PHLD Class Proteins: A Family of New Players in the p53 Network


Abstract
1. Introduction
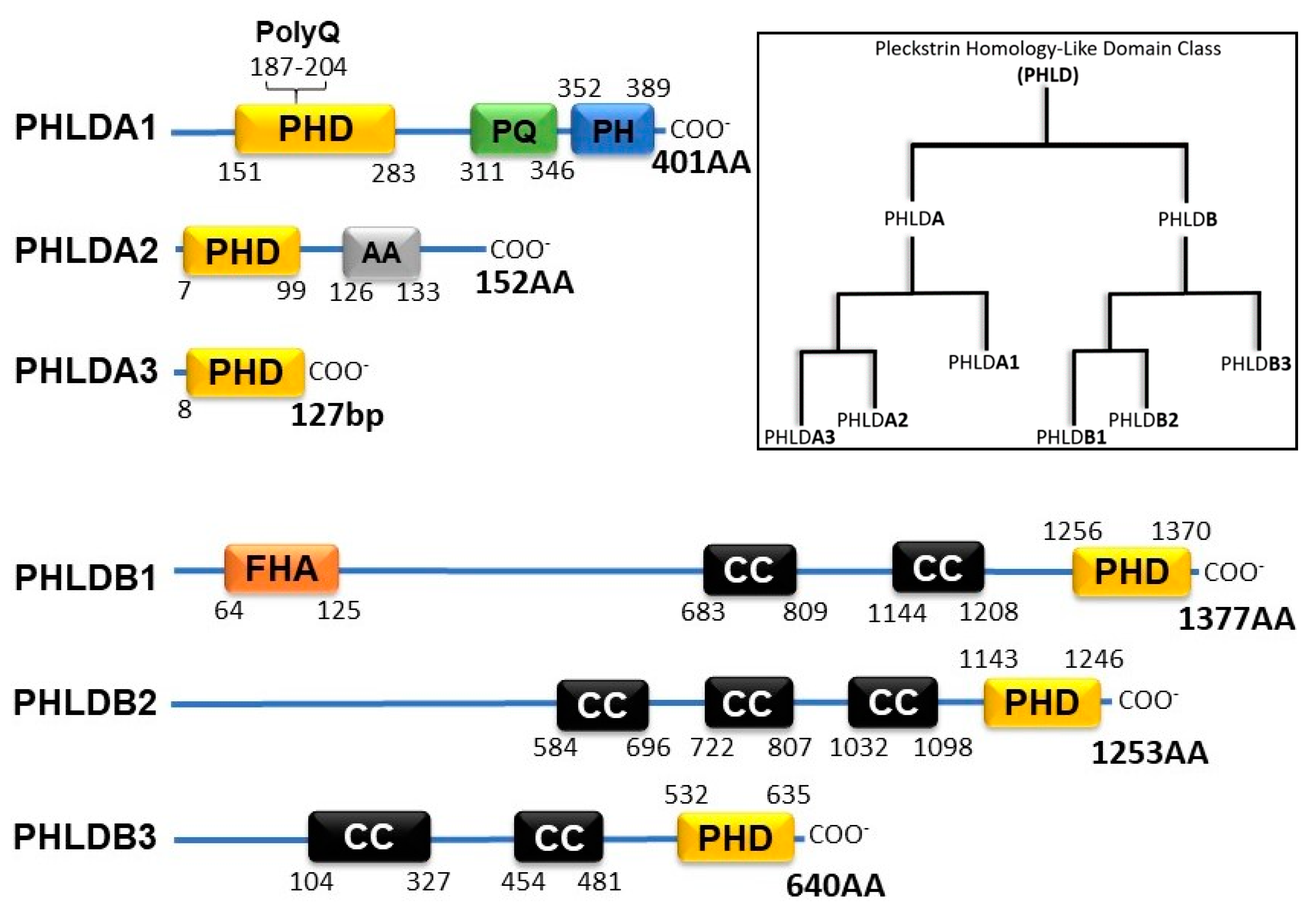
2. PHLD Class Proteins and Cancer
p53 and the PHLD Class Proteins
3. Prospects and More Questions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Haslam, R.J.; Koide, H.B.; Hemmings, B.A. Pleckstrin domain homology. Nature 1993, 363, 309–310. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A. Domains and Phosphoinositides. Biochem. Soc. Symp. 2007, 74, 81–93. [Google Scholar] [CrossRef]
- Dowler, S.; Currie, R.A.; Campbell, D.G.; Deak, M.; Kular, G.; Downes, C.P.; Alessi, D.R. Identification of pleckstrin-homology-domain-containing proteins with novel phosphoinositide-binding specificities. Biochem. J. 2000, 351, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Durocher, D.; Jackson, S.P. The FHA domain. FEBS Lett. 2002, 513, 58–66. [Google Scholar] [CrossRef]
- Park, C.G.; Lee, S.Y.; Kandala, G.; Lee, S.Y.; Choi, Y. A Novel Gene Product that Couples TCR Signaling to Fas(CD95) Expression in Activation-Induced Cell Death. Immunity 1996, 4, 583–591. [Google Scholar] [CrossRef]
- Frank, D.; Mendelsohn, C.L.; Ciccone, E.; Svensson, K.; Ohlsson, R.; Tycko, B. A novel pleckstrin homology-related gene family defined by Ipl/Tssc3, TDAG51, and Tih1: Tissue-specific expression, chromosomal location, and parental imprinting. Mamm. Genome 1999, 10, 1150–1159. [Google Scholar] [CrossRef]
- Nagai, M.A. Pleckstrin homology-like domain, family A, member 1 (PHLDA1) and cancer. Biomed. Rep. 2016, 4, 275–281. [Google Scholar] [CrossRef]
- Ren, L.; Mendoza, A.; Zhu, J.; Briggs, J.W.; Halsey, C.; Hong, E.S.; Burkett, S.S.; Morrow, J.J.; Lizardo, M.M.; Osborne, T.; et al. Characterization of the metastatic phenotype of a panel of established osteosarcoma cells. Oncotarget 2015, 6, 29469–29481. [Google Scholar] [CrossRef]
- Liu, L.; Shi, Y.; Shi, J.; Wang, H.; Sheng, Y.; Jiang, Q.; Chen, H.; Li, X.; Dong, J. The long non-coding RNA SNHG1 promotes glioma progression by competitively binding to miR-194 to regulate PHLDA1 expression. Cell Death Dis. 2019, 10, 463. [Google Scholar] [CrossRef]
- Totzeck, F.; Andrade-Navarro, M.A.; Mier, P. The Protein Structure Context of PolyQ Regions. PLOS ONE 2017, 12, e0170801. [Google Scholar] [CrossRef]
- Chen, Y.; Takikawa, M.; Tsutsumi, S.; Yamaguchi, Y.; Okabe, A.; Shimada, M.; Kawase, T.; Sada, A.; Ezawa, I.; Takano, Y.; et al. PHLDA1, another PHLDA family protein that inhibits Akt. Cancer Sci. 2018, 109, 3532–3542. [Google Scholar] [CrossRef] [PubMed]
