Antioxidant and Neuroprotective Effects Induced by Cannabidiol and Cannabigerol in Rat CTX-TNA2 Astrocytes and Isolated Cortexes

 ,
, 


 ,
, 

 ,
,  , and
, and 
Abstract
1. Introduction
2. Results
2.1. Antioxidant Activity
2.2. In Vitro Model
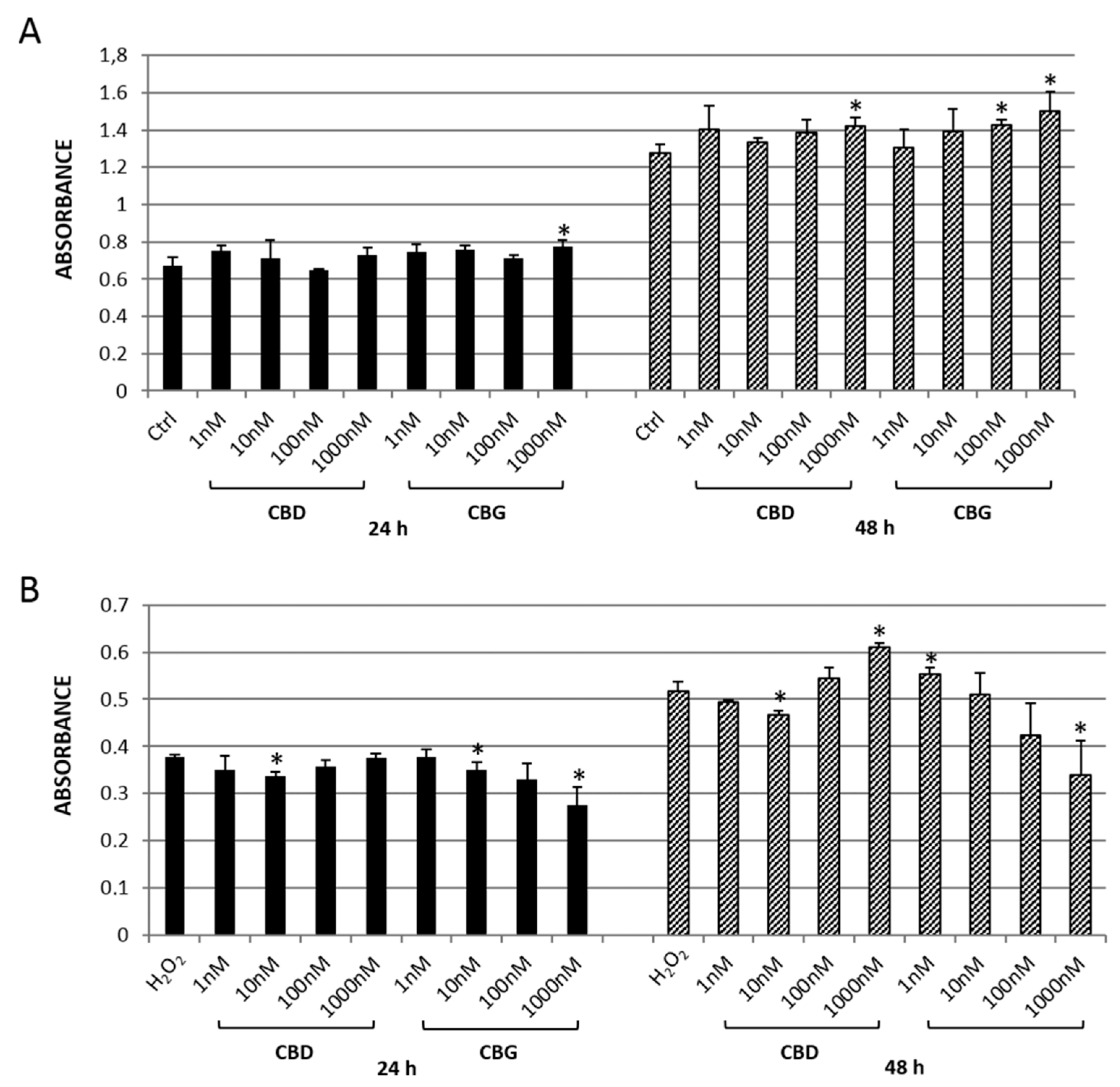
2.2.1. MTT Assay
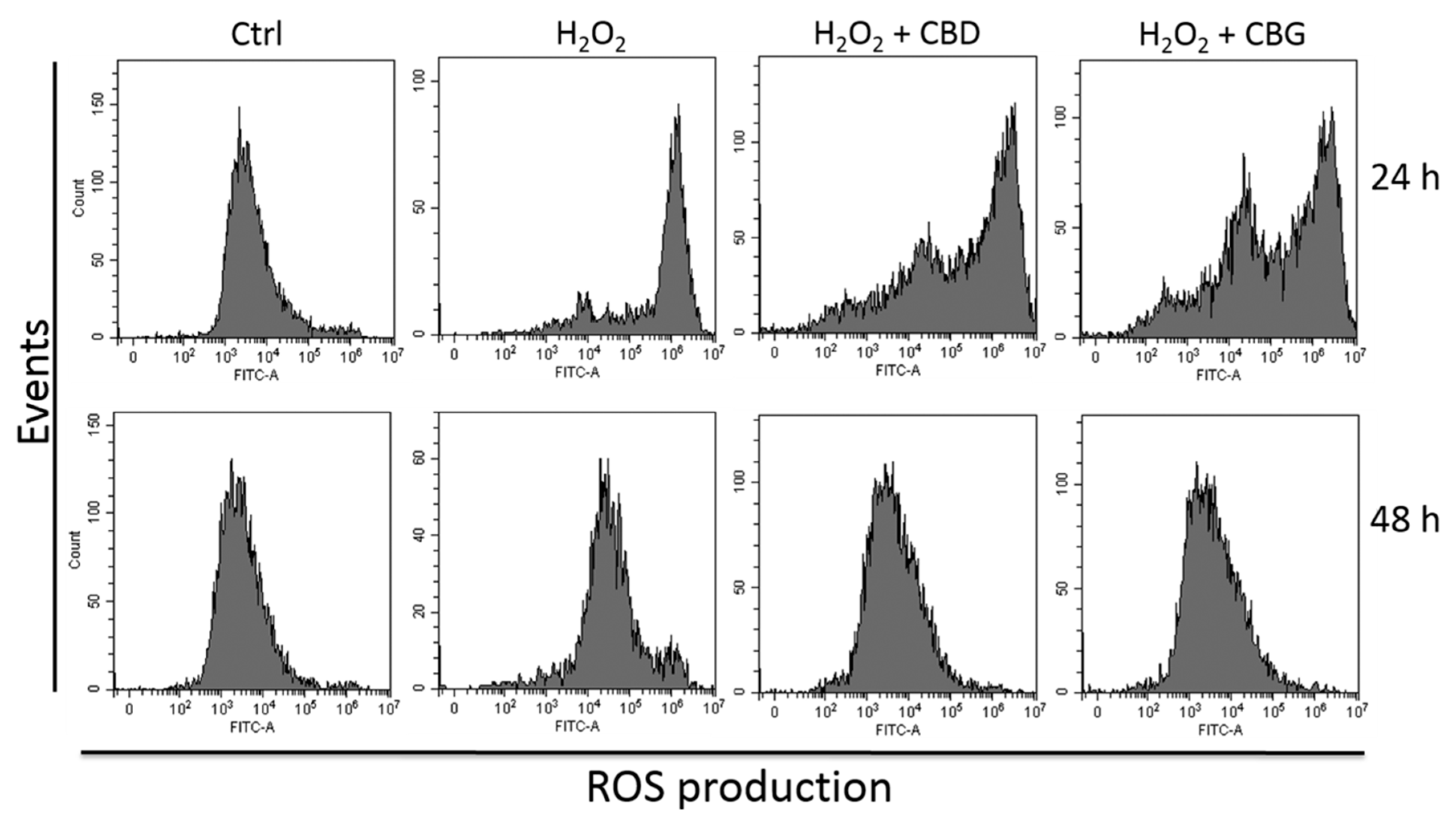
2.2.2. Reactive Oxygen Species (ROS) Production
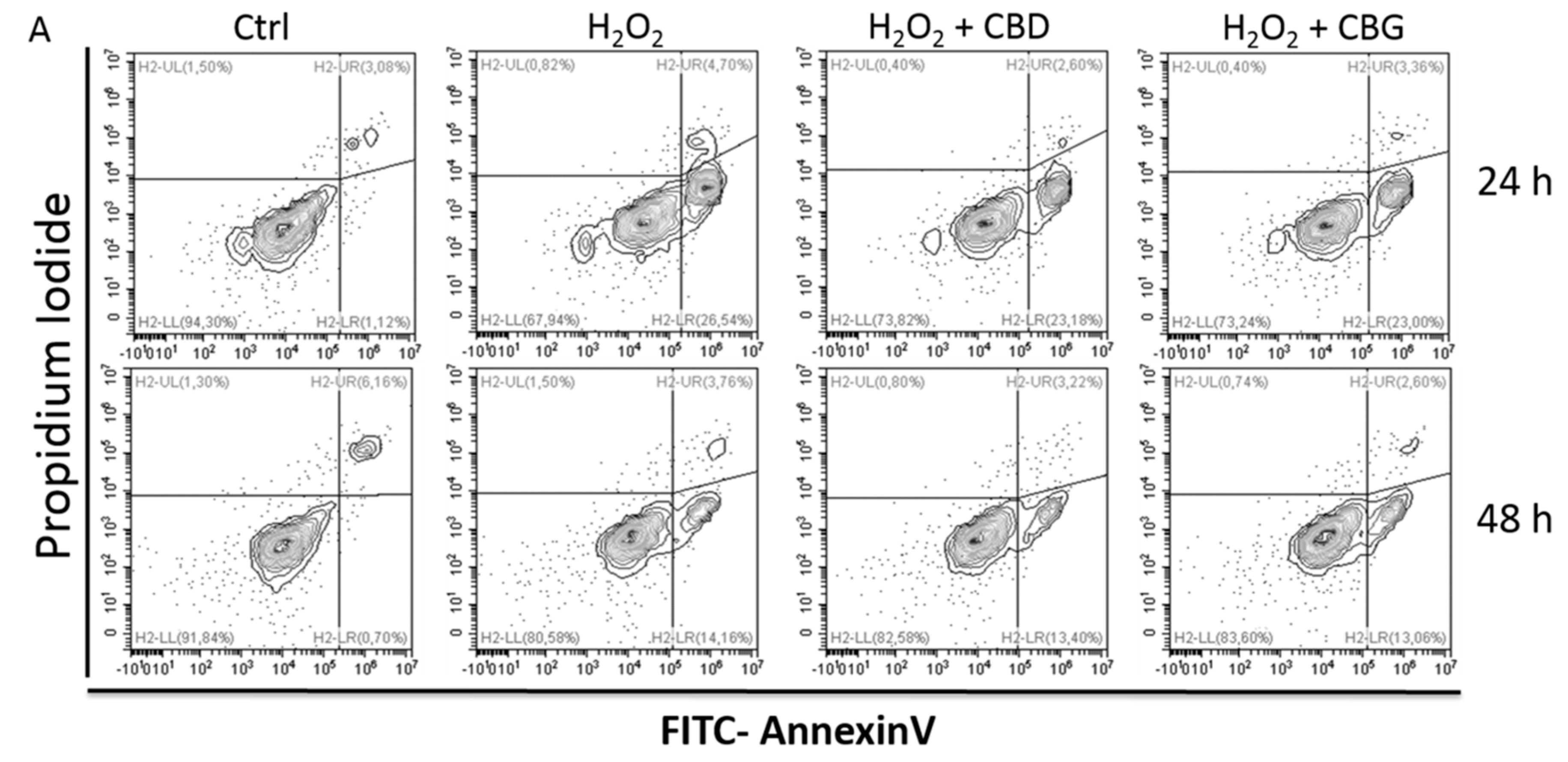
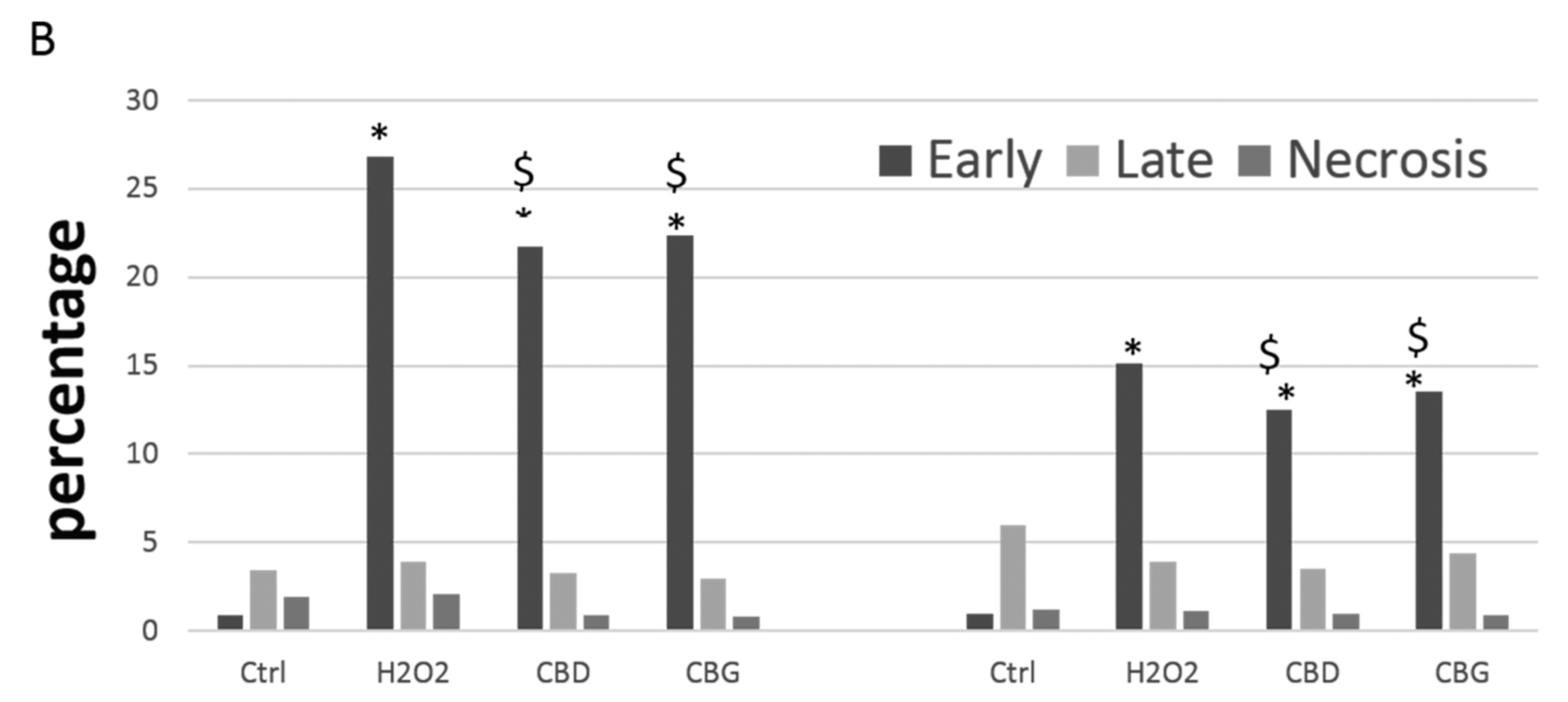
2.2.3. Apoptosis Occurrence
2.2.4. Western Blotting Analysis
2.3. Ex Vivo Studies
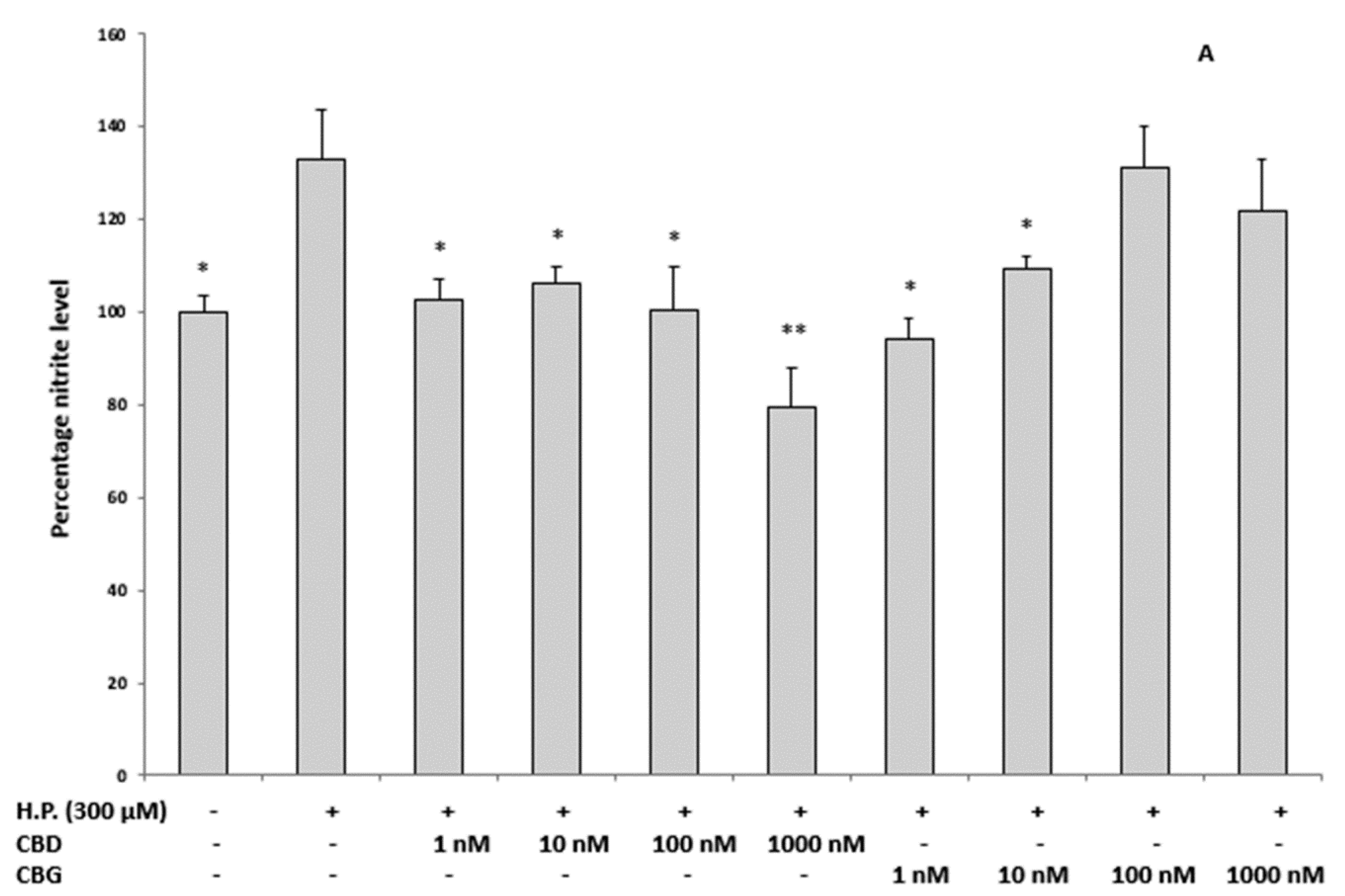
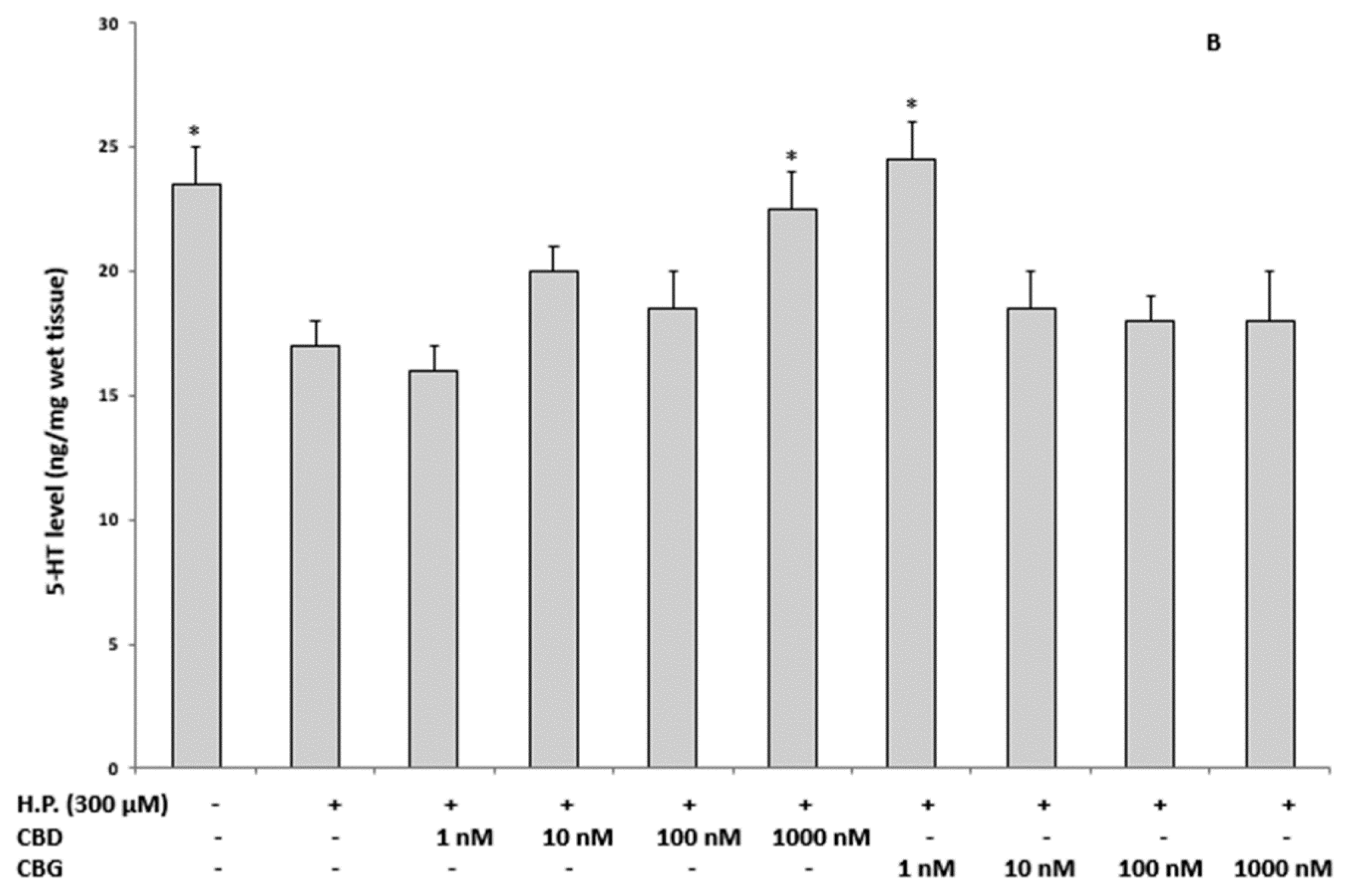
2.3.1. Oxidative Stress and Nitrite Production and Serotonin (5-HT) Concentration
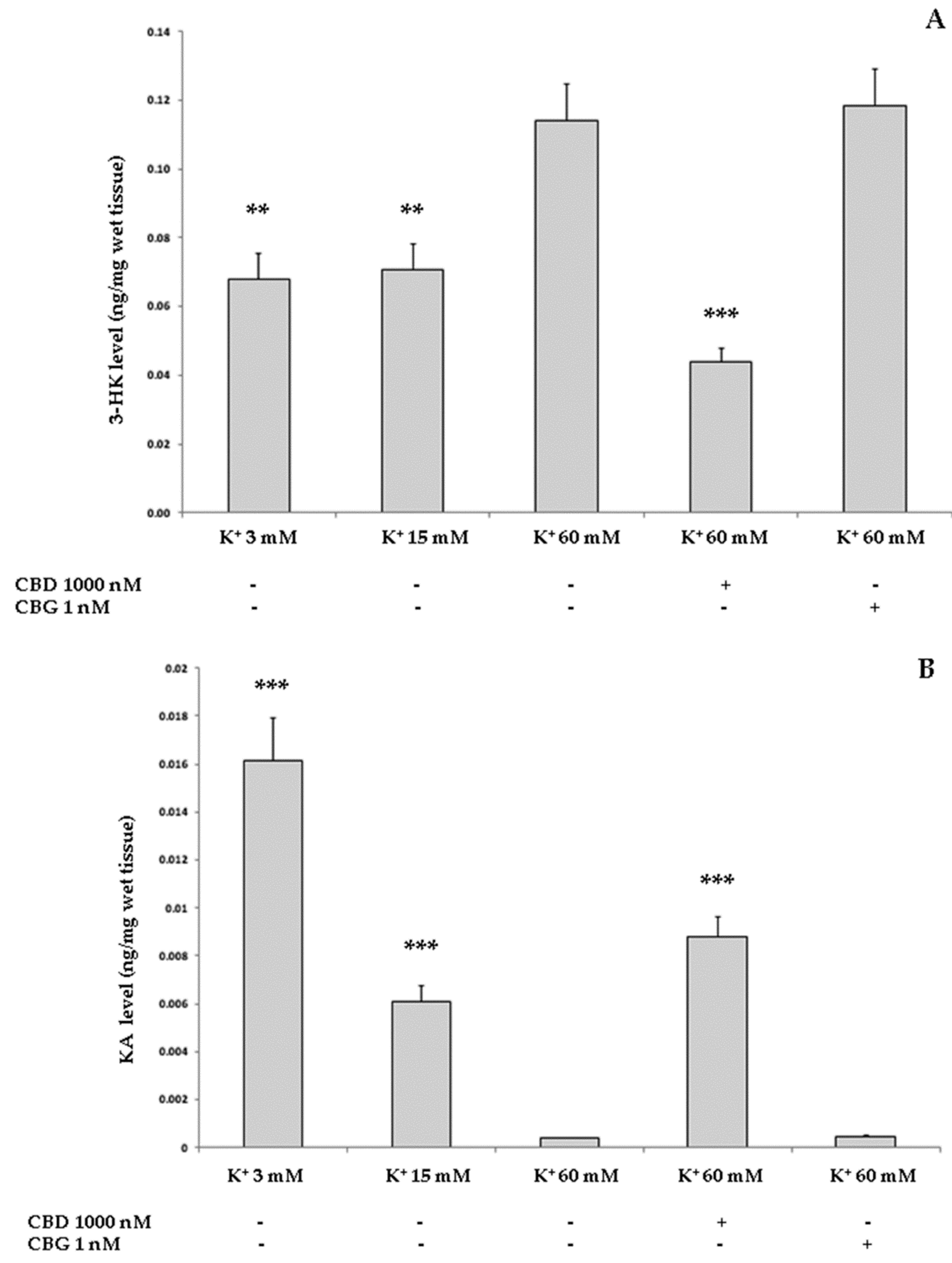
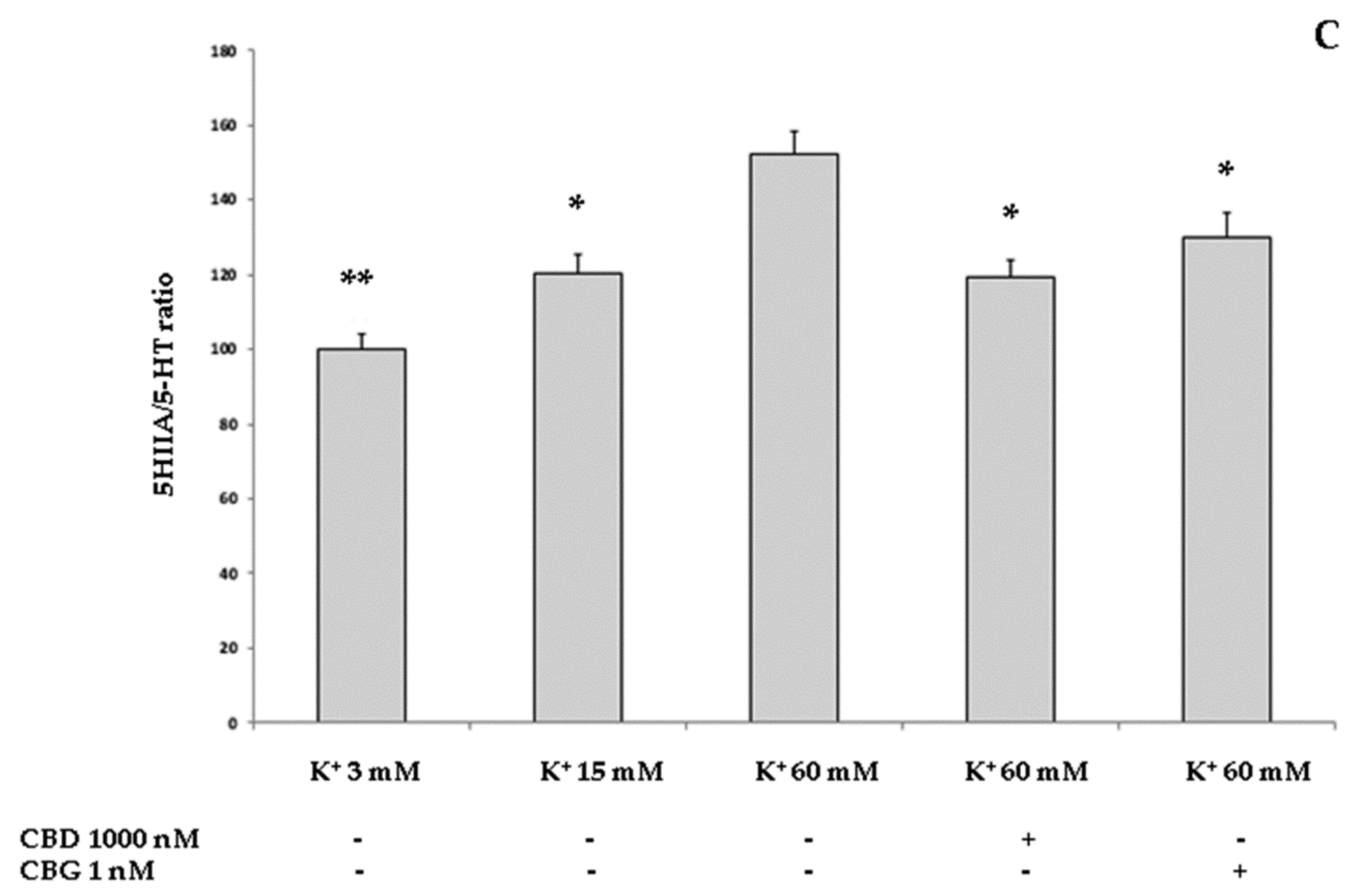
2.3.2. Neurotoxic Stimulus and 3-Hydrokinurenine (3-HK) and Kynurenic acid (KA) Production, and 5-HT Turnover
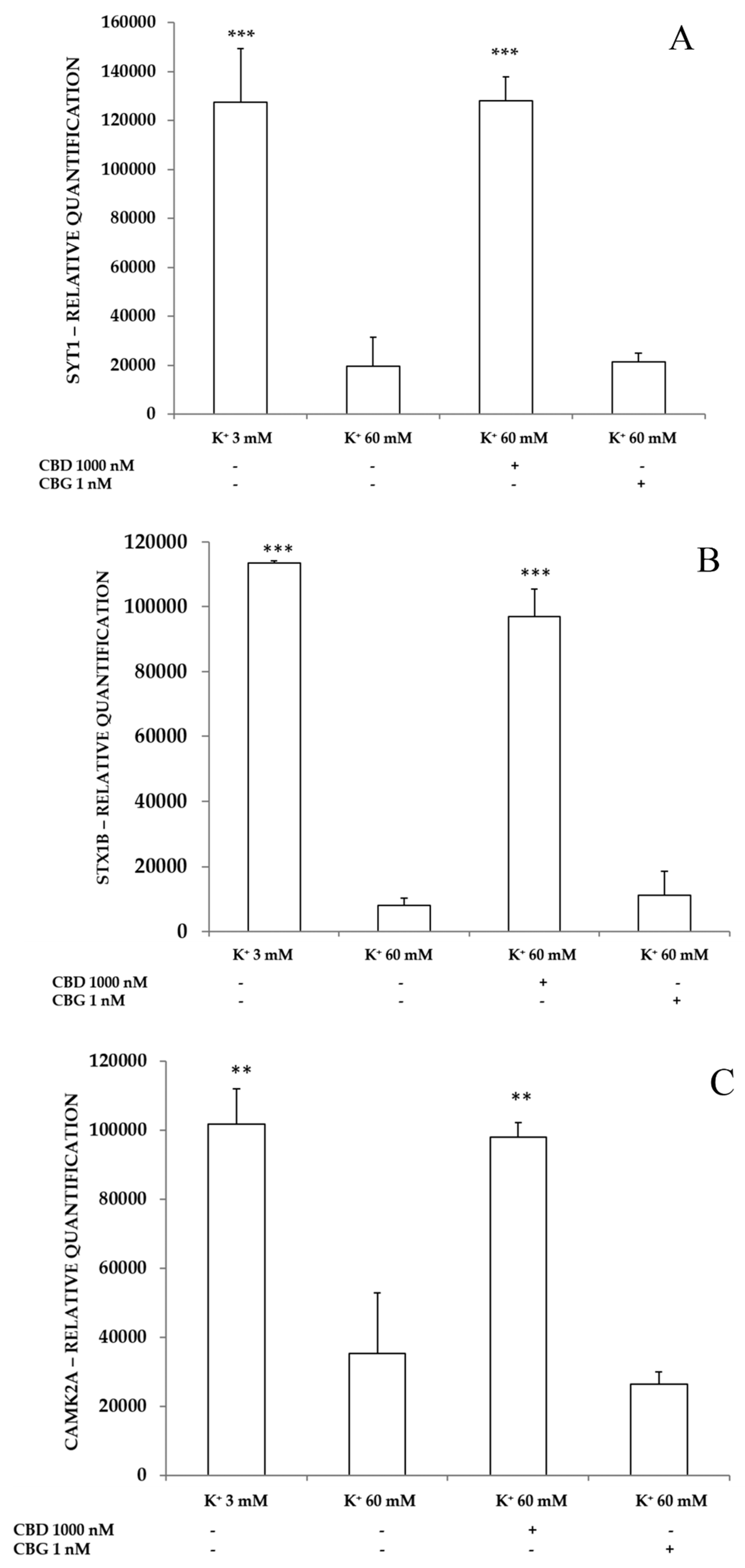
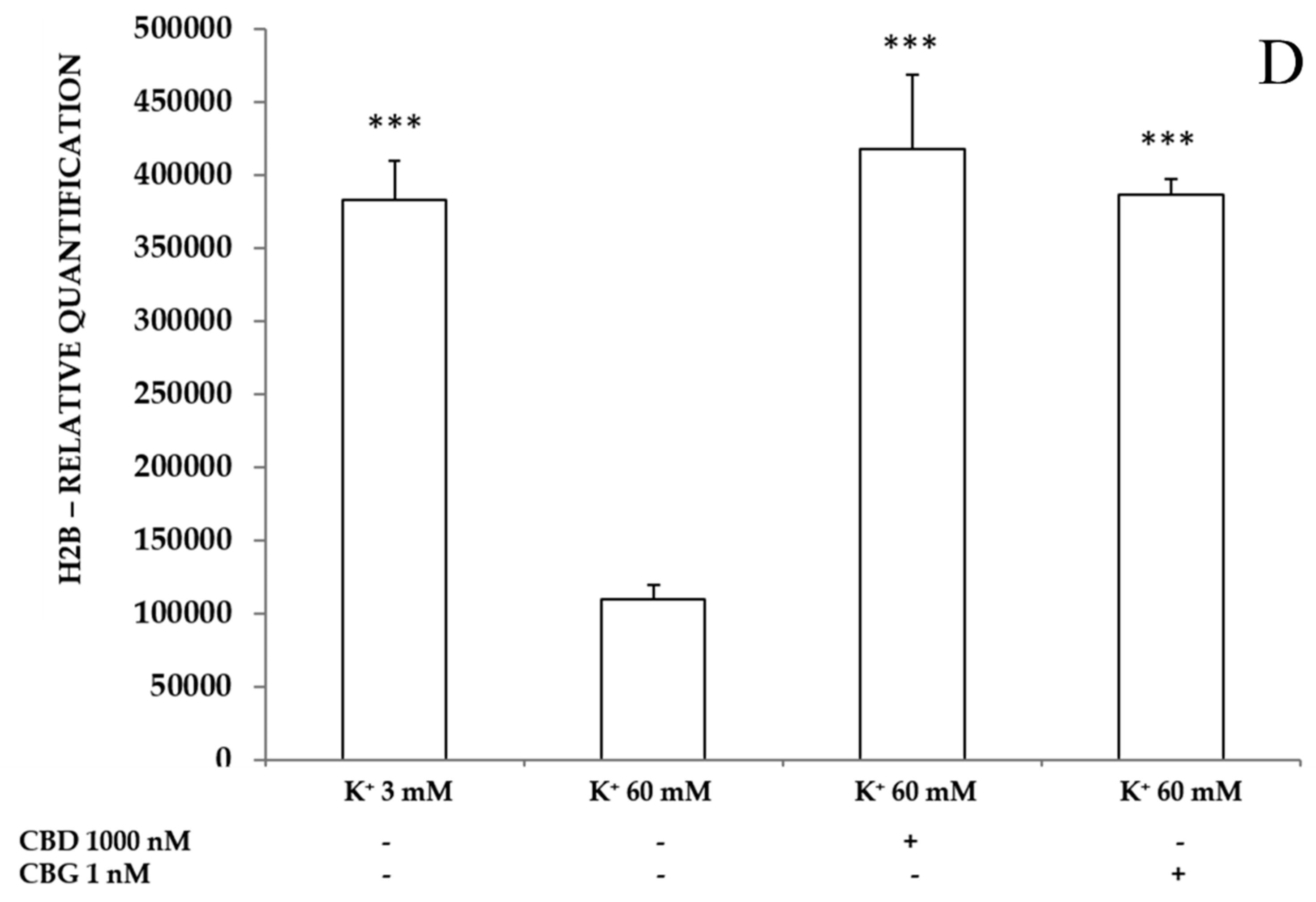
2.3.3. Neurotoxic Stimulus and Proteomic Analysis
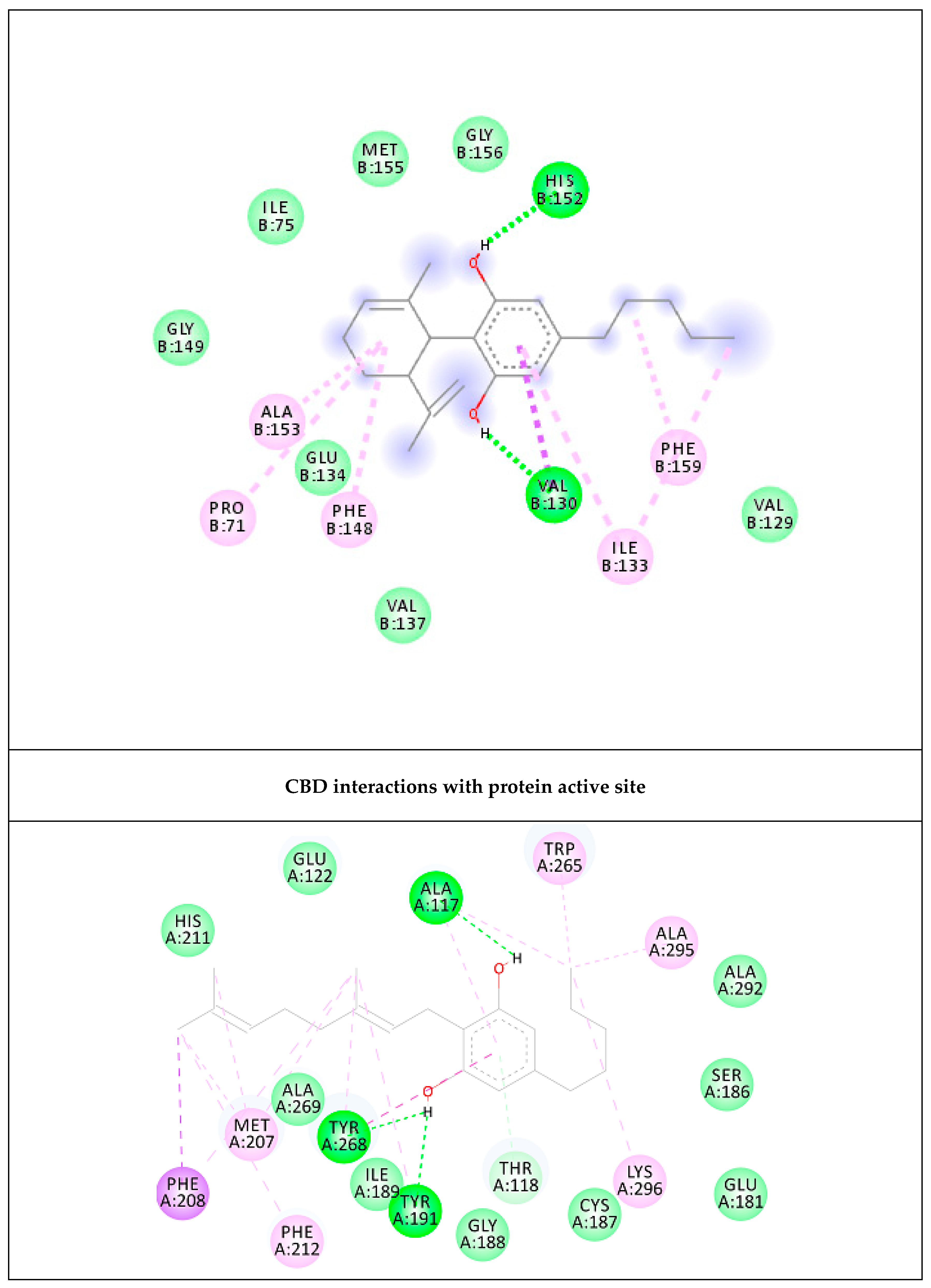
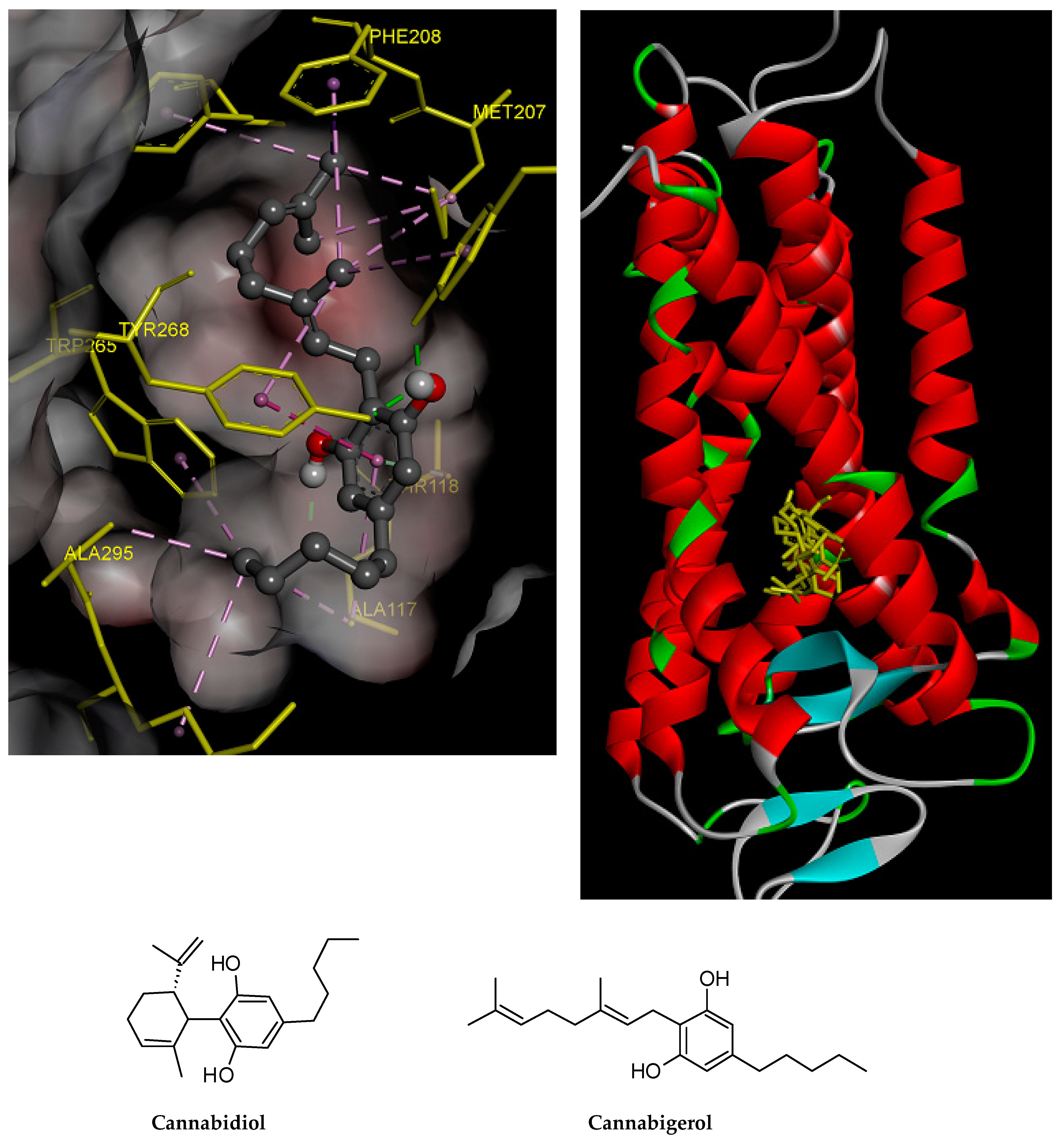
2.4. Prediction of Putative Target and Results of Docking Analysis
3. Discussion
4. Materials and Methods
4.1. Drugs
4.2. Determination of Antioxidant and Enzyme Inhibitory Effects
4.3. In Vitro Studies
4.3.1. Cell Culture and Treatment
4.3.2. MTT Assay
4.3.3. Flow Cytometry Analysis of Reactive Oxygen Species (ROS) Production
4.3.4. Flow Cytometry Apoptosis Detection
4.3.5. Western Blotting Analysis
4.4. Ex Vivo Studies
4.4.1. Oxidative Stress and Nitrite Production
4.4.2. A Neurotoxicity Paradigm
4.4.3. High Performance Liquid Chromatography (HPLC) Determination of Serotonin (5-HT), 5-Hydroxyindoleacetic (5HIIA) and 3-Hydroxykinurenine (3-HK)
4.4.4. HPLC–Fluorimetric Determination of Kinurenic Acid (KA)
4.4.5. Proteomic Analysis
4.5. Bioinformatic and Docking Studies
4.5.1. Bioinformatic Analysis: Prediction of Putative Target Proteins
4.5.2. Docking Calculations
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fasinu, P.S.; Phillips, S.; ElSohly, M.A.; Walker, L.A. Current status and prospects for cannabidiol preparations as new therapeutic agents. Pharmacotherapy 2016, 36, 781–796. [Google Scholar] [CrossRef] [PubMed]
