski, P.; Mohn, K.L.; Cressman, D.E.; Hsu, J.C.; Bravo, R.; Taub, R. Rapid Induction in Regenerating Liver of Rl/If-1 (an I-Kappa-B That Inhibits Nf-Kappa-B, Relb-P50, and C-Rel-P50) and Phf, a Novel Kappa-B Site-Binding Complex. Mol. Cell Biol. 1992, 12, 2898–2908. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, M.J.; Webber, E.M.; Donovan, J.R.; Fausto, N. Rapid DNA-Binding by Nuclear Factor Kappa-B in Hepatocytes at the Start of Liver-Regeneration. Cell Growth Differ. 1995, 6, 417–427. [Google Scholar]
- Black, D.; Lyman, S.; Heider, T.R.; Behrns, K.E. Molecular and cellular features of hepatic regeneration. J. Surg. Res. 2004, 117, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Iimuro, Y.; Nishiura, T.; Hellerbrand, C.; Behrns, K.E.; Schoonhoven, R.; Grisham, J.W.; Brenner, D.A. NFkappaB prevents apoptosis and liver dysfunction during liver regeneration. J. Clin. Invest. 1998, 101, 802–811. [Google Scholar] [CrossRef]
- Loyer, P.; Cariou, S.; Glaise, D.; Bilodeau, M.; Baffet, G.; GuguenGuillouzo, C. Growth factor dependence of progression through G(1) and S phases of adult rat hepatocytes in vitro—Evidence of a mitogen restriction point in mid-late G(1). J. Biol. Chem. 1996, 271, 11484–11492. [Google Scholar] [CrossRef]
- Albrecht, J.H.; Hansen, L.K. Cyclin D1 promotes mitogen-independent cell cycle progression in hepatocytes. Cell Growth Differ. 1999, 10, 397–404. [Google Scholar]
- Huh, C.G.; Factor, V.M.; Sanchez, A.; Uchida, K.; Conner, E.A.; Thorgeirsson, S.S. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc. Natl. Acad. Sci. USA 2004, 101, 4477–4482. [Google Scholar] [CrossRef]
- Nakamura, T.; Mizuno, S. The discovery of Hepatocyte Growth Factor (HGF) and its significance for cell biology, life sciences and clinical medicine. Proc. Jpn. Acad. B-Phys. 2010, 86, 588–610. [Google Scholar] [CrossRef]
- Michalopoulos, G. HGF and liver regeneration. Gastroenterol. Jpn. 1993, 28 (Suppl. 4), 36–39. [Google Scholar] [CrossRef] [PubMed]
- Lindroos, P.M.; Zarnegar, R.; Michalopoulos, G.K. Hepatocyte growth factor (hepatopoietin A) rapidly increases in plasma before DNA synthesis and liver regeneration stimulated by partial hepatectomy and carbon tetrachloride administration. Hepatology 1991, 13, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Zambreg, I.; Assouline, B.; Housset, C.; Schiffer, E. Overexpression of TGF-α and EGFR, a key event in liver carcinogenesis, is induced by hypoxia specifically in hepatocytes. Gastroenterol. Hepatol. 2019, 4, 1–4. [Google Scholar] [CrossRef]
- Michalopoulos, G.K.; Khan, Z. Liver regeneration, growth factors, and amphiregulin. Gastroenterology 2005, 128, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Tomiya, T.; Ogata, I.; Fujiwara, K. Transforming growth factor alpha levels in liver and blood correlate better than hepatocyte growth factor with hepatocyte proliferation during liver regeneration. Am. J. Pathol. 1998, 153, 955–961. [Google Scholar] [CrossRef]
- Mead, J.E.; Fausto, N. Transforming growth factor alpha may be a physiological regulator of liver regeneration by means of an autocrine mechanism. Proc. Natl. Acad. Sci. USA 1989, 86, 1558–1562. [Google Scholar] [CrossRef] [PubMed]