- Fearon, A.E.; Carter, E.P.; Clayton, N.S.; Wilkes, E.H.; Baker, A.-M.; Kapitonova, E.; Bakhouche, B.A.; Tanner, Y.; Wang, J.; Gadaleta, E.; et al. PHLDA1 Mediates Drug Resistance in Receptor Tyrosine Kinase-Driven Cancer. Cell Rep. 2018, 22, 2469–2481. [Google Scholar] [CrossRef] [PubMed]
- Kawase, T.; Ohki, R.; Shibata, T.; Tsutsumi, S.; Kamimura, N.; Inazawa, J.; Ohta, T.; Ichikawa, H.; Aburatani, H.; Tashiro, F.; et al. PH Domain-Only Protein PHLDA3 Is a p53-Regulated Repressor of Akt. Cell 2009, 136, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, G.; Koul, S.; Ohki, R.; Maurer, M.; Borczuk, A.; Halmos, B. PHLDA2 is a key oncogene-induced negative feedback inhibitor of EGFR/ErbB2 signaling via interference with AKT signaling. Oncotarget 2015, 9, 24914–24926. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wang, X.; Hibshoosh, H.; Jin, C.; Halmos, B. Modulation of ErbB2 Blockade in ErbB2-Positive Cancers: The Role of ErbB2 Mutations and PHLDA1. PLOS ONE 2014, 9, e106349. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Lu, Y.; Liu, L. Correlation of decreased expression of PHLDA1 protein with malignant phenotype of gastric adenocarcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 5230–5235. [Google Scholar]
- Chiu, S.-T.; Hsieh, F.-J.; Chen, S.-W.; Shu, H.-F.; Li, H. Clinicopathologic Correlation of Up-regulated Genes Identified Using cDNA Microarray and Real-time Reverse Transcription-PCR in Human Colorectal Cancer. Cancer Epidemiol. Biomark. Prev. 2005, 14, 437–443. [Google Scholar] [CrossRef]
- Li, Y.; Song, X.; Liu, Z.; Li, Q.; Huang, M.; Su, B.; Mao, Y.; Wang, Y.; Mo, W.; Chen, H. Upregulation of miR-214 Induced Radioresistance of Osteosarcoma by Targeting PHLDA2 via PI3K/Akt Signaling. Front. Oncol. 2019, 9, 298. [Google Scholar] [CrossRef]
- Takikawa, M.; Ohki, R. A vicious partnership between AKT and PHLDA3 to facilitate neuroendocrine tumors. Cancer Sci. 2017, 108, 1101–1108. [Google Scholar] [CrossRef]
- Ohki, R.; Saito, K.; Chen, Y.; Kawase, T.; Hiraoka, N.; Saigawa, R.; Minegishi, M.; Aita, Y.; Yanai, G.; Shimizu, H.; et al. PHLDA3 is a novel tumor suppressor of pancreatic neuroendocrine tumors. Proc. Natl. Acad. Sci. USA 2014, 111, E2404–E2413. [Google Scholar] [CrossRef]
- Li, X.; Cao, H.; Liu, Y. Genetic epidemiology and risk factors for brain tumors. J. Cent. South Univ. Med. Sci. 2018, 43, 345–353. [Google Scholar] [CrossRef]