- Citti, C.; Linciano, P.; Russo, F.; Luongo, L.; Iannotta, M.; Maione, S.; Laganà, A.; Capriotti, A.L.; Forni, F.; Vandelli, M.A.; et al. A novel phytocannabinoid isolated from Cannabis sativa L. with an in vivo cannabimimetic activity higher than Δ9-tetrahydrocannabinol: Δ9-Tetrahydrocannabiphorol. Sci. Rep. 2019, 9, 20335. [Google Scholar] [CrossRef] [PubMed]
- Marchalant, Y.; Brothers, H.M.; Norman, G.J.; Karelina, K.; DeVries, A.C.; Wenk, G.L. Cannabinoids attenuate the effects of aging upon neuroinflammation and neurogenesis. Neurobiol. Dis. 2009, 34, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Aso, E.; Ferrer, I. Cannabinoids for treatment of Alzheimer’s disease: Moving toward the clinic. Front. Pharmacol. 2014, 5, 37. [Google Scholar] [CrossRef]
- Muller, C.; Morales, P.; Reggio, P.H. Cannabinoid ligands targeting TRP channels. Front. Mol. Neurosci. 2018, 11, 487. [Google Scholar] [CrossRef]
- Bi, G.H.; Galaj, E.; He, Y.; Xi, Z.X. Cannabidiol inhibits sucrose self-administration by CB1 and CB2 receptor mechanisms in rodents. Addict. Biol. 2019, e12783. [Google Scholar] [CrossRef]
- Devinsky, O.; Cilio, M.R.; Cross, H.; Fernandez-Ruiz, J.; French, J.; Hill, C.; Katz, R.; Di Marzo, V.; Jutras-Aswad, D.; Notcutt, W.G.; et al. Cannabidiol: Pharmacology and potential therapeutic role in epilepsy and other neuropsychiatric disorders. Epilepsia 2014, 55, 791–802. [Google Scholar] [CrossRef]
- Devinsky, O.; Marsh, E.; Friedman, D.; Thiele, E.; Laux, L.; Sullivan, J.; Miller, I.; Flamini, R.; Wilfong, A.; Filloux, F.; et al. Cannabidiol in patients with treatment-resistant epilepsy: An open-label interventional trial. Lancet Neurol. 2016, 15, 270–278. [Google Scholar] [CrossRef]
- Atalay, S.; Jarocka-Karpowicz, I.; Skrzydlewska, E. Antioxidative and Anti-Inflammatory Properties of Cannabidiol. Antioxidants (Basel) 2019, 9, 21. [Google Scholar] [CrossRef]
- Cascio, M.G.; Gauson, L.A.; Stevenson, L.A.; Ross, R.A.; Pertwee, R.G. Evidence that the plant cannabinoid cannabigerol is a highly potent alpha2-adrenoceptor agonist and moderately potent 5HT1A receptor antagonist. Br. J. Pharmacol. 2010, 159, 129–141. [Google Scholar] [CrossRef]