- Breitkopf, K.; Godoy, P.; Ciuclan, L.; Singer, M.V.; Dooley, S. TGF-beta/Smad signaling in the injured liver. Zeitschrift für Gastroenterol. 2006, 44, 57–66. [Google Scholar] [CrossRef]
- Tao, Y.C.; Wang, M.L.; Chen, E.Q.; Tang, H. Liver Regeneration: Analysis of the Main Relevant Signaling Molecules. Mediat Inflamm 2017, 2017, 4256352. [Google Scholar] [CrossRef]
- Schon, H.-T.; Weiskirchen, R. Immunomodulatory effects of transforming growth factor-β in the liver. Hepatobiliary Surg. Nutr. 2014, 3, 386–406. [Google Scholar]
- Braun, L.; Mead, J.E.; Panzica, M.; Mikumo, R.; Bell, G.I.; Fausto, N. Transforming growth factor beta mRNA increases during liver regeneration: A possible paracrine mechanism of growth regulation. Proc. Natl. Acad. Sci. USA 1988, 85, 1539–1543. [Google Scholar] [CrossRef]
- Macias-Silva, M.; Li, W.; Leu, J.I.; Crissey, M.A.S.; Taub, R. Up-regulated transcriptional repressors SnoN and Ski bind Smad proteins to antagonize transforming growth factor-beta signals during liver regeneration. J. Biol. Chem. 2002, 277, 28483–28490. [Google Scholar] [CrossRef] [PubMed]
- Werner, S.; Alzheimer, C. Roles of activin in tissue repair, fibrosis, and inflammatory disease. Cytokine Growth Factor Rev. 2006, 17, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Fischer, G.; Hyvönen, M. Structure and activation of pro-activin A. Nat. Commun. 2016, 7, 12052. [Google Scholar] [CrossRef]
- Zimmermann, A. Regulation of liver regeneration. Nephrol. Dial. Transplant. 2004, 19 (Suppl. 4), iv6–iv10. [Google Scholar] [CrossRef] [PubMed]
- Takamura, K.; Tsuchida, K.; Miyake, H.; Tashiro, S.; Sugino, H. Activin and activin receptor expression changes in liver regeneration in rat. J. Surg. Res. 2005, 126, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, W.; Liang, H.-F.; Zhou, Q.-F.; Ding, Z.-Y.; Yang, H.-Q.; Liu, W.-B.; Wu, Y.-H.; Man, Q.; Zhang, B.-X.; et al. Activin A induces growth arrest through a SMAD-dependent pathway in hepatic progenitor cells. Cell Commun. Signal. 2014, 12, 18. [Google Scholar] [CrossRef]
- Hully, J.R.; Chang, L.; Schwall, R.H.; Widmer, H.R.; Terrell, T.G.; Gillett, N.A. Induction of Apoptosis in the Murine Liver with Recombinant Human Activin-A. Hepatology 1994, 20, 854–862. [Google Scholar] [CrossRef]
- Schwall, R.H.; Robbins, K.; Jardieu, P.; Chang, L.; La, C.; Terrel, T.G. Activin induces cell death in hepatocytes in vivo and in vitro. Hepatology 1993, 18, 347–356. [Google Scholar] [CrossRef]
- Wang, X.; Fu, S.; Wang, Y.; Yu, P.; Hu, J.; Gu, W.; Xu, X.-M.; Lu, P. Interleukin-1β mediates proliferation and differentiation of multipotent neural precursor cells through the activation of SAPK/JNK pathway. Mol. Cell. Neurosci. 2007, 36, 343–354. [Google Scholar] [CrossRef]
- Goss, J.A.; Mangino, M.J.; Flye, M.W. Kupffer cell autoregulation of IL-1 production by PGE2 during hepatic regeneration. J. Surg. Res. 1992, 52, 422–428. [Google Scholar] [CrossRef]
- Boulton, R.; Woodman, A.; Calnan, D.; Selden, C.; Tam, F.; Hodgson, H. Nonparenchymal cells from regenerating rat liver generate interleukin- 1alpha and -1beta: A mechanism of negative regulation of hepatocyte proliferation. Hepatology 1997, 26, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Arakaki, R.; Ichihara, A. Interleukin-1-Beta Is a Potent Growth Inhibitor of Adult-Rat Hepatocytes in Primary Culture. Exp. Cell Res. 1988, 179, 488–497. [Google Scholar] [CrossRef]
