Current Molecular Markers of Melanoma and Treatment Targets

 and
and 
Abstract
1. Introduction
2. Molecular Pathways of Melanoma Formation
3. Molecular Markers
3.1. Prognostic or Diagnostic Markers
3.1.1. GNAQ/GNA11
3.1.2. CDKN2A
3.1.3. BAP1
3.1.4. SF3B1
3.1.5. EIF1AX
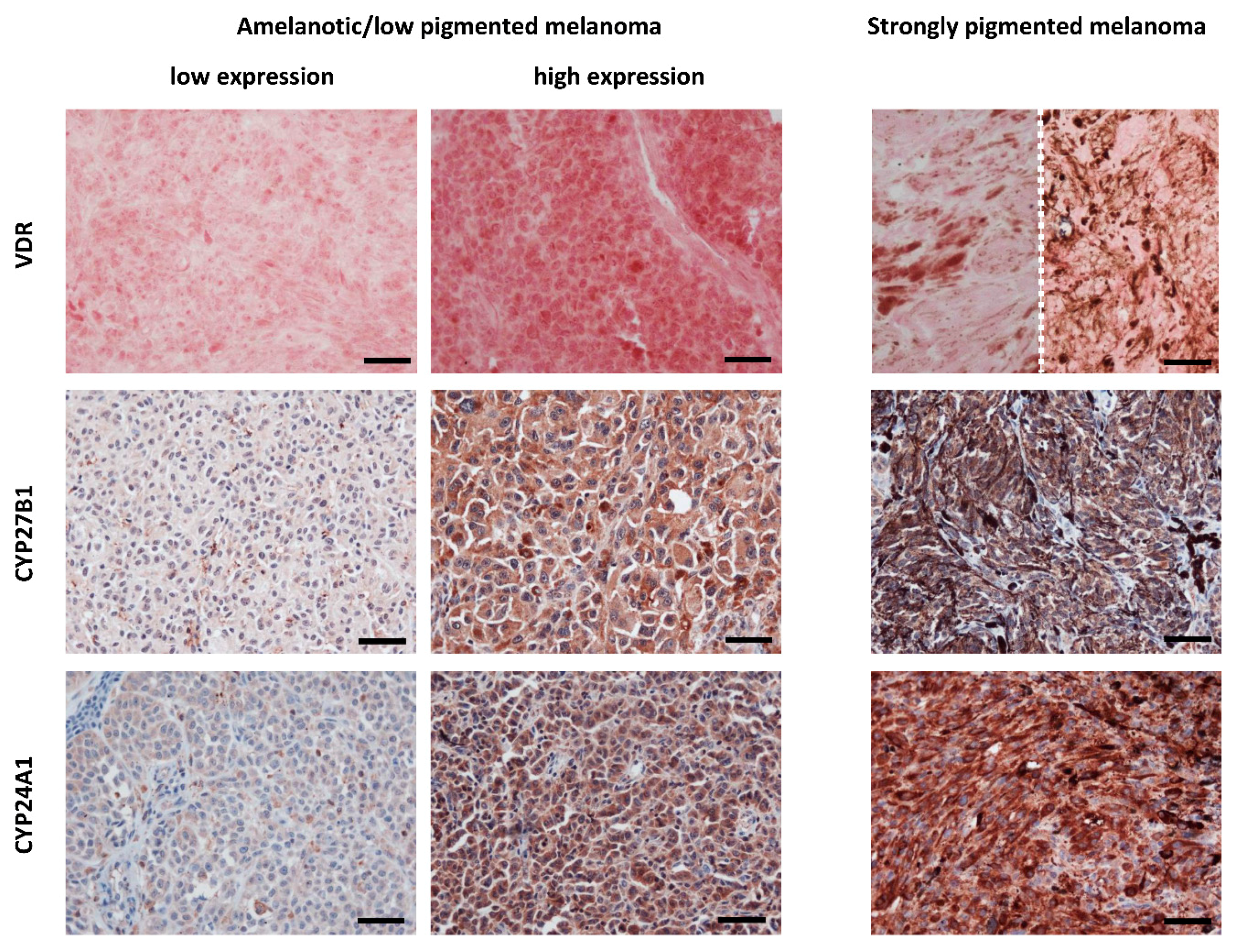
3.1.6. VDR
3.1.7. MC1R
3.1.8. MITF
3.1.9. HAPLN1
3.2. Members of the Melanin Synthesis Pathway
3.2.1. Melanin
3.2.2. Melanogenesis Related Proteins (TYR, TRP1, TRP2)
3.3. Therapeutic Targets
3.3.1. B-raf
3.3.2. N-ras
3.3.3. C-KIT
3.3.4. Immune Checkpoint (CTLA-4, PD-1, PD-L1)
4. Summary and Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Cancer Facts & Figures; American Cancer Society: Atlanta, GA, USA, 2019.
- Rastrelli, M.; Tropea, S.; Rossi, C.R.; Alaibac, M. Melanoma: Epidemiology, Risk Factors, Pathogenesis, Diagnosis and Classification. In Vivo 2014, 28, 1005–1011. [Google Scholar] [PubMed]
- Sample, A.; He, Y.-Y. Mechanisms and Prevention of Uv-Induced Melanoma. Photodermatol. Photoimmunol. Photomed. 2018, 34, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Guy, G.P., Jr.; Ekwueme, D.U.; Tangka, F.K.; Richardson, L.C. Melanoma Treatment Costs: A Systematic Review of the Literature, 1990–2011. Am. J. Prev. Med. 2012, 43, 537–545. [Google Scholar] [CrossRef]
- Elder, D.E.; Bastian, B.C.; Cree, I.A.; Massi, D.; Scolyer, R.A. The 2018 World Health Organization Classification of Cutaneous, Mucosal, and Uveal Melanoma: Detailed Analysis of 9 Distinct Subtypes Defined by Their Evolutionary Pathway. Arch. Pathol. Lab. Med. 2020, 144, 500–522. [Google Scholar] [CrossRef]
- Pan, Y.; Adler, N.R.; Wolfe, R.; McLean, C.A.; Kelly, J.W. Nodular Melanoma Is Less Likely Than Superficial Spreading Melanoma to Be Histologically Associated with a Naevus. Med. J. Aust. 2017, 207, 333–338. [Google Scholar] [CrossRef]
- Bolognia, J.L.S.; Julie, V.; Duncan, K.O.; Ko, C.J. Cutaneous Melanoma. In Dermatology Essentials; Elsevier Inc.: Amsterdam, The Netherlands, 2014. [Google Scholar]
- McLaughlin, C.C.; Wu, X.C.; Jemal, A.; Martin, H.J.; Roche, L.M.; Chen, V.W. Incidence of Noncutaneous Melanomas in the U.S. Cancer 2005, 103, 1000–1007. [Google Scholar] [CrossRef]
- Krantz, B.A.; Dave, N.; Komatsubara, K.M.; Marr, B.P.; Carvajal, R.D. Uveal Melanoma: Epidemiology, Etiology, and Treatment of Primary Disease. Clin. Ophthalmol. 2017, 11, 279–289. [Google Scholar] [CrossRef]
- Kaliki, S.; Shields, C.L. Uveal Melanoma: Relatively Rare but Deadly Cancer. Eye (London) 2017, 31, 241–257. [Google Scholar] [CrossRef]
- Shah, C.P.; Weis, E.; Lajous, M.; Shields, J.A.; Shields, C.L. Intermittent and Chronic Ultraviolet Light Exposure and Uveal Melanoma: A Meta-Analysis. Ophthalmology 2005, 112, 1599–1607. [Google Scholar] [CrossRef]
- Weis, E.; Shah, C.P.; Lajous, M.; Shields, J.A.; Shields, C.L. The Association between Host Susceptibility Factors and Uveal Melanoma: A Meta-Analysis. Arch. Ophthalmol. 2006, 124, 54–60. [Google Scholar] [CrossRef]
- Jager, M.J.; Shields, C.L.; Cebulla, C.M.; Abdel-Rahman, M.H.; Grossniklaus, H.E.; Stern, M.-H.; Carvajal, R.D.; Belfort, R.N.; Jia, R.; Shields, J.A.; et al. Uveal Melanoma. Nat. Rev. Dis. Prim. 2020, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the Braf Gene in Human Cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Melis, C.; Rogiers, A.; Bechter, O.; van den Oord, J.J. Molecular Genetic and Immunotherapeutic Targets in Metastatic Melanoma. Virchows Arch. 2017, 471, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Pracht, M.; Mogha, A.; Lespagnol, A.; Fautrel, A.; Mouchet, N.; Le Gall, F.; Paumier, V.; Lefeuvre-Plesse, C.; Rioux-Leclerc, N.; Mosser, J.; et al. Prognostic and Predictive Values of Oncogenic Braf, Nras, C-Kit and Mitf in Cutaneous and Mucous Melanoma. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 1530–1538. [Google Scholar] [CrossRef]
- Jakob, J.A.; Bassett, R.L., Jr.; Ng, C.S.; Curry, J.L.; Joseph, R.W.; Alvarado, G.C.; Rohlfs, M.L.; Richard, J.; Gershenwald, J.E.; Kim, K.B.; et al. Nras Mutation Status Is an Independent Prognostic Factor in Metastatic Melanoma. Cancer 2012, 118, 4014–4023. [Google Scholar] [CrossRef]
- Lee, J.H.; Choi, J.W.; Kim, Y.S. Frequencies of Braf and Nras Mutations Are Different in Histological Types and Sites of Origin of Cutaneous Melanoma: A Meta-Analysis. Br. J. Dermatol. 2011, 164, 776–784. [Google Scholar] [CrossRef]
- Beadling, C.; Jacobson-Dunlop, E.; Hodi, F.S.; Le, C.; Warrick, A.; Patterson, J.; Town, A.; Harlow, A.; Cruz, F., 3rd; Azar, S.; et al. Kit Gene Mutations and Copy Number in Melanoma Subtypes. Clin. Cancer Res. 2008, 14, 6821–6828. [Google Scholar] [CrossRef]
- Goldstein, A.M.; Chan, M.; Harland, M.; Hayward, N.K.; Demenais, F.; Timothy Bishop, D.; Azizi, E.; Bergman, W.; Bianchi-Scarra, G.; Bruno, W.; et al. Features Associated with Germline Cdkn2a Mutations: A Genomel Study of Melanoma-Prone Families from Three Continents. J. Med. Genet. 2007, 44, 99–106. [Google Scholar] [CrossRef]
- Aoude, L.G.; Wadt, K.A.W.; Pritchard, A.L.; Hayward, N.K. Genetics of Familial Melanoma: 20 Years after Cdkn2a. Pigment Cell Melanoma Res. 2015, 28, 148–160. [Google Scholar] [CrossRef]
- Florell, S.R.; Meyer, L.J.; Boucher, K.M.; Porter-Gill, P.A.; Hart, M.; Erickson, J.; Cannon-Albright, L.A.; Pershing, L.K.; Harris, R.M.; Samlowski, W.E.; et al. Longitudinal Assessment of the Nevus Phenotype in a Melanoma Kindred. J. Investig. Dermatol. 2004, 123, 576–582. [Google Scholar] [CrossRef]