- di Giacomo, V.; Chiavaroli, A.; Orlando, G.; Cataldi, A.; Rapino, M.; Di Valerio, V.; Leone, S.; Brunetti, L.; Menghini, L.; Recinella, L.; et al. Neuroprotective and Neuromodulatory Effects Induced by Cannabidiol and Cannabigerol in Rat Hypo-E22 cells and Isolated Hypothalamus. Antioxidants (Basel) 2020, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Boison, D.; Steinhäuser, C. Epilepsy and astrocyte energy metabolism. Glia 2018, 66, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Qi, G.; Mi, Y.; Yin, F. Cellular Specificity and Inter-cellular Coordination in the Brain Bioenergetic System: Implications for Aging and Neurodegeneration. Front. Physiol. 2020, 8, 1531. [Google Scholar] [CrossRef] [PubMed]
- Baxter, P.S.; Hardingham, G.E. Adaptive regulation of the brain’s antioxidant defences by neurons and astrocytes. Free Radic. Biol. Med. 2016, 100, 147–152. [Google Scholar] [CrossRef]
- Gupta, K.; Patani, R.; Baxter, P.; Serio, A.; Story, D.; Tsujita, T.; Hayes, J.D.; Pedersen, R.A.; Hardingham, G.E.; Chandran, S. Human embryonic stem cell derived astrocytes mediate non-cell-autonomous neuroprotection through endogenous and drug-induced mechanisms. Cell Death Differ. 2012, 19, 779–787. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef]
- Vázquez, C.; Tolón, R.M.; Pazos, M.R.; Moreno, M.; Koester, E.C.; Cravatt, B.F.; Hillard, C.J.; Romero, J. Endocannabinoids regulate the activity of astrocytic hemichannels and the microglial response against an injury: In vivo studies. Neurobiol. Dis. 2015, 79, 41–50. [Google Scholar] [CrossRef]
- Ceprián, M.; Jiménez-Sánchez, L.; Vargas, C.; Barata, L.; Hind, W.; Martínez-Orgado, J. Cannabidiol reduces brain damage and improves functional recovery in a neonatal rat model of arterial ischemic stroke. Neuropharmacology 2017, 116, 151–159. [Google Scholar] [CrossRef]
- Lafuente, H.; Alvarez, F.J.; Pazos, M.R.; Alvarez, A.; Rey-Santano, M.C.; Mielgo, V.; Murgia-Esteve, X.; Hilario, E.; Martinez-Orgado, J. Cannabidiol reduces brain damage and improves functional recovery after acute hypoxia-ischemia in newborn pigs. Pediatr. Res. 2011, 70, 272–277. [Google Scholar] [CrossRef]
- Rodríguez-Cueto, C.; Santos-García, I.; García-Toscano, L.; Espejo-Porras, F.; Bellido, M.; Fernández-Ruiz, J.; Muñoz, E.; de Lago, E. Neuroprotective effects of the cannabigerol quinone derivative VCE-003.2 in SOD1G93A transgenic mice, an experimental model of amyotrophic lateral sclerosis. Biochem. Pharmacol. 2018, 157, 217–226. [Google Scholar] [CrossRef]