- Gan, L.; Chitturi, S.; Farrell, G.C. Mechanisms and implications of age-related changes in the liver: Nonalcoholic Fatty liver disease in the elderly. Curr. Gerontol. Geriatr. Res. 2011, 2011, 831536. [Google Scholar] [CrossRef]
- Schmucker, D.L. Aging and the liver: An update. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 1998, 53, B315–B321. [Google Scholar] [CrossRef] [PubMed]
- Schmucker, D.L.; Sanchez, H. Liver regeneration and aging: A current perspective. Curr. Gerontol. Geriatr. Res. 2011, 2011, 526379. [Google Scholar] [CrossRef] [PubMed]
- Evarts, R.P.; Nagy, P.; Marsden, E.; Thorgeirsson, S.S. A precursor—Product relationship exists between oval cells and hepatocytes in rat liver. Carcinogenesis 1987, 8, 1737–1740. [Google Scholar] [CrossRef]
- Cheng, Y.; Wang, X.; Wang, B.; Zhou, H.; Dang, S.; Shi, Y.; Hao, L.; Luo, Q.; Jin, M.; Zhou, Q.; et al. Aging-associated oxidative stress inhibits liver progenitor cell activation in mice. Aging 2017, 9, 1359–1374. [Google Scholar] [CrossRef]
- Itoh, T.; Miyajima, A. Liver regeneration by stem/progenitor cells. Hepatology 2014, 59, 1617–1626. [Google Scholar] [CrossRef]
- Thorgeirsson, S.S. Hepatic stem cells in liver regeneration. FASEB J. 1996, 10, 1249–1256. [Google Scholar] [CrossRef]
- Than, N.N.; Newsome, P.N. Stem cells for liver regeneration. QJM Mon. J. Assoc. Physicians 2014, 107, 417–421. [Google Scholar] [CrossRef]
- Schaub, J.R.; Malato, Y.; Gormond, C.; Willenbring, H. Evidence against a Stem Cell Origin of New Hepatocytes in a Common Mouse Model of Chronic Liver Injury. Cell Rep. 2014, 8, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Gilgenkrantz, H.; Collin de l’Hortet, A. Understanding Liver Regeneration: From Mechanisms to Regenerative Medicine. Am. J. Pathol. 2018, 188, 1316–1327. [Google Scholar] [CrossRef] [PubMed]
- Fausto, N. Liver regeneration: From laboratory to clinic. Liver Transplant. 2001, 7, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Viebahn, C.S.; Yeoh, G.C. What fires prometheus? The link between inflammation and regeneration following chronic liver injury. Int. J. Biochem. Cell Biol. 2008, 40, 855–873. [Google Scholar] [CrossRef] [PubMed]
- Best, J.; Dollé, L.; Manka, P.; Coombes, J.; van Grunsven, L.; Syn, W.-K. Role of liver progenitors in acute liver injury. Front. Physiol. 2013, 4, 258. [Google Scholar] [CrossRef]
- Drosos, I.; Kolios, G. Stem Cells in Liver Regeneration and Their Potential Clinical Applications. Stem Cell Rev. Rep. 2013, 9, 668–684. [Google Scholar] [CrossRef]
- Best, J.; Manka, P.; Syn, W.-K.; Dollé, L.; van Grunsven, L.A.; Canbay, A. Role of liver progenitors in liver regeneration. Hepatobiliary Surg. Nutr. 2015, 4, 48–58. [Google Scholar]
- Tanaka, M.; Miyajima, A. Liver regeneration and fibrosis after inflammation. Inflamm. Regen. 2016, 36, 19. [Google Scholar] [CrossRef]
- Lukacs-Kornek, V.; Lammert, F. The progenitor cell dilemma: Cellular and functional heterogeneity in assistance or escalation of liver injury. J. Hepatol. 2017, 66, 619–630. [Google Scholar] [CrossRef]