- Brożyna, A.A.; Jozwicki, W.; Janjetovic, Z.; Slominski, A.T. Expression of Vitamin D Receptor Decreases During Progression of Pigmented Skin Lesions. Hum. Pathol. 2011, 42, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Brożyna, A.A.; Jóźwicki, W.; Slominski, A.T. Decreased Vdr Expression in Cutaneous Melanomas as Marker of Tumor Progression: New Data and Analyses. Anticancer Res. 2014, 34, 2735–2743. [Google Scholar] [PubMed]
- Tagliabue, E.; Gandini, S.; Bellocco, R.; Maisonneuve, P.; Newton-Bishop, J.; Polsky, D.; Lazovich, D.; Kanetsky, P.A.; Ghiorzo, P.; Gruis, N.A.; et al. Mc1r Variants as Melanoma Risk Factors Independent of at-Risk Phenotypic Characteristics: A Pooled Analysis from the M-Skip Project. Cancer Manag. Res. 2018, 10, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, S.; Sera, F.; Gandini, S.; Iodice, S.; Caini, S.; Maisonneuve, P.; Fargnoli, M.C. Mc1r Variants, Melanoma and Red Hair Color Phenotype: A Meta-Analysis. Int. J. Cancer 2008, 122, 2753–2760. [Google Scholar] [CrossRef]
- Naffouje, S.; Naffouje, R.; Bhagwandin, S.; Salti, G.I. Microphthalmia Transcription Factor in Malignant Melanoma Predicts Occult Sentinel Lymph Node Metastases and Survival. Melanoma Res. 2015, 25, 496–502. [Google Scholar] [CrossRef]
- Yokoyama, S.; Woods, S.L.; Boyle, G.M.; Aoude, L.G.; MacGregor, S.; Zismann, V.; Gartside, M.; Cust, A.E.; Haq, R.; Harland, M.; et al. A Novel Recurrent Mutation in Mitf Predisposes to Familial and Sporadic Melanoma. Nature 2011, 480, 99–103. [Google Scholar] [CrossRef]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in Gna11 in Uveal Melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef]
- Kalirai, H.; Dodson, A.; Faqir, S.; Damato, B.E.; Coupland, S.E. Lack of Bap1 Protein Expression in Uveal Melanoma Is Associated with Increased Metastatic Risk and Has Utility in Routine Prognostic Testing. Br. J. Cancer 2014, 111, 1373–1380. [Google Scholar] [CrossRef]
- Rai, K.; Pilarski, R.; Boru, G.; Rehman, M.; Saqr, A.H.; Massengill, J.B.; Singh, A.; Marino, M.J.; Davidorf, F.H.; Cebulla, C.M.; et al. Germline Bap1 Alterations in Familial Uveal Melanoma. Genes Chromosom. Cancer 2017, 56, 168–174. [Google Scholar] [CrossRef]
- Garfield, E.M.; Walton, K.E.; Quan, V.L.; VandenBoom, T.; Zhang, B.; Kong, B.Y.; Isales, M.C.; Panah, E.; Kim, G.; Gerami, P. Histomorphologic Spectrum of Germline-Related and Sporadic Bap1-Inactivated Melanocytic Tumors. J. Am. Acad. Dermatol. 2018, 79, 525–534. [Google Scholar] [CrossRef]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 2017, 32, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Smit, K.N.; Jager, M.J.; de Klein, A.; Kiliҫ, E. Uveal Melanoma: Towards a Molecular Understanding. Prog. Retin. Eye Res. 2019, 75, 100800. [Google Scholar] [CrossRef] [PubMed]
- Yavuzyigitoglu, S.; Koopmans, A.E.; Verdijk, R.M.; Vaarwater, J.; Eussen, B.; van Bodegom, A.; Paridaens, D.; Kiliç, E.; de Klein, A.; Rotterdam Ocular Melanoma Study Group. Uveal Melanomas with Sf3b1 Mutations: A Distinct Subclass Associated with Late-Onset Metastases. Ophthalmology 2016, 123, 1118–1128. [Google Scholar] [CrossRef]
- Dono, M.; Angelini, G.; Cecconi, M.; Amaro, A.; Esposito, A.I.; Mirisola, V.; Maric, I.; Lanza, F.; Nasciuti, F.; Viaggi, S.; et al. Mutation Frequencies of Gnaq, Gna11, Bap1, Sf3b1, Eif1ax and Tert in Uveal Melanoma: Detection of an Activating Mutation in the Tert Gene Promoter in a Single Case of Uveal Melanoma. Br. J. Cancer 2014, 110, 1058–1065. [Google Scholar] [CrossRef]
- Martin, M.; Maßhöfer, L.; Temming, P.; Rahmann, S.; Metz, C.; Bornfeld, N.; van de Nes, J.; Klein-Hitpass, L.; Hinnebusch, A.G.; Horsthemke, B.; et al. Exome Sequencing Identifies Recurrent Somatic Mutations in Eif1ax and Sf3b1 in Uveal Melanoma with Disomy 3. Nat. Genet. 2013, 45, 933–936. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib Plus Binimetinib Versus Vemurafenib or Encorafenib in Patients with Braf-Mutant Melanoma (Columbus): A Multicentre, Open-Label, Randomised Phase 3 Trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in Braf-Mutated Metastatic Melanoma: A Multicentre, Open-Label, Phase 3 Randomised Controlled Trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Sarkisian, S.; Davar, D. Mek Inhibitors for the Treatment of Nras Mutant Melanoma. Drug Des. Dev. Ther. 2018, 12, 2553–2565. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A Landscape of Driver Mutations in Melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef]
- Smalley, K.S.M.; Eisen, T.G. Farnesyl Transferase Inhibitor Sch66336 Is Cytostatic, Pro-Apoptotic and Enhances Chemosensitivity to Cisplatin in Melanoma Cells. Int. J. Cancer 2003, 105, 165–175. [Google Scholar] [CrossRef]