- Gugliandolo, A.; Pollastro, F.; Grassi, G.; Bramanti, P.; Mazzon, E. In Vitro Model of Neuroinflammation: Efficacy of Cannabigerol, a Non-Psychoactive Cannabinoid. Int. J. Mol. Sci. 2018, 19, 1992. [Google Scholar] [CrossRef] [PubMed]
- Di Giacomo, V.; Ferrante, C.; Ronci, M.; Cataldi, A.; Di Valerio, V.; Rapino, M.; Recinella, L.; Chiavaroli, A.; Leone, S.; Vladimir-Knežević, S.; et al. Multiple pharmacological and toxicological investigations on Tanacetum parthenium and Salix alba extracts: Focus on potential application as anti-migraine agents. Food Chem. Toxicol. 2019, 133, 110783. [Google Scholar] [CrossRef] [PubMed]
- Raiteri, L.; Stigliani, S.; Zedda, L.; Raiteri, M.; Bonanno, G. Multiple mechanisms of transmitter release evoked by “pathologically” elevated extracellular [K+]: Involvement of transporter reversal and mitochondrial calcium. J. Neurochem. 2002, 80, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Maddison, D.C.; Giorgini, F. The kynurenine pathway and neurodegenerative disease. Semin. Cell. Dev. Biol. 2015, 40, 134–141. [Google Scholar] [CrossRef]
- Hamelink, C.; Hampson, A.; Wink, D.A.; Eiden, L.E.; Eskay, R.L. Comparison of cannabidiol, antioxidants, and diuretics in reversing binge ethanol-induced neurotoxicity. J. Pharmacol. Exp. Ther. 2005, 314, 780–788. [Google Scholar] [CrossRef]
- Öztaskın, N.; Taslimi, P.; Maraş, A.; Gülcin, İ.; Göksu, S. Novel antioxidant bromophenols with acetylcholinesterase, butyrylcholinesterase and carbonic anhydrase inhibitory actions. Bioorg. Chem. 2017, 74, 104–114. [Google Scholar] [CrossRef]
- Borrelli, F.; Aviello, G.; Romano, B.; Orlando, P.; Capasso, R.; Maiello, F.; Guadagno, F.; Petrosino, S.; Capasso, F.; Di Marzo, V.; et al. Cannabidiol, a safe and non-psychotropic ingredient of the marijuana plant Cannabis sativa, is protective in a murine model of colitis. J. Mol. Med. (Berl.) 2009, 87, 1111–1121. [Google Scholar] [CrossRef]
- Juknat, A.; Pietr, M.; Kozela, E.; Rimmerman, N.; Levy, R.; Gao, F.; Coppola, G.; Geschwind, D.; Vogel, Z. Microarray and pathway analysis reveal distinct mechanisms underlying cannabinoid-mediated modulation of LPS-induced activation of BV-2 microglial cells. PLoS ONE 2013, 8, e61462. [Google Scholar] [CrossRef]
- Juknat, A.; Pietr, M.; Kozela, E.; Rimmerman, N.; Levy, R.; Coppola, G.; Geschwind, D.; Vogel, Z. Differential transcriptional profiles mediated by exposure to the cannabinoids cannabidiol and D9-tetrahydrocannabinol in BV-2 microglial cells. Br. J. Pharmacol. 2012, 165, 2512–2528. [Google Scholar] [CrossRef]