- Ma, Z.; Li, F.; Chen, L.; Gu, T.; Zhang, Q.; Qu, Y.; Xu, M.; Cai, X.; Lu, L. Autophagy promotes hepatic differentiation of hepatic progenitor cells by regulating the Wnt/β-catenin signaling pathway. J. Mol. Histol. 2019, 50, 75–90. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.Q.; et al. Regulation of Mammalian Autophagy in Physiology and Pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef]
- Zhi-Hong Liu, J.C.H. Podocytopathy. Contrib. Nephrol. Basel Karger 2014, 183, 83–100. [Google Scholar]
- Feng, Y.; Klionsky, D.J. Autophagic membrane delivery through ATG9. Cell Res. 2017, 27, 161–162. [Google Scholar] [CrossRef]
- Mizushima, N. The pleiotropic role of autophagy: From protein metabolism to bactericide. Cell Death Differ. 2005, 12, 1535–1541. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef]
- Su, Z.Y.; Yang, Z.Z.; Xu, Y.Q.; Chen, Y.B.; Yu, Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol. Cancer 2015, 14, 48. [Google Scholar] [CrossRef]
- Shen, H.M.; Codogno, P. Autophagic cell death Loch Ness monster or endangered species? Autophagy 2011, 7, 457–465. [Google Scholar] [CrossRef]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef]
- Ke, P.-Y. Diverse Functions of Autophagy in Liver Physiology and Liver Diseases. Int. J. Mol. Sci. 2019, 20, 300. [Google Scholar] [CrossRef] [PubMed]
- Czaja, M.J.; Ding, W.X.; Donohue, T.M.; Friedman, S.L.; Kim, J.S.; Komatsu, M.; Lemasters, J.J.; Lemoine, A.; Lin, J.D.; Ou, J.H.J.; et al. Functions of autophagy in normal and diseased liver. Autophagy 2013, 9, 1131–1158. [Google Scholar] [CrossRef] [PubMed]
- Ezaki, J.; Matsumoto, N.; Takeda-Ezaki, M.; Komatsu, M.; Takahashi, K.; Hiraoka, Y.; Taka, H.; Fujimura, T.; Takehana, K.; Yoshida, M.; et al. Liver autophagy contributes to the maintenance of blood glucose and amino acid levels. Autophagy 2011, 7, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lopez, N.; Singh, R. Autophagy and Lipid Droplets in the Liver. Annu. Rev. Nutr. 2015, 35, 215–237. [Google Scholar] [CrossRef]
- Sato, T.; Yamashina, S.; Izumi, K.; Ueno, T.; Koike, M.; Ikejima, K.; Peters, C.; Watanabe, S. Cathepsin L-deficiency enhances liver regeneration after partial hepatectomy. Life Sci. 2019, 221, 293–300. [Google Scholar] [CrossRef]
- Hales, K.G. Mitochondrial Fusion and Division. Nat. Educ. 2010, 3, 12. [Google Scholar]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar]
- Eisner, V.; Picard, M.; Hajnóczky, G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat Cell Biol. 2018, 20, 755–765. [Google Scholar] [CrossRef]
- Lackner, L.L. Shaping the dynamic mitochondrial network. BMC Biol. 2014, 12, 35. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.Y. Mitophagy in the Pathogenesis of Liver Diseases. Cells 2020, 9, 831. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Schmucker, D.L.; Sachs, H. Quantifying dense bodies and lipofuscin during aging: A morphologist’s perspective. Arch. Gerontol. Geriatr. 2002, 34, 249–261. [Google Scholar] [CrossRef]
- Terman, A.; Brunk, U.T. Lipofuscin: Mechanisms of formation and increase with age. Acta Pathol. Microbiol. Et Immunol. Scand. 1998, 106, 265–276. [Google Scholar] [CrossRef]
- Terman, A.; Kurz, T.; Navratil, M.; Arriaga, E.A.; Brunk, U.T. Mitochondrial Turnover and Aging of Long-Lived Postmitotic Cells: The Mitochondrial-Lysosomal Axis Theory of Aging. Antioxid. Redox. Sign. 2010, 12, 503–535. [Google Scholar] [CrossRef]