- Niessner, H.; Beck, D.; Sinnberg, T.; Lasithiotakis, K.; Maczey, E.; Gogel, J.; Venturelli, S.; Berger, A.; Mauthe, M.; Toulany, M.; et al. The Farnesyl Transferase Inhibitor Lonafarnib Inhibits Mtor Signaling and Enforces Sorafenib-Induced Apoptosis in Melanoma Cells. J. Investig. Dermatol. 2011, 131, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.A.; Busam, K.; Pinkel, D.; Bastian, B.C. Somatic Activation of Kit in Distinct Subtypes of Melanoma. J. Clin. Oncol. 2006, 24, 4340–4346. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Antonescu, C.R.; Wolchok, J.D.; Chapman, P.B.; Roman, R.-A.; Teitcher, J.; Panageas, K.S.; Busam, K.J.; Chmielowski, B.; Lutzky, J.; et al. Kit as a Therapeutic Target in Metastatic Melanoma. JAMA 2011, 305, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Lawrence, D.P.; Weber, J.S.; Gajewski, T.F.; Gonzalez, R.; Lutzky, J.; O’Day, S.J.; Hamid, O.; Wolchok, J.D.; Chapman, P.B.; et al. Phase Ii Study of Nilotinib in Melanoma Harboring Kit Alterations Following Progression to Prior Kit Inhibition. Clin. Cancer Res. 2015, 21, 2289–2296. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Carvajal, R.D.; Dummer, R.; Hauschild, A.; Daud, A.; Bastian, B.C.; Markovic, S.N.; Queirolo, P.; Arance, A.; Berking, C.; et al. Efficacy and Safety of Nilotinib in Patients with Kit-Mutated Metastatic or Inoperable Melanoma: Final Results from the Global, Single-Arm, Phase Ii Team Trial. Ann. Oncol. 2017, 28, 1380–1387. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Hannon, G.J.; Beach, D. A New Regulatory Motif in Cell-Cycle Control Causing Specific Inhibition of Cyclin D/Cdk4. Nature 1993, 366, 704–707. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiong, Y.; Yarbrough, W.G. Arf Promotes Mdm2 Degradation and Stabilizes P53: Arf-Ink4a Locus Deletion Impairs Both the Rb and P53 Tumor Suppression Pathways. Cell 1998, 92, 725–734. [Google Scholar] [CrossRef]
- Soura, E.; Eliades, P.J.; Shannon, K.; Stratigos, A.J.; Tsao, H. Hereditary Melanoma: Update on Syndromes and Management: Genetics of Familial Atypical Multiple Mole Melanoma Syndrome. J. Am. Acad. Dermatol. 2016, 74, 395–410. [Google Scholar] [CrossRef]
- Burdette-Radoux, S.; Tozer, R.G.; Lohmann, R.C.; Quirt, I.; Ernst, D.S.; Walsh, W.; Wainman, N.; Colevas, A.D.; Eisenhauer, E.A. Phase Ii Trial of Flavopiridol, a Cyclin Dependent Kinase Inhibitor, in Untreated Metastatic Malignant Melanoma. Investig. New Drugs 2004, 22, 315–322. [Google Scholar] [CrossRef]
- Baker, A.R.; McDonnell, D.P.; Hughes, M.; Crisp, T.M.; Mangelsdorf, D.J.; Haussler, M.R.; Pike, J.W.; Shine, J.; O’Malley, B.W. Cloning and Expression of Full-Length Cdna Encoding Human Vitamin D Receptor. Proc. Natl. Acad. Sci. USA 1988, 85, 3294–3298. [Google Scholar] [CrossRef]
- Demay, M.B.; Kiernan, M.S.; DeLuca, H.F.; Kronenberg, H.M. Sequences in the Human Parathyroid Hormone Gene That Bind the 1,25-Dihydroxyvitamin D3 Receptor and Mediate Transcriptional Repression in Response to 1,25-Dihydroxyvitamin D3. Proc. Natl. Acad. Sci. USA 1992, 89, 8097–8101. [Google Scholar] [CrossRef] [PubMed]
- Colnot, S.; Lambert, M.; Blin, C.; Thomasset, M.; Perret, C. Identification of DNA Sequences That Bind Retinoid X Receptor-1,25(Oh)2d3-Receptor Heterodimers with High Affinity. Mol. Cell. Endocrinol. 1995, 113, 89–98. [Google Scholar] [CrossRef]
- Saw, R.P.M.; Armstrong, B.K.; Mason, R.S.; Morton, R.L.; Shannon, K.F.; Spillane, A.J.; Stretch, J.R.; Thompson, J.F. Adjuvant Therapy with High Dose Vitamin D Following Primary Treatment of Melanoma at High Risk of Recurrence: A Placebo Controlled Randomised Phase Ii Trial (Anzmtg 02.09 Mel-D). BMC Cancer 2014, 14, 780. [Google Scholar] [CrossRef] [PubMed]
- De Smedt, J.; Van Kelst, S.; Boecxstaens, V.; Stas, M.; Bogaerts, K.; Vanderschueren, D.; Aura, C.; Vandenberghe, K.; Lambrechts, D.; Wolter, P.; et al. Vitamin D Supplementation in Cutaneous Malignant Melanoma Outcome (Vidme): A Randomized Controlled Trial. BMC Cancer 2017, 17, 562. [Google Scholar] [CrossRef]
- Levy, C.; Khaled, M.; Fisher, D.E. Mitf: Master Regulator of Melanocyte Development and Melanoma Oncogene. Trends Mol. Med. 2006, 12, 406–414. [Google Scholar] [CrossRef]
- Price, E.R.; Horstmann, M.A.; Wells, A.G.; Weilbaecher, K.N.; Takemoto, C.M.; Landis, M.W.; Fisher, D.E. Alpha-Melanocyte-Stimulating Hormone Signaling Regulates Expression of Microphthalmia, a Gene Deficient in Waardenburg Syndrome. J. Biol. Chem. 1998, 273, 33042–33047. [Google Scholar] [CrossRef]
- Bertolotto, C.; Abbe, P.; Hemesath, T.J.; Bille, K.; Fisher, D.E.; Ortonne, J.P.; Ballotti, R. Microphthalmia Gene Product as a Signal Transducer in Camp-Induced Differentiation of Melanocytes. J. Cell Biol. 1998, 142, 827–835. [Google Scholar] [CrossRef]
- Rózanowska, M.; Sarna, T.; Land, E.J.; Truscott, T.G. Free Radical Scavenging Properties of Melanin Interaction of Eu- and Pheo-Melanin Models with Reducing and Oxidising Radicals. Free Radic. Biol. Med. 1999, 26, 518–525. [Google Scholar]
- Slominski, A.; Tobin, D.J.; Shibahara, S.; Wortsman, J. Melanin Pigmentation in Mammalian Skin and Its Hormonal Regulation. Physiol. Rev. 2004, 84, 1155–1228. [Google Scholar] [CrossRef]
- Slominski, R.M.; Zmijewski, M.A.; Slominski, A.T. The Role of Melanin Pigment in Melanoma. Exp. Dermatol. 2015, 24, 258–259. [Google Scholar] [CrossRef]
- Slominski, A.; Zmijewski, M.A.; Pawelek, J. L-Tyrosine and L-Dihydroxyphenylalanine as Hormone-Like Regulators of Melanocyte Functions. Pigment Cell Melanoma Res. 2012, 25, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Ito, S. The Ifpcs Presidential Lecture: A Chemist’s View of Melanogenesis. Pigment Cell Res. 2003, 16, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Ecker, B.L.; Douglass, S.M.; Kugel, C.H.; Webster, M.R.; Almeida, F.V.; Somasundaram, R.; Hayden, J.; Ban, E.; Ahmadzadeh, H.; et al. Remodeling of the Collagen Matrix in Aging Skin Promotes Melanoma Metastasis and Affects Immune Cell Motility. Cancer Discov. 2019, 9, 64–81. [Google Scholar] [CrossRef] [PubMed]