- Abnosi, M.H.; Yari, S. The toxic effect of gallic acid on biochemical factors, viability and proliferation of rat bone marrow mesenchymal stem cells was compensated by boric acid. J. Trace Elem. Med. Biol. 2018, 48, 246–253. [Google Scholar] [CrossRef]
- Halliwell, B.; Clement, M.V.; Ramalingam, J.; Long, L.H. Hydrogen peroxide. Ubiquitous in cell culture and in vivo? IUBMB Life. 2000, 50, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.; Leibfritz, D.; Richter-Landsberg, C. Oxidative stress-induced metabolic alterations in rat brain astrocytes studied by multinuclear NMR spectroscopy. J. Neurosci. Res. 1999, 58, 576–585. [Google Scholar] [CrossRef]
- Gómez Del Pulgar, T.; De Ceballos, M.L.; Guzmán, M.; Velasco, G. Cannabinoids protect astrocytes from ceramide-induced apoptosis through the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 2002, 277, 36527–36533. [Google Scholar] [CrossRef] [PubMed]
- Giacoppo, S.; Soundara Rajan, T.; Galuppo, M.; Pollastro, F.; Grassi, G.; Bramanti, P.; Mazzon, E. Purified Cannabidiol, the main non-psychotropic component of Cannabis sativa, alone, counteracts neuronal apoptosis in experimental multiple sclerosis. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 4906–4919. [Google Scholar]
- Da Silva, V.K.; de Freitas, B.S.; Garcia, R.C.L.; Monteiro, R.T.; Hallak, J.E.; Zuardi, A.W.; Crippa, J.A.S.; Schröder, N. Antiapoptotic effects of cannabidiol in an experimental model of cognitive decline induced by brain iron overload. Transl. Psychiatr. 2018, 8, 176. [Google Scholar] [CrossRef]
- Chung-Ha, O.D.; Kim, K.-Y.; Bushong, E.A.; Mills, E.A.; Boassa, D.; Shih, T.; Kinebuchi, M.; Phan, S.; Zhou, Y.; Bihlmeyer, N.A.; et al. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. USA 2014, 111, 9633–9638. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef]
- Borrelli, F.; Fasolino, I.; Romano, B.; Capasso, R.; Maiello, F.; Coppola, D.; Orlando, P.; Battista, G.; Pagano, E.; Di Marzo, V.; et al. Beneficial effect of the non-psychotropic plant cannabinoid cannabigerol on experimental inflammatory bowel disease. Biochem. Pharmacol. 2013, 85, 1306–1316. [Google Scholar] [CrossRef]