- Perse, M.; Injac, R.; Erman, A. Oxidative Status and Lipofuscin Accumulation in Urothelial Cells of Bladder in Aging Mice. PLoS ONE 2013, 8, e59638. [Google Scholar] [CrossRef]
- Terman, A.; Gustafsson, B.; Brunk, U.T. Autophagy, organelles and ageing. J. Pathol. 2007, 211, 134–143. [Google Scholar] [CrossRef]
- Moreno-Garcia, A.; Kun, A.; Calero, O.; Medina, M.; Calero, M. An Overview of the Role of Lipofuscin in Age-Related Neurodegeneration. Front. Neurosci. 2018, 12, 464. [Google Scholar] [CrossRef]
- Yuan, Y.J.; Chen, Y.N.; Peng, T.Q.; Li, L.; Zhu, W.Z.; Liu, F.; Liu, S.Y.; An, X.X.; Luo, R.X.; Cheng, J.Q.; et al. Mitochondrial ROS-induced lysosomal dysfunction impairs autophagic flux and contributes to M1 macrophage polarization in a diabetic condition. Clin Sci. 2019, 133, 1759–1777. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Invest. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.; Noda, T.; Yoshimori, T.; Rubinsztein, D.C. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat. Chem. Biol. 2011, 7, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Leidal, A.M.; Levine, B.; Debnath, J. Autophagy and the cell biology of age-related disease. Nat. Cell Biol. 2018, 20, 1338–1348. [Google Scholar] [CrossRef]
- Byun, S.; Lee, E.; Lee, K.W. Therapeutic Implications of Autophagy Inducers in Immunological Disorders, Infection, and Cancer. Int. J. Mol. Sci. 2017, 18, 1959. [Google Scholar] [CrossRef]
- Vicencio, J.M.; Ortiz, C.; Criollo, A.; Jones, A.W.E.; Kepp, O.; Galluzzi, L.; Joza, N.; Vitale, I.; Morselli, E.; Tailler, M.; et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ. 2009, 16, 1006–1017. [Google Scholar] [CrossRef]
- Levine, B.; Packer, M.; Codogno, P. Development of autophagy inducers in clinical medicine. J. Clin. Invest. 2015, 125, 14–24. [Google Scholar] [CrossRef]
- Sarkar, S.; Ravikumar, B.; Floto, R.A.; Rubinsztein, D.C. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009, 16, 46–56. [Google Scholar] [CrossRef]
- Li, Y.; Inoki, K.; Yeung, R.; Guan, K.L. Regulation of TSC2 by 14-3-3 binding. J. Biol. Chem. 2002, 277, 44593–44596. [Google Scholar] [CrossRef]
- Nellist, M.; Goedbloed, M.A.; de Winter, C.; Verhaaf, B.; Jankie, A.; Reuser, A.J.J.; van den Ouweland, A.M.W.; van der Sluijs, P.; Halley, D.J.J. Identification and characterization of the interaction between tuberin and 14-3-3 zeta. J. Biol. Chem. 2002, 277, 39417–39424. [Google Scholar] [CrossRef]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Corradetti, M.N.; Guan, K.L. Upstream of the mammalian target of rapamycin: Do all roads pass through mTOR? Oncogene 2006, 25, 6347–6360. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 127, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-Activated Protein Kinase Connects Energy Sensing to Mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer. 2009, 9, 563–575. [Google Scholar] [CrossRef]
- Ganley, I.G.; du Lam, H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 Complexes Mediate mTOR Signaling to the Autophagy Machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell. 2009, 20, 1981–1991. [Google Scholar] [CrossRef]
- Kamada, Y.; Yoshino, K.; Kondo, C.; Kawamata, T.; Oshiro, N.; Yonezawa, K.; Ohsumi, Y. Tor Directly Controls the Atg1 Kinase Complex to Regulate Autophagy. Mol. Cell Biol. 2010, 30, 1049–1058. [Google Scholar] [CrossRef]