- Kim, L.H.; Kim, J.-H.; Namgoong, S.; Cheong, H.S.; Yoon, S.-J.; Kim, E.H.; Kim, S.H.; Kim, S.H.; Chang, J.H.; Shin, H.D. A PHLDB1 variant associated with the nonfunctional pituitary adenoma. J. Neuro-Oncology 2019, 142, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhou, T.; Li, Y.; Yu, Z.; Sun, L. p53 target miR-29c-3p suppresses colon cancer cell invasion and migration through inhibition of PHLDB2. Biochem. Biophys. Res. Commun. 2017, 487, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Chao, T.; Zhou, X.; Cao, B.; Liao, P.; Liu, H.; Chen, Y.; Park, H.-W.; Zeng, S.X.; Lu, H. Pleckstrin homology domain-containing protein PHLDB3 supports cancer growth via a negative feedback loop involving p53. Nat. Commun. 2016, 7, 13755. [Google Scholar] [CrossRef]
- Wang, K.-W.; Dong, M. Role of circular RNAs in gastric cancer: Recent advances and prospects. World J. Gastrointest. Oncol. 2019, 11, 459–469. [Google Scholar] [CrossRef]
- Gomes, A.N.D.M.; Nagai, M.A.; Lourenço, S.V.; Coutinho-Camillo, C.M. Apoptosis and proliferation during human salivary gland development. J. Anat. 2019, 234, 830–838. [Google Scholar] [CrossRef]
- Viana-Pereira, M.; Moreno, D.A.; Linhares, P.; Amorim, J.; Nabiço, R.; Costa, S.; Vaz, R.; Reis, R.M. Replication of GWAS identifies RTEL1, CDKN2A/B, and PHLDB1 SNPs as risk factors in Portuguese gliomas patients. Mol. Boil. Rep. 2019, 47, 877–886. [Google Scholar] [CrossRef]
- Chen, G.; Zhou, T.; Ma, T.; Cao, T.; Yu, Z. Oncogenic effect of PHLDB2 is associated with epithelial-mesenchymal transition and E-cadherin regulation in colorectal cancer. Cancer Cell Int. 2019, 19, 184. [Google Scholar] [CrossRef]
- Seetharaman, S.; Etienne-Manneville, S. Microtubules at focal adhesions - a double-edged sword. J. Cell Sci. 2019, 132, jcs232843. [Google Scholar] [CrossRef]
- Qin, A.; Johnson, A.; Ross, J.S.; Miller, V.A.; Ali, S.M.; Schrock, A.B.; Gadgeel, S.M. Detection of Known and Novel FGFR Fusions in Non–Small Cell Lung Cancer by Comprehensive Genomic Profiling. J. Thorac. Oncol. 2019, 14, 54–62. [Google Scholar] [CrossRef]
- Nesterov, A.; Lu, X.; Johnson, M.; Miller, G.J.; Ivashchenko, Y.; Kraft, A.S. Elevated Akt Activity Protects the Prostate Cancer Cell Line LNCaP from TRAIL-induced Apoptosis. J. Boil. Chem. 2001, 276, 10767–10774. [Google Scholar] [CrossRef] [PubMed]
- Braun, C.J.; Zhang, X.; Savelyeva, I.; Wolff, S.; Moll, U.M.; Schepeler, T.; Ørntoft, T.F.; Andersen, C.L.; Dobbelstein, M. p53-Responsive micrornas 192 and 215 are capable of inducing cell cycle arrest. Cancer Res. 2008, 68, 10094–10104. [Google Scholar] [CrossRef] [PubMed]