- Ramis, M.R.; Sarubbo, F.; Terrasa, J.L.; Moranta, D.; Aparicio, S.; Miralles, A.; Esteban, S. Chronic α-Tocopherol Increases Central Monoamines Synthesis and Improves Cognitive and Motor Abilities in Old Rats. Rejuvenation Res. 2016, 19, 159–171. [Google Scholar] [CrossRef]
- Karl, T.; Garner, B.; Cheng, D. The therapeutic potential of the phytocannabinoid cannabidiol for Alzheimer’s disease. Behav. Pharmacol. 2017, 28, 142–160. [Google Scholar] [CrossRef] [PubMed]
- Hughes, B.; Herron, C.E. Cannabidiol Reverses Deficits in Hippocampal LTP in a Model of Alzheimer’s Disease. Neurochem. Res. 2019, 44, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Hu, S.; Kang, X.; Wang, C. Synaptotagmins: Beyond Presynaptic Neurotransmitter Release. Neuroscientist 2020, 26, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Gerber, S.H.; Rah, J.C.; Min, S.W.; Liu, X.; de Wit, H.; Dulubova, I.; Meyer, A.C.; Rizo, J.; Arancillo, M.; Hammer, R.E.; et al. Conformational switch of syntaxin-1 controls synaptic vesicle fusion. Science 2008, 321, 1507–1510. [Google Scholar] [CrossRef] [PubMed]
- Hell, J.W. CaMKII: Claiming center stage in postsynaptic function and organization. Neuron 2014, 81, 249–265. [Google Scholar] [CrossRef] [PubMed]
- Sales, A.J.; Crestani, C.C.; Guimarães, F.S.; Joca, S.R.L. Antidepressant-like effect induced by Cannabidiol is dependent on brain serotonin levels. Prog. Neuropsychopharmacol. Biol. Psychiatr. 2018, 86, 255–261. [Google Scholar] [CrossRef]
- Thomas, E.A.; D’Mello, S.R. Complex neuroprotective and neurotoxic effects of histone deacetylases. J. Neurochem. 2018, 145, 96–110. [Google Scholar] [CrossRef]
- Zwergel, C.; Valente, S.; Jacob, C.; Mai, A. Emerging approaches for histone deacetylase inhibitor drug discovery. Expert Opin. Drug. Discov. 2015, 10, 599–613. [Google Scholar] [CrossRef]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinlammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef]
- Röder, C.; Bellmann, R.; McCarson, K.E.; Krause, J.E.; Sperk, G. Kainic acid induced seizures cause a marked increase in the expression of neurokinin-3 receptor mRNA in the rat cerebellum. Neurosci. Lett. 1994, 181, 158–160. [Google Scholar] [CrossRef]
- Tooney, P.A.; Au, G.G.; Chahl, L.A. Tachykinin NK1 and NK3 receptors in the prefrontal cortex of the human brain. Clin. Exp. Pharmacol. Physiol. 2000, 27, 947–949. [Google Scholar] [CrossRef] [PubMed]