- Fong, L.; Small, E.J. Anti-Cytotoxic T-Lymphocyte Antigen-4 Antibody: The First in an Emerging Class of Immunomodulatory Antibodies for Cancer Treatment. J. Clin. Oncol. 2008, 26, 5275–5283. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, Durable Tumor Remission, and Long-Term Safety in Patients with Advanced Melanoma Receiving Nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Landis, C.A.; Masters, S.B.; Spada, A.; Pace, A.M.; Bourne, H.R.; Vallar, L. Gtpase Inhibiting Mutations Activate the Alpha Chain of Gs and Stimulate Adenylyl Cyclase in Human Pituitary Tumours. Nature 1989, 340, 692–696. [Google Scholar] [CrossRef]
- Kalinec, G.; Nazarali, A.J.; Hermouet, S.; Xu, N.; Gutkind, J.S. Mutated Alpha Subunit of the Gq Protein Induces Malignant Transformation in Nih 3t3 Cells. Mol. Cell. Biol. 1992, 12, 4687–4693. [Google Scholar] [CrossRef]
- Amaro, A.; Gangemi, R.; Piaggio, F.; Angelini, G.; Barisione, G.; Ferrini, S.; Pfeffer, U. The Biology of Uveal Melanoma. Cancer Metastasis Rev. 2017, 36, 109–140. [Google Scholar] [CrossRef]
- Pan, H.; Jia, R.; Zhang, L.; Xu, S.; Wu, Q.; Song, X.; Zhang, H.; Ge, S.; Leon Xu, X.; Fan, X. Bap1 Regulates Cell Cycle Progression through E2f1 Target Genes and Mediates Transcriptional Silencing Via H2a Monoubiquitination in Uveal Melanoma Cells. Int. J. Biochem. Cell Biol. 2015, 60, 176–184. [Google Scholar] [CrossRef]
- Vivet-Noguer, R.; Tarin, M.; Roman-Roman, S.; Alsafadi, S. Emerging Therapeutic Opportunities Based on Current Knowledge of Uveal Melanoma Biology. Cancers 2019, 11, 1019. [Google Scholar] [CrossRef] [PubMed]
- Alsafadi, S.; Houy, A.; Battistella, A.; Popova, T.; Wassef, M.; Henry, E.; Tirode, F.; Constantinou, A.; Piperno-Neumann, S.; Roman-Roman, S.; et al. Cancer-Associated Sf3b1 Mutations Affect Alternative Splicing by Promoting Alternative Branchpoint Usage. Nat. Commun. 2016, 7, 10615. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, J.; Si, K.; Maitra, U. Function of Eukaryotic Translation Initiation Factor 1a (Eif1a) (Formerly Called Eif-4c) in Initiation of Protein Synthesis. J. Biol. Chem. 1997, 272, 7883–7891. [Google Scholar] [CrossRef]
- Blanchard, D.A.; Mouhamad, S.; Auffredou, M.-T.; Pesty, A.; Bertoglio, J.; Leca, G.; Vazquez, A. Cdk2 Associates with Map Kinase in Vivo and Its Nuclear Translocation Is Dependent on Map Kinase Activation in Il-2-Dependent Kit 225 T Lymphocytes. Oncogene 2000, 19, 4184–4189. [Google Scholar] [CrossRef][Green Version]
- Seger, R.; Seger, D.; Reszka, A.A.; Munar, E.S.; Eldar-Finkelman, H.; Dobrowolska, G.; Jensen, A.M.; Campbell, J.S.; Fischer, E.H.; Krebs, E.G. Overexpression of Mitogen-Activated Protein Kinase Kinase (Mapkk) and Its Mutants in Nih 3t3 Cells. Evidence That Mapkk Involvement in Cellular Proliferation Is Regulated by Phosphorylation of Serine Residues in Its Kinase Subdomains Vii and Viii. J. Biol. Chem. 1994, 269, 25699–25709. [Google Scholar]
- Lavoie, J.N.; L’Allemain, G.; Brunet, A.; Müller, R.; Pouysségur, J. Cyclin D1 Expression Is Regulated Positively by the P42/P44mapk and Negatively by the P38/Hogmapk Pathway. J. Biol. Chem. 1996, 271, 20608–20616. [Google Scholar] [CrossRef]
- Raman, M.; Chen, W.; Cobb, M.H. Differential Regulation and Properties of Mapks. Oncogene 2007, 26, 3100–3112. [Google Scholar] [CrossRef]
- Dent, P.; Haser, W.; Haystead, T.A.; Vincent, L.A.; Roberts, T.M.; Sturgill, T.W. Activation of Mitogen-Activated Protein Kinase Kinase by V-Raf in Nih 3t3 Cells and in Vitro. Science 1992, 257, 1404–1407. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; App, H.; Zhang, X.F.; Banerjee, P.; Brautigan, D.L.; Rapp, U.R.; Avruch, J. Raf-1 Activates Map Kinase-Kinase. Nature 1992, 358, 417–421. [Google Scholar] [CrossRef]
- Sigoillot, F.D.; Evans, D.R.; Guy, H.I. Growth-Dependent Regulation of Mammalian Pyrimidine Biosynthesis by the Protein Kinase a and Mapk Signaling Cascades. J. Biol. Chem. 2002, 277, 15745–15751. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Harrison, J.K.; Vincent, L.A.; Haystead, C.; Haystead, T.A.; Michel, H.; Hunt, D.F.; Lynch, K.R.; Sturgill, T.W. Molecular Structure of a Protein-Tyrosine/Threonine Kinase Activating P42 Mitogen-Activated Protein (Map) Kinase: Map Kinase Kinase. Proc. Natl. Acad. Sci. USA 1993, 90, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghì, A.; Spandidos, D.A.; McCubrey, J.A.; Candido, S.; Libra, M. Cutaneous Melanoma: From Pathogenesis to Therapy (Review). Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Burotto, M.; Chiou, V.L.; Lee, J.-M.; Kohn, E.C. The Mapk Pathway across Different Malignancies: A New Perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt Phosphorylation of Bad Couples Survival Signals to the Cell-Intrinsic Death Machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Mayo, L.D.; Donner, D.B. A Phosphatidylinositol 3-Kinase/Akt Pathway Promotes Translocation of Mdm2 from the Cytoplasm to the Nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 11598–11603. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. Tsc2 Is Phosphorylated and Inhibited by Akt and Suppresses Mtor Signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Yuan, T.L.; Cantley, L.C. Pi3k Pathway Alterations in Cancer: Variations on a Theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef]