- Saraon, P.; Grozavu, I.; Lim, S.H.; Snider, J.; Yao, Z.; Stagljar, I. Detecting Membrane Protein-protein Interactions Using the Mammalian Membrane Two-hybrid (MaMTH) Assay. Curr. Protoc. Chem. Boil. 2017, 9, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, E.; Kalli, A.C.; Yasuoka, K.; Sansom, M.S. Interactions of Pleckstrin Homology Domains with Membranes: Adding Back the Bilayer via High-Throughput Molecular Dynamics. Struct. 2016, 24, 1421–1431. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.L.; Jiang, Z.Y.; Mabardy, A.S.; Del Campo, C.M.; Lambright, D.G.; Holik, J.; Fogarty, K.E.; Straubhaar, J.; Nicoloro, S.; Chawla, A.; et al. A Novel Pleckstrin Homology Domain-containing Protein Enhances Insulin-stimulated Akt Phosphorylation and GLUT4 Translocation in Adipocytes*. J. Boil. Chem. 2010, 285, 27581–27589. [Google Scholar] [CrossRef]
- Robey, R.B.; Hay, N. Is Akt the “Warburg kinase”?—Akt-energy metabolism interactions and oncogenesis. Semin. Cancer Boil. 2008, 19, 25–31. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Expression Level in Normal Tissues * | Expression Level in Disease | References | |
|---|---|---|---|
| PHLDA1 | All Tissues ↑ Salivary Glands | ↓ Breast Cancer | [7,11,12,15] |
| ↓ Ovarian Cancer | [7,11] | ||
| ↓ Endometrial Cancer | [7,12] | ||
| ↓ Lung Cancer | [15] | ||
| ↓ Gastric Cancer | [7,16] | ||
| ↑ Colorectal Cancer | [7,17] | ||
| ↑ Osteosarcoma | [7,8] | ||
| PHLDA2 | Many Tissues ↑ Placenta | ↓ Breast Cancer | [14] |
| ↓ Lung Cancer | [14] | ||
| ↓ Osteosarcoma | [18] | ||
| PHLDA3 | All Tissues | ↓ Lung Cancer (Neuroendocrine Tumor) | [13] |
| ↓ Pancreatic Cancer (Neuroendocrine Tumor) | [19,20] | ||
| PHLDB1 | Many Tissues ↑ Brain | SNPs ↑ Brain Cancer | [21,22] |
| PHLDB2 | Many Tissues ↑ Placenta | ↑ Colorectal Cancer | [23] |
| PHLDB3 | All Tissues | ↑ Colorectal Cancer | [24] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuselier, T.T.; Lu, H. PHLD Class Proteins: A Family of New Players in the p53 Network. Int. J. Mol. Sci. 2020, 21, 3543. https://doi.org/10.3390/ijms21103543
Fuselier TT, Lu H. PHLD Class Proteins: A Family of New Players in the p53 Network. International Journal of Molecular Sciences. 2020; 21(10):3543. https://doi.org/10.3390/ijms21103543
Chicago/Turabian StyleFuselier, Taylor T., and Hua Lu. 2020. "PHLD Class Proteins: A Family of New Players in the p53 Network" International Journal of Molecular Sciences 21, no. 10: 3543. https://doi.org/10.3390/ijms21103543
APA StyleFuselier, T. T., & Lu, H. (2020). PHLD Class Proteins: A Family of New Players in the p53 Network. International Journal of Molecular Sciences, 21(10), 3543. https://doi.org/10.3390/ijms21103543