- Primi, M.C.; Maltarollo, V.G.; Magalhães, J.G.; de Sá, M.M.; Rangel-Yagui, C.O.; Trossini, G.H. Convergent QSAR studies on a series of NK₃ receptor antagonists for schizophrenia treatment. J. Enzyme Inhib. Med. Chem. 2016, 31, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Oishi, S.; Fujii, N. Neuropeptide derivatives to regulate the reproductive axis: Kisspeptin receptor (KISS1R) ligands and neurokinin-3 receptor (NK3R) ligands. Biopolymers 2016, 106, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Orlando, G.; Leone, S.; Ferrante, C.; Chiavaroli, A.; Mollica, A.; Stefanucci, A.; Macedonio, G.; Dimmito, M.P.; Leporini, L.; Menghini, L.; et al. Effects of Kisspeptin-10 on Hypothalamic Neuropeptides and Neurotransmitters Involved in Appetite Control. Molecules 2018, 23, 3071. [Google Scholar] [CrossRef]
- Kojima, S.; Tohei, A.; Ikeda, M.; Anzai, N. An Endogenous Tachykinergic NK2/NK3 Receptor Cascade System Controlling the Release of Serotonin from Colonic Mucosa. Curr. Neuropharmacol. 2015, 13, 830–835. [Google Scholar] [CrossRef]
- Giampietro, L.; Gallorini, M.; De Filippis, B.; Amoroso, R.; Cataldi, A.; di Giacomo, V. PPAR-γ agonist GL516 reduces oxidative stress and apoptosis occurrence in a rat astrocyte cell line. Neurochem. Int. 2019, 126, 239–245. [Google Scholar] [CrossRef]
- Rapino, M.; Di Valerio, V.; Zara, S.; Gallorini, M.; Marconi, G.D.; Sancilio, S.; Marsich, E.; Ghinassi, B.; di Giacomo, V.; Cataldi, A. Chitlac-coated Thermosets Enhance Osteogenesis and Angiogenesis in a Co-culture of Dental Pulp Stem Cells and Endothelial Cells. Nanomaterials (Basel) 2019, 9, 928. [Google Scholar] [CrossRef]
- Gallorini, M.; Sancilio, S.; Zara, S.; De Colli, M.; Di Giulio, M.; Cataldi, A.; di Giacomo, V. Involvement of mitochondrial signalling pathway in HGFs/S. mitis coculture response to TEGDMA treatment. J. Biomed. Mater. Res. A 2014, 102, 3931–3938. [Google Scholar] [CrossRef]
- Sancilio, S.; Gallorini, M.; Cataldi, A.; Sancillo, L.; Rana, R.A.; di Giacomo, V. Modifications in Human Oral Fibroblast Ultrastructure, Collagen Production, and Lysosomal Compartment in Response to Electronic Cigarette Fluids. J. Periodontol. 2017, 88, 673–680. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AChE Inhibition | BChE Inhibition | |
|---|---|---|
| CBD | 1.04 ± 0.02 a | 1.88 ± 0.11 a |
| CBG | 1.15 ± 0.03 b | 1.48 ± 0.11 b |
| Galantamine | 0.01 ± 0.001 c | 0.02 ± 0.002 c |