- Rodriguez-Viciana, P.; Warne, P.H.; Dhand, R.; Vanhaesebroeck, B.; Gout, I.; Fry, M.J.; Waterfield, M.D.; Downward, J. Phosphatidylinositol-3-Oh Kinase as a Direct Target of Ras. Nature 1994, 370, 527–532. [Google Scholar] [CrossRef]
- Carpenter, C.L.; Auger, K.R.; Chanudhuri, M.; Yoakim, M.; Schaffhausen, B.; Shoelson, S.; Cantley, L.C. Phosphoinositide 3-Kinase Is Activated by Phosphopeptides That Bind to the Sh2 Domains of the 85-Kda Subunit. J. Biol. Chem. 1993, 268, 9478–9483. [Google Scholar]
- Klippel, A.; Kavanaugh, W.M.; Pot, D.; Williams, L.T. A Specific Product of Phosphatidylinositol 3-Kinase Directly Activates the Protein Kinase Akt through Its Pleckstrin Homology Domain. Mol. Cell. Biol. 1997, 17, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Rameh, L.E.; Tolias, K.F.; Duckworth, B.C.; Cantley, L.C. A New Pathway for Synthesis of Phosphatidylinositol-4,5-Bisphosphate. Nature 1997, 390, 192–196. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Maiello, M.R.; D’Alessio, A.; Pergameno, M.; Normanno, N. The Ras/Raf/Mek/Erk and the Pi3k/Akt Signalling Pathways: Role in Cancer Pathogenesis and Implications for Therapeutic Approaches. Expert Opin. Ther. Targets 2012, 16 (Suppl. 2), S17–S27. [Google Scholar] [CrossRef] [PubMed]
- Maehama, T.; Dixon, J.E. The Tumor Suppressor, Pten/Mmac1, Dephosphorylates the Lipid Second Messenger, Phosphatidylinositol 3,4,5-Trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. Yap1 Increases Organ Size and Expands Undifferentiated Progenitor Cells. Curr. Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef]
- Xin, M.; Kim, Y.; Sutherland, L.B.; Qi, X.; McAnally, J.; Schwartz, R.J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Regulation of Insulin-Like Growth Factor Signaling by Yap Governs Cardiomyocyte Proliferation and Embryonic Heart Size. Sci. Signal. 2011, 4, ra70. [Google Scholar] [CrossRef]
- George, N.M.; Day, C.E.; Boerner, B.P.; Johnson, R.L.; Sarvetnick, N.E. Hippo Signaling Regulates Pancreas Development through Inactivation of Yap. Mol. Cell. Biol. 2012, 32, 5116–5128. [Google Scholar] [CrossRef]
- Hossain, Z.; Ali, S.M.; Ko, H.L.; Xu, J.; Ng, C.P.; Guo, K.; Qi, Z.; Ponniah, S.; Hong, W.; Hunziker, W. Glomerulocystic Kidney Disease in Mice with a Targeted Inactivation of Wwtr1. Proc. Natl. Acad. Sci. USA 2007, 104, 1631–1636. [Google Scholar] [CrossRef]
- Yu, F.X.; Luo, J.; Mo, J.S.; Liu, G.; Kim, Y.C.; Meng, Z.; Zhao, L.; Peyman, G.; Ouyang, H.; Jiang, W.; et al. Mutant Gq/11 Promote Uveal Melanoma Tumorigenesis by Activating Yap. Cancer Cell 2014, 25, 822–830. [Google Scholar] [CrossRef]
- Field, M.G.; Durante, M.A.; Anbunathan, H.; Cai, L.Z.; Decatur, C.L.; Bowcock, A.M.; Kurtenbach, S.; Harbour, J.W. Punctuated Evolution of Canonical Genomic Aberrations in Uveal Melanoma. Nat. Commun. 2018, 9, 116. [Google Scholar] [CrossRef]
- Pleasance, E.D.; Cheetham, R.K.; Stephens, P.J.; McBride, D.J.; Humphray, S.J.; Greenman, C.D.; Varela, I.; Lin, M.L.; Ordóñez, G.R.; Bignell, G.R.; et al. A Comprehensive Catalogue of Somatic Mutations from a Human Cancer Genome. Nature 2010, 463, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-Genome Landscapes of Major Melanoma Subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Berger, A.C.; Davidson, R.S.; Poitras, J.K.; Chabra, I.; Hope, R.; Brackeen, A.; Johnson, C.E.; Maetzold, D.J.; Middlebrook, B.; Oelschlager, K.M.; et al. Clinical Impact of a 31-Gene Expression Profile Test for Cutaneous Melanoma in 156 Prospectively and Consecutively Tested Patients. Curr. Med. Res. Opin. 2016, 32, 1599–1604. [Google Scholar] [CrossRef] [PubMed]
- Cassarino, D.S.; Lewine, N.; Cole, D.; Wade, B.; Gustavsen, G. Budget Impact Analysis of a Novel Gene Expression Assay for the Diagnosis of Malignant Melanoma. J. Med. Econ. 2014, 17, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Ferris, L.K.; Jansen, B.; Ho, J.; Busam, K.J.; Gross, K.; Hansen, D.D.; Alsobrook, J.P., 2nd; Yao, Z.; Peck, G.L.; Gerami, P. Utility of a Noninvasive 2-Gene Molecular Assay for Cutaneous Melanoma and Effect on the Decision to Biopsy. JAMA Dermatol. 2017, 153, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Lian, C.G. Molecular Testing for Cutaneous Melanoma: An Update and Review. Arch. Pathol. Lab. Med. 2019, 143, 811–820. [Google Scholar] [CrossRef]
- Griewank, K.G.; Schilling, B.; Scholz, S.L.; Metz, C.H.; Livingstone, E.; Sucker, A.; Moller, I.; Reis, H.; Franklin, C.; Cosgarea, I.; et al. Oncogene Status as a Diagnostic Tool in Ocular and Cutaneous Melanoma. Eur. J. Cancer 2016, 57, 112–117. [Google Scholar] [CrossRef]
- Patel, S.P.; Kim, D.W.; Lacey, C.L.; Hwu, P. Gna11 Mutation in a Patient with Cutaneous Origin Melanoma: A Case Report. Medicine 2016, 95, e2336. [Google Scholar] [CrossRef]
- Koopmans, A.E.; Vaarwater, J.; Paridaens, D.; Naus, N.C.; Kilic, E.; de Klein, A. Patient Survival in Uveal Melanoma Is Not Affected by Oncogenic Mutations in Gnaq and Gna11. Br. J. Cancer 2013, 109, 493–496. [Google Scholar] [CrossRef]
- Staby, K.M.; Gravdal, K.; Mork, S.J.; Heegaard, S.; Vintermyr, O.K.; Krohn, J. Prognostic Impact of Chromosomal Aberrations and Gnaq, Gna11 and Bap1 Mutations in Uveal Melanoma. Acta Ophthalmol. 2018, 96, 31–38. [Google Scholar] [CrossRef]