- Rosso, P.; Fioramonti, M.; Fracassi, A.; Marangoni, M.; Taglietti, V.; Siteni, S.; Segatto, M. AMPK in the central nervous system: Physiological roles and pathological implications. Res. Rep. Biol. 2016, 7, 1–13. [Google Scholar]
- Inoki, K.; Zhu, T.Q.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Galluzzi, L.; Morselli, E.; Kepp, O.; Malik, S.A.; Kroemer, G. Autophagy regulation by p53. Curr. Opin. Cell Biol. 2010, 22, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Baroja-Mazo, A.; Revilla-Nuin, B.; Ramírez, P.; Pons, J.A. Immunosuppressive potency of mechanistic target of rapamycin inhibitors in solid-organ transplantation. World J. Transpl. 2016, 6, 183–192. [Google Scholar] [CrossRef]
- Noda, T.; Ohsumi, Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 1998, 273, 3963–3966. [Google Scholar] [CrossRef]
- Palmes, D.; Zibert, A.; Budny, T.; Bahde, R.; Minin, E.; Kebschull, L.; Spiegel, H.U. Impact of rapamycin on liver regeneration. Virchows Arch. Int. J. Pathol. 2008, 452, 545–557. [Google Scholar] [CrossRef]
- Fouraschen, S.M.; de Ruiter, P.E.; Kwekkeboom, J.; de Bruin, R.W.; Kazemier, G.; Metselaar, H.J.; Tilanus, H.W.; van der Laan, L.J.; de Jonge, J. mTOR signaling in liver regeneration: Rapamycin combined with growth factor treatment. World J. Transpl. 2013, 3, 36–47. [Google Scholar] [CrossRef]
- Jiang, Y.P.; Ballou, L.M.; Lin, R.Z. Rapamycin-insensitive regulation of 4e-BP1 in regenerating rat liver. J. Biol. Chem. 2001, 276, 10943–10951. [Google Scholar] [CrossRef]
- Roach, P.J. AMPK -> ULK1 -> Autophagy. Mol. Cell Biol. 2011, 31, 3082–3084. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Guha, P.; Tyagi, R.; Chowdhury, S.; Reilly, L.; Fu, C.; Xu, R.; Resnick, A.C.; Snyder, S.H. IPMK Mediates Activation of ULK Signaling and Transcriptional Regulation of Autophagy Linked to Liver Inflammation and Regeneration. Cell Rep. 2019, 26, 2692–2703.e7. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia-Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Di Malta, C.; Cinque, L.; Settembre, C. Transcriptional Regulation of Autophagy: Mechanisms and Diseases. Front Cell. Dev. Biol. 2019, 7, 114. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, G.; Han, D.H.; Lee, M.; Kim, I.; Kim, B.; Kim, K.H.; Song, Y.M.; Yoo, J.E.; Wang, H.J.; et al. Ezetimibe ameliorates steatohepatitis via AMP activated protein kinase-TFEB-mediated activation of autophagy and NLRP3 inflammasome inhibition. Autophagy 2017, 13, 1767–1781. [Google Scholar] [CrossRef]
- Sarkar, S.; Floto, R.A.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.J.; Rubinsztein, D.C. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 2005, 170, 1101–1111. [Google Scholar] [CrossRef]
- Kania, E.; Roest, G.; Vervliet, T.; Parys, J.B.; Bultynck, G. IP3 Receptor-Mediated Calcium Signaling and Its Role in Autophagy in Cancer. Front. Oncol. 2017, 7, 140. [Google Scholar] [CrossRef]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.-H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Shpilka, T.; Elazar, Z. Mechanisms of Autophagosome Biogenesis. Curr. Biol. 2012, 22, R29–R34. [Google Scholar] [CrossRef]