| DPPH Scavenging Ability Test | ABTS Scavenging Ability Test | Ferric Reducing Antioxidant Power (FRAP) | Cupric Reducing Antioxidant Capacity (CUPRAC) | |
|---|---|---|---|---|
| CBD | 1.87 ± 0.04 b | 1.04 ± 0.01 a | >2 | 0.94 ± 0.01 a |
| CBG | 1.95 ± 0.01 a | 1.03 ± 0.01 b | >2 | 0.59 ± 0.01 b |
| Trolox | 0.21 ± 0.01 c | 0.41 ± 0.01 c | 0.18 ± 0.01 | 0.50 ± 0.01 c |
| Targets | ∆G (Ki) | Key Residues | No. of HB |
|---|---|---|---|
| CBD | −6.72 (12.04 µM) | His152 (HB), Val130 (HB), Ala153, Ile133, Pro71, Phe159, Phe148 | 2 |
| CBG | −10.36 (25.5 nM) | Tyr191 (HB), Ala117 (HB), Tyr268 (HB & pi–pi), Trp265, Ala295, Lys296, Phe212, Met207, Phe208 | 3 |
| Control RET | −10.81 (11.8 nM) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
di Giacomo, V.; Chiavaroli, A.; Recinella, L.; Orlando, G.; Cataldi, A.; Rapino, M.; Di Valerio, V.; Ronci, M.; Leone, S.; Brunetti, L.; et al. Antioxidant and Neuroprotective Effects Induced by Cannabidiol and Cannabigerol in Rat CTX-TNA2 Astrocytes and Isolated Cortexes. Int. J. Mol. Sci. 2020, 21, 3575. https://doi.org/10.3390/ijms21103575
di Giacomo V, Chiavaroli A, Recinella L, Orlando G, Cataldi A, Rapino M, Di Valerio V, Ronci M, Leone S, Brunetti L, et al. Antioxidant and Neuroprotective Effects Induced by Cannabidiol and Cannabigerol in Rat CTX-TNA2 Astrocytes and Isolated Cortexes. International Journal of Molecular Sciences. 2020; 21(10):3575. https://doi.org/10.3390/ijms21103575
Chicago/Turabian Styledi Giacomo, Viviana, Annalisa Chiavaroli, Lucia Recinella, Giustino Orlando, Amelia Cataldi, Monica Rapino, Valentina Di Valerio, Maurizio Ronci, Sheila Leone, Luigi Brunetti, and et al. 2020. "Antioxidant and Neuroprotective Effects Induced by Cannabidiol and Cannabigerol in Rat CTX-TNA2 Astrocytes and Isolated Cortexes" International Journal of Molecular Sciences 21, no. 10: 3575. https://doi.org/10.3390/ijms21103575
APA Styledi Giacomo, V., Chiavaroli, A., Recinella, L., Orlando, G., Cataldi, A., Rapino, M., Di Valerio, V., Ronci, M., Leone, S., Brunetti, L., Menghini, L., Zengin, G., Ak, G., Abdallah, H. H., & Ferrante, C. (2020). Antioxidant and Neuroprotective Effects Induced by Cannabidiol and Cannabigerol in Rat CTX-TNA2 Astrocytes and Isolated Cortexes. International Journal of Molecular Sciences, 21(10), 3575. https://doi.org/10.3390/ijms21103575