- Nobori, T.; Miura, K.; Wu, D.J.; Lois, A.; Takabayashi, K.; Carson, D.A. Deletions of the Cyclin-Dependent Kinase-4 Inhibitor Gene in Multiple Human Cancers. Nature 1994, 368, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Kannan, K.; Xu, J.; Bosenberg, M.W.; Chin, L. Both Products of the Mouse Ink4a/Arf Locus Suppress Melanoma Formation in Vivo. Oncogene 2003, 22, 5055–5059. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Read, J.; Wadt, K.A.; Hayward, N.K. Melanoma Genetics. J. Med. Genet. 2016, 53, 1–14. [Google Scholar] [CrossRef]
- Sargen, M.R.; Kanetsky, P.A.; Newton-Bishop, J.; Hayward, N.K.; Mann, G.J.; Gruis, N.A.; Tucker, M.A.; Goldstein, A.M.; Bianchi-Scarra, G.; Puig, S.; et al. Histologic Features of Melanoma Associated with Cdkn2a Genotype. J. Am. Acad. Dermatol. 2015, 72, 496–507.e7. [Google Scholar] [CrossRef]
- Helgadottir, H.; Höiom, V.; Tuominen, R.; Nielsen, K.; Jönsson, G.; Olsson, H.; Hansson, J. Germline Cdkn2a Mutation Status and Survival in Familial Melanoma Cases. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Dalmasso, B.; Pastorino, L.; Ciccarese, G.; Andreotti, V.; Grillo, F.; Mastracci, L.; Spagnolo, F.; Ballestrero, A.; Queirolo, P.; Bruno, W.; et al. Cdkn2a Germline Mutations Are Not Associated with Poor Survival in an Italian Cohort of Melanoma Patients. J. Am. Acad. Dermatol. 2019, 80, 1263–1271. [Google Scholar] [CrossRef]
- Yu, H.; Mashtalir, N.; Daou, S.; Hammond-Martel, I.; Ross, J.; Sui, G.; Hart, G.W.; Rauscher, F.J., 3rd; Drobetsky, E.; Milot, E.; et al. The Ubiquitin Carboxyl Hydrolase Bap1 Forms a Ternary Complex with Yy1 and Hcf-1 and Is a Critical Regulator of Gene Expression. Mol. Cell. Biol. 2010, 30, 5071–5085. [Google Scholar] [CrossRef]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.O.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent Mutation of Bap1 in Metastasizing Uveal Melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef]
- Koopmans, A.E.; Verdijk, R.M.; Brouwer, R.W.W.; van den Bosch, T.P.P.; van den Berg, M.M.P.; Vaarwater, J.; Kockx, C.E.M.; Paridaens, D.; Naus, N.C.; Nellist, M.; et al. Clinical Significance of Immunohistochemistry for Detection of Bap1 Mutations in Uveal Melanoma. Mod. Pathol. 2014, 27, 1321–1330. [Google Scholar] [CrossRef]
- Decatur, C.L.; Ong, E.; Garg, N.; Anbunathan, H.; Bowcock, A.M.; Field, M.G.; Harbour, J.W. Driver Mutations in Uveal Melanoma: Associations with Gene Expression Profile and Patient Outcomes. JAMA Ophthalmol. 2016, 134, 728–733. [Google Scholar] [CrossRef]
- Shain, A.H.; Bagger, M.M.; Yu, R.; Chang, D.; Liu, S.; Vemula, S.; Weier, J.F.; Wadt, K.; Heegaard, S.; Bastian, B.C.; et al. The Genetic Evolution of Metastatic Uveal Melanoma. Nat. Genet. 2019, 51, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.P.; Lane, A.M.; DeAngelis, M.M.; Mayne, K.; Crabtree, M.; Gragoudas, E.S.; Kim, I.K. Clinical Characteristics of Uveal Melanoma in Patients with Germline Bap1 Mutations. JAMA Ophthalmol. 2015, 133, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Ewens, K.G.; Lalonde, E.; Richards-Yutz, J.; Shields, C.L.; Ganguly, A. Comparison of Germline Versus Somatic Bap1 Mutations for Risk of Metastasis in Uveal Melanoma. BMC Cancer 2018, 18, 1172. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, T.; Obenauf, A.C.; Murali, R.; Fried, I.; Griewank, K.G.; Ulz, P.; Windpassinger, C.; Wackernagel, W.; Loy, S.; Wolf, I.; et al. Germline Mutations in Bap1 Predispose to Melanocytic Tumors. Nat. Genet. 2011, 43, 1018–1021. [Google Scholar] [CrossRef]
- Njauw, C.N.; Kim, I.; Piris, A.; Gabree, M.; Taylor, M.; Lane, A.M.; DeAngelis, M.M.; Gragoudas, E.; Duncan, L.M.; Tsao, H. Germline Bap1 Inactivation Is Preferentially Associated with Metastatic Ocular Melanoma and Cutaneous-Ocular Melanoma Families. PLoS ONE 2012, 7, e35295. [Google Scholar] [CrossRef]
- Kumar, R.; Taylor, M.; Miao, B.; Ji, Z.; Njauw, J.C.N.; Jönsson, G.; Frederick, D.T.; Tsao, H. Bap1 Has a Survival Role in Cutaneous Melanoma. J. Investig. Dermatol. 2015, 135, 1089–1097. [Google Scholar] [CrossRef]
- Atkinson, A.; Linos, K.; Yan, S.; Tsongalis, G.; Lefferts, J. Molecular Diagnostic Techniques Used to Confirm an Unusual Case of Bapoma. FASEB J. 2017, 31 (Suppl. 1), 807.17. [Google Scholar]
- Yu, H.; Pak, H.; Hammond-Martel, I.; Ghram, M.; Rodrigue, A.; Daou, S.; Barbour, H.; Corbeil, L.; Hébert, J.; Drobetsky, E.; et al. Tumor Suppressor and Deubiquitinase Bap1 Promotes DNA Double-Strand Break Repair. Proc. Natl. Acad. Sci. USA 2014, 111, 285–290. [Google Scholar] [CrossRef]
- de Koning, L.; Decaudin, D.; El Botty, R.; Nicolas, A.; Carita, G.; Schuller, M.; Ouine, B.; Cartier, A.; Naguez, A.; Fleury, J.; et al. Parp Inhibition Increases the Response to Chemotherapy in Uveal Melanoma. Cancers 2019, 11, 751. [Google Scholar] [CrossRef]
- Harbour, J.W.; Roberson, E.D.; Anbunathan, H.; Onken, M.D.; Worley, L.A.; Bowcock, A.M. Recurrent Mutations at Codon 625 of the Splicing Factor Sf3b1 in Uveal Melanoma. Nat. Genet. 2013, 45, 133–135. [Google Scholar] [CrossRef]
- Kong, Y.; Krauthammer, M.; Halaban, R. Rare Sf3b1 R625 Mutations in Cutaneous Melanoma. Melanoma Res. 2014, 24, 332–334. [Google Scholar] [CrossRef]