- Schiebler, M.; Brown, K.; Hegyi, K.; Newton, S.M.; Renna, M.; Hepburn, L.; Klapholz, C.; Coulter, S.; Obregon-Henao, A.; Tamayo, M.H.; et al. Functional drug screening reveals anticonvulsants as enhancers of mTOR-independent autophagic killing of Mycobacterium tuberculosis through inositol depletion. EMBO Mol. Med. 2015, 7, 127–139. [Google Scholar] [CrossRef]
- Zhang, J.-J.; Zhou, Q.-M.; Chen, S.; Le, W.-D. Repurposing carbamazepine for the treatment of amyotrophic lateral sclerosis in SOD1-G93A mouse model. CNS Neurosci. 2018, 24, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Bootman, M.; Chehab, T.; Bultynck, G.; Parys, J.; Rietdorf, K. The regulation of autophagy by calcium signals: Do we have a consensus? Cell Calcium 2017, 70, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, M.; Chen, D.H.; Gao, W.T.; Guan, J.L.; Komatsu, M.; Yin, X.M. Autophagy Induced by Calcium Phosphate Precipitates Involves Endoplasmic Reticulum Membranes in Autophagosome Biogenesis. PLoS ONE 2012, 7, e52347. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, X.; Matei, N.; McBride, D.; Tang, J.; Yan, M.; Zhang, J.H. Ezetimibe, a NPC1L1 inhibitor, attenuates neuronal apoptosis through AMPK dependent autophagy activation after MCAO in rats. Exp. Neurol. 2018, 307, 12–23. [Google Scholar] [CrossRef]
- Chang, E.; Kim, L.; Park, S.E.; Rhee, E.J.; Lee, W.Y.; Oh, K.W.; Park, C.Y. Ezetimibe improves hepatic steatosis in relation to autophagy in obese and diabetic rats. World J. Gastroenterol. 2015, 21, 7754–7763. [Google Scholar] [CrossRef]
- Sarkar, S.; Krishna, G.; Imarisio, S.; Saiki, S.; O’Kane, C.J.; Rubinsztein, D.C. A rational mechanism for combination treatment of Huntington’s disease using lithium and rapamycin. Hum. Mol. Genet. 2007, 17, 170–178. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Modes of Liver Regeneration | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Year | Author | Conditions for 1st Line Regeneration | Conditions and Mechanism for 2nd Line Regeneration | Main Cell Type for 2nd Line of Regeneration (as Indicated by Author) | |||||
| Mild Liver Injury | CCL4 ΔΔ | PH | Severe Liver Injury | Impaired Proliferative Capacity of Mature HCs 🗶 | Chronic Liver Injury | Delayed Response to Hepatic Injury | |||
| 1996 | Snorri [91] | YΔ | Y | OCs * | |||||
| 2001 | Nelson [95] | Y | Y | HSCs **/LPCs | |||||
| 2008 | Viebahn [96] | Y | Y | LPCs | |||||
| 2013 | Jan [97] | Not given | Y | LPCs | |||||
| 2013 | Ioannis [98] | Y | Y | Y | Y | Y | PCs *** | ||
| 2014 | THAN [92] | Y | Y | Y | Y | Y | LPCs | ||
| 2014 | Itoh [90] | Y | Y | Y | Y | LPCs | |||
| 2015 | Jan [99] | Y | Y | Y | LPCs | ||||
| 2016 | Minoru [100] | Y | Y | Y | LPCs | ||||
| 2017 | Veronika [101] | Not given | Y | LPCs | |||||
| Commonly Used Autophagy Inducers | ||
|---|---|---|
| Autophagy Inducers | Mode of ACTION | mTOR-Dependent |
| Rapamycin | mTOR inhibitor | Yes |
| Everolimus | mTOR inhibitor | Yes |
| Temsirolimus | mTOR inhibitor | Yes |
| Torins | mTOR inhibitor | Yes |
| Perifosine | AKT inhibitor | Yes |
| Ezetimibe | AMPK activator; MAPK/ERK inhibitor | No |
| Carbamazepine | Ins and IP3 * inhibitor | No |
| Sodium valproate | Ins and IP3 * inhibitor | No |
| Xestospongin B | IP3 receptor inhibitor | No |
| Xestospongin C | IP3 receptor inhibitor | No |
| Lithium chloride | IMPase ** inhibitor | No |
| Trehalose | Glucose transporter inhibitor; AMPK activator | No |