- Bikle, D.D. Vitamin D Metabolism and Function in the Skin. Mol. Cell. Endocrinol. 2011, 347, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Verlinden, L.; Verstuyf, A.; Convents, R.; Marcelis, S.; Van Camp, M.; Bouillon, R. Action of 1,25(Oh)2d3 on the Cell Cycle Genes, Cyclin D1, P21 and P27 in Mcf-7 Cells. Mol. Cell. Endocrinol. 1998, 142, 57–65. [Google Scholar] [CrossRef]
- Hershberger, P.A.; Modzelewski, R.A.; Shurin, Z.R.; Rueger, R.M.; Trump, D.L.; Johnson, C.S. 1,25-Dihydroxycholecalciferol (1,25-D3) Inhibits the Growth of Squamous Cell Carcinoma and Down-Modulates P21(Waf1/Cip1) in Vitro and in Vivo. Cancer Res. 1999, 59, 2644–2649. [Google Scholar]
- Danielsson, C.; Fehsel, K.; Polly, P.; Carlberg, C. Differential Apoptotic Response of Human Melanoma Cells to 1 Alpha,25-Dihydroxyvitamin D3 and Its Analogues. Cell Death Differ. 1998, 5, 946–952. [Google Scholar] [CrossRef]
- Cordero, J.B.; Cozzolino, M.; Lu, Y.; Vidal, M.; Slatopolsky, E.; Stahl, P.D.; Barbieri, M.A.; Dusso, A. 1,25-Dihydroxyvitamin D Down-Regulates Cell Membrane Growth- and Nuclear Growth-Promoting Signals by the Epidermal Growth Factor Receptor. J. Biol. Chem. 2002, 277, 38965–38971. [Google Scholar] [CrossRef]
- Pinczewski, J.; Slominski, A. The Potential Role of Vitamin D in the Progression of Benign and Malignant Melanocytic Neoplasms. Exp. Dermatol. 2010, 19, 860–864. [Google Scholar] [CrossRef]
- Slominski, A.T.; Brożyna, A.A.; Skobowiat, C.; Zmijewski, M.A.; Kim, T.K.; Janjetovic, Z.; Oak, A.S.; Jozwicki, W.; Jetten, A.M.; Mason, R.S.; et al. On the Role of Classical and Novel Forms of Vitamin D in Melanoma Progression and Management. J. Steroid Biochem. Mol. Biol. 2018, 177, 159–170. [Google Scholar] [CrossRef]
- Slominski, A.T.; Brożyna, A.A.; Zmijewski, M.A.; Jóźwicki, W.; Jetten, A.M.; Mason, R.S.; Tuckey, R.C.; Elmets, C.A. Vitamin D Signaling and Melanoma: Role of Vitamin D and Its Receptors in Melanoma Progression and Management. Lab. Investig. 2017, 97, 706–724. [Google Scholar] [CrossRef]
- Del Puerto, C.; Navarrete-Dechent, C.; Molgó, M.; Camargo, C.A., Jr.; Borzutzky, A.; González, S. Immunohistochemical Expression of Vitamin D Receptor in Melanocytic Naevi and Cutaneous Melanoma: A Case-Control Study. Br. J. Dermatol. 2018, 179, 95–100. [Google Scholar] [CrossRef]
- Brozyna, A.A.; Jozwicki, W.; Janjetovic, Z.; Slominski, A.T. Expression of the Vitamin D-Activating Enzyme 1alpha-Hydroxylase (Cyp27b1) Decreases During Melanoma Progression. Hum. Pathol. 2013, 44, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Brozyna, A.A.; Jochymski, C.; Janjetovic, Z.; Jozwicki, W.; Tuckey, R.C.; Slominski, A.T. Cyp24a1 Expression Inversely Correlates with Melanoma Progression: Clinic-Pathological Studies. Int. J. Mol. Sci. 2014, 15, 19000–19017. [Google Scholar] [CrossRef] [PubMed]
- Orlow, I.; Roy, P.; Reiner, A.S.; Yoo, S.; Patel, H.; Paine, S.; Armstrong, B.K.; Kricker, A.; Marrett, L.D.; Millikan, R.C.; et al. Vitamin D Receptor Polymorphisms in Patients with Cutaneous Melanoma. Int. J. Cancer 2012, 130, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Orlow, I.; Reiner, A.S.; Thomas, N.E.; Roy, P.; Kanetsky, P.A.; Luo, L.; Paine, S.; Armstrong, B.K.; Kricker, A.; Marrett, L.D.; et al. Vitamin D Receptor Polymorphisms and Survival in Patients with Cutaneous Melanoma: A Population-Based Study. Carcinogenesis 2016, 37, 30–38. [Google Scholar] [CrossRef]
- Orlow, I.; Shi, Y.; Kanetsky, P.A.; Thomas, N.E.; Luo, L.; Corrales-Guerrero, S.; Cust, A.E.; Sacchetto, L.; Zanetti, R.; Rosso, S.; et al. The Interaction between Vitamin D Receptor Polymorphisms and Sun Exposure around Time of Diagnosis Influences Melanoma Survival. Pigment Cell Melanoma Res. 2018, 31, 287–296. [Google Scholar] [CrossRef]
- Newton-Bishop, J.A.; Beswick, S.; Randerson-Moor, J.; Chang, Y.M.; Affleck, P.; Elliott, F.; Chan, M.; Leake, S.; Karpavicius, B.; Haynes, S.; et al. Serum 25-Hydroxyvitamin D3 Levels Are Associated with Breslow Thickness at Presentation and Survival from Melanoma. J. Clin. Oncol. 2009, 27, 5439–5444. [Google Scholar] [CrossRef]
- Vasilovici, A.F.; Grigore, L.E.; Ungureanu, L.; Fechete, O.; Candrea, E.; Trifa, A.P.; Vișan, S.; Șenilă, S.; Cosgarea, R. Vitamin D Receptor Polymorphisms and Melanoma. Oncol. Lett. 2019, 17, 4162–4169. [Google Scholar] [CrossRef]
- Markiewicz, A.; Brożyna, A.A.; Podgórska, E.; Elas, M.; Urbańska, K.; Jetten, A.M.; Slominski, A.T.; Jóźwicki, W.; Orłowska-Heitzman, J.; Dyduch, G.; et al. Vitamin D Receptors (Vdr), Hydroxylases Cyp27b1 and Cyp24a1 and Retinoid-Related Orphan Receptors (Ror) Level in Human Uveal Tract and Ocular Melanoma with Different Melanization Levels. Sci. Rep. 2019, 9, 9142. [Google Scholar] [CrossRef]
- Colston, K.; Colston, M.J.; Feldman, D. 1,25-Dihydroxyvitamin D3 and Malignant Melanoma: The Presence of Receptors and Inhibition of Cell Growth in Culture. Endocrinology 1981, 108, 1083–1086. [Google Scholar] [CrossRef]
- Frampton, R.J.; Omond, S.A.; Eisman, J.A. Inhibition of Human Cancer Cell Growth by 1,25-Dihydroxyvitamin D3 Metabolites. Cancer Res. 1983, 43, 4443. [Google Scholar]
- Skobowiat, C.; Oak, A.S.; Kim, T.K.; Yang, C.H.; Pfeffer, L.M.; Tuckey, R.C.; Slominski, A.T. Noncalcemic 20-Hydroxyvitamin D3 Inhibits Human Melanoma Growth in in Vitro and in Vivo Models. Oncotarget 2017, 8, 9823–9834. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, A.; Wierzbicka, J.; Nadkarni, S.; Brown, G.; Kutner, A.; Żmijewski, M.A. Antiproliferative Activity of Double Point Modified Analogs of 1,25-Dihydroxyvitamin D2 against Human Malignant Melanoma Cell Lines. Int. J. Mol. Sci. 2016, 17, 76. [Google Scholar] [CrossRef] [PubMed]