| Amiodarone | Calcium channel blocker | No |
| (a). Scientific Evidence that Activating Autophagy Is Promoting Liver Regeneration | |||||||
|---|---|---|---|---|---|---|---|
| Author Year | Research Model | Pathway | Autophagy Modulation | Parameters Indicating | |||
| Enhanced Autophagy | Enhanced Regeneration | Reduced Autophagy | Reduced Regeneration | ||||
| Takeo [13] 2014 | Mice | Not investigated | Atg5-KO | LC3-II: --- p62: +++ | BrdU: --- | ||
| Lin [26] 2015 | C57BL/6 mice, Male | mTOR- independent | Amiodarone | LC3-II: +++ p62: --- | LBWR *: +++ Ki-67: +++ | ||
| Atg7-KD | LC3-II: --- | LBWR *: --- Ki-67: --- | |||||
| Cheng [29] 2015 | Liver progenitor cells | Not investigated | Overexpression of Beclin1 | LC3-II: +++ | PAS ×: +++ | ||
| Beclin1-KD; Atg5-KD | LC3-II: --- p62: +++ | CCK-8: --- PAS: --- | |||||
| Liu [5] 2018 | SD rats, Male, 3m, 22m; Primary rat hepatocytes | Not investigated | Young plasma | LC3-II: +++ p62: --- | Ki-67: +++ | ||
| 3-Methyladenine; Wortmannin | LC3-II: --- | Ki-67: --- | |||||
| Jia [28] 2019 | SD rats, Male, 5w | Not investigated | 70% Portal Vein Ligation | LC3-II: +++ | Cyclin D1: +++ | ||
| Guha [164] 2019 | Mice; MEFs; HEK293T cells. | IPMK-AMPK-ULK1; IPMK-AMPK-SIRT1 | IPMK-KO | LC3-II: --- | Ki-67: --- Edu: --- | ||
| (b). Scientific Evidence that Inducing Autophagy Is Inhibiting Liver Regeneration | |||||||
| Author Year | Research Model | Pathway | Autophagy Modulation | Parameters Indicating | |||
| Enhanced Autophagy | Reduced Regeneration | Reduced Autophagy | Enhanced Regeneration | ||||
| Jiang [161] 2001 | Harlan SD rats, Male | mTOR- dependent | Rapamycin | Not investigated | LWRR **: --- | ||
| Palme [159] 2008 | Lewis rats, Male | mTOR- dependent | Rapamycin | Not investigated | Ki-67: --- | ||
| Fouraschen [160] 2013 | C57BL/6 mice Male, 12–15w | mTOR- dependent | Rapamycin and Steroid dexamethasone | LC3-II: +++ | BrdU: --- PCNA: --- LWRR **: --- | ||
| Kawaguchi [27] 2013 | C57BL/6J mice, Male, 6–8w | mTOR- dependent | Temsirolimus | Not investigated | LBWR *: --- PCNA: --- | ||
| Shi [25] 2018 | Balb/c mice, 6–8w | mTOR- dependent | Rapamycin | LC3-II: +++ | LBWR *: --- PCNA: --- | ||
| 3-Methyladenine; | LC3-II: --- | LBWR *: +++ PCNA: +++ | |||||
| ASPP2-haploinsufficient | LC3-II: --- p62: +++ | LBWR *: +++ PCNA: +++ | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, F.; Hua, C.; Tautenhahn, H.-M.; Dirsch, O.; Dahmen, U. The Role of Autophagy for the Regeneration of the Aging Liver. Int. J. Mol. Sci. 2020, 21, 3606. https://doi.org/10.3390/ijms21103606
Xu F, Hua C, Tautenhahn H-M, Dirsch O, Dahmen U. The Role of Autophagy for the Regeneration of the Aging Liver. International Journal of Molecular Sciences. 2020; 21(10):3606. https://doi.org/10.3390/ijms21103606
Chicago/Turabian StyleXu, Fengming, Chuanfeng Hua, Hans-Michael Tautenhahn, Olaf Dirsch, and Uta Dahmen. 2020. "The Role of Autophagy for the Regeneration of the Aging Liver" International Journal of Molecular Sciences 21, no. 10: 3606. https://doi.org/10.3390/ijms21103606
APA StyleXu, F., Hua, C., Tautenhahn, H.-M., Dirsch, O., & Dahmen, U. (2020). The Role of Autophagy for the Regeneration of the Aging Liver. International Journal of Molecular Sciences, 21(10), 3606. https://doi.org/10.3390/ijms21103606