- Brożyna, A.A.; Hoffman, R.M.; Slominski, A.T. Relevance of Vitamin D in Melanoma Development, Progression and Therapy. Anticancer Res. 2020, 40, 473–489. [Google Scholar] [CrossRef]
- Harant, H.; Spinner, D.; Reddy, G.S.; Lindley, I.J. Natural Metabolites of 1alpha,25-Dihydroxyvitamin D(3) Retain Biologic Activity Mediated through the Vitamin D Receptor. J. Cell. Biochem. 2000, 78, 112–120. [Google Scholar] [CrossRef]
- Wasiewicz, T.; Piotrowska, A.; Wierzbicka, J.; Slominski, A.T.; Zmijewski, M.A. Antiproliferative Activity of Non-Calcemic Vitamin D Analogs on Human Melanoma Lines in Relation to Vdr and Pdia3 Receptors. Int. J. Mol. Sci. 2018, 19, 2583. [Google Scholar] [CrossRef]
- Wasiewicz, T.; Szyszka, P.; Cichorek, M.; Janjetovic, Z.; Tuckey, R.C.; Slominski, A.T.; Zmijewski, M.A. Antitumor Effects of Vitamin D Analogs on Hamster and Mouse Melanoma Cell Lines in Relation to Melanin Pigmentation. Int. J. Mol. Sci. 2015, 16, 6645–6667. [Google Scholar] [CrossRef]
- Slominski, A.T.; Kim, T.-K.; Janjetovic, Z.; Tuckey, R.C.; Bieniek, R.; Yue, J.; Li, W.; Chen, J.; Nguyen, M.N.; Tang, E.K.Y.; et al. 20-Hydroxyvitamin D2 Is a Noncalcemic Analog of Vitamin D with Potent Antiproliferative and Prodifferentiation Activities in Normal and Malignant Cells. Am. J. Physiol. Cell Physiol. 2011, 300, C526–C541. [Google Scholar] [CrossRef]
- Slominski, A.T.; Janjetovic, Z.; Kim, T.-K.; Wright, A.C.; Grese, L.N.; Riney, S.J.; Nguyen, M.N.; Tuckey, R.C. Novel Vitamin D Hydroxyderivatives Inhibit Melanoma Growth and Show Differential Effects on Normal Melanocytes. Anticancer Res. 2012, 32, 3733–3742. [Google Scholar]
- Janjetovic, Z.; Brozyna, A.A.; Tuckey, R.C.; Kim, T.K.; Nguyen, M.N.; Jozwicki, W.; Pfeffer, S.R.; Pfeffer, L.M.; Slominski, A.T. High Basal Nf-Κb Activity in Nonpigmented Melanoma Cells Is Associated with an Enhanced Sensitivity to Vitamin D3 Derivatives. Br. J. Cancer 2011, 105, 1874–1884. [Google Scholar] [CrossRef]
- Slominski, A.T.; Kim, T.K.; Li, W.; Postlethwaite, A.; Tieu, E.W.; Tang, E.K.Y.; Tuckey, R.C. Detection of Novel Cyp11a1-Derived Secosteroids in the Human Epidermis and Serum and Pig Adrenal Gland. Sci. Rep. 2015, 5, 14875. [Google Scholar] [CrossRef]
- Tuckey, R.C.; Cheng, C.Y.S.; Slominski, A.T. The Serum Vitamin D Metabolome: What We Know and What Is Still to Discover. J. Steroid Biochem. Mol. Biol. 2019, 186, 4–21. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Xu, W.; Chen, Y.; Liu, X.; Xi, M. Abnormal Apoptosis of Trophoblastic Cells Is Related to the up-Regulation of Cyp11a Gene in Placenta of Preeclampsia Patients. PLoS ONE 2013, 8, e59609. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Zjawiony, J.; Wortsman, J.; Semak, I.; Stewart, J.; Pisarchik, A.; Sweatman, T.; Marcos, J.; Dunbar, C.; C Tuckey, R. A Novel Pathway for Sequential Transformation of 7-Dehydrocholesterol and Expression of the P450scc System in Mammalian Skin. Eur. J. Biochem. 2004, 271, 4178–4188. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Semak, I.; Zjawiony, J.; Wortsman, J.; Li, W.; Szczesniewski, A.; Tuckey, R.C. The Cytochrome P450scc System Opens an Alternate Pathway of Vitamin D3 Metabolism. FEBS J. 2005, 272, 4080–4090. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.R.; Sennoune, S.R.; Martinez-Zaguilan, R.; Slominski, A.T.; Pruitt, K. Regulation of Retinoid Mediated Cholesterol Efflux Involves Liver X Receptor Activation in Mouse Macrophages. Biochem. Biophys. Res. Commun. 2015, 464, 312–317. [Google Scholar] [CrossRef]
- Schedel, M.; Jia, Y.; Michel, S.; Takeda, K.; Domenico, J.; Joetham, A.; Ning, F.; Strand, M.; Han, J.; Wang, M.; et al. 1,25d3 Prevents Cd8(+)Tc2 Skewing and Asthma Development through Vdr Binding Changes to the Cyp11a1 Promoter. Nat. Commun. 2016, 7, 10213. [Google Scholar] [CrossRef]
- Manna, P.R.; Stetson, C.L.; Daugherty, C.; Shimizu, I.; Syapin, P.J.; Garrel, G.; Cohen-Tannoudji, J.; Huhtaniemi, I.; Slominski, A.T.; Pruitt, K.; et al. Up-Regulation of Steroid Biosynthesis by Retinoid Signaling: Implications for Aging. Mech. Ageing Dev. 2015, 150, 74–82. [Google Scholar] [CrossRef]
- Slominski, R.M.; Tuckey, R.C.; Manna, P.R.; Jetten, A.M.; Postlethwaite, A.; Raman, C.; Slominski, A.T. Extra-Adrenal Glucocorticoid Biosynthesis: Implications for Autoimmune and Inflammatory Disorders. Genes Immun. 2020. [Google Scholar] [CrossRef]
- Slominski, A.T.; Manna, P.R.; Tuckey, R.C. On the Role of Skin in the Regulation of Local and Systemic Steroidogenic Activities. Steroids 2015, 103, 72–88. [Google Scholar] [CrossRef]
- Asgari, M.M.; Maruti, S.S.; Kushi, L.H.; White, E. A Cohort Study of Vitamin D Intake and Melanoma Risk. J. Investig. Dermatol. 2009, 129, 1675–1680. [Google Scholar] [CrossRef]
- Weinstein, D.; Leininger, J.; Hamby, C.; Safai, B. Diagnostic and Prognostic Biomarkers in Melanoma. J. Clin. Aesthet. Dermatol. 2014, 7, 13–24. [Google Scholar] [PubMed]
- Abdel-Malek, Z.A.; Ruwe, A.; Kavanagh-Starner, R.; Kadekaro, A.L.; Swope, V.; Haskell-Luevano, C.; Koikov, L.; Knittel, J.J. Alpha-Msh Tripeptide Analogs Activate the Melanocortin 1 Receptor and Reduce Uv-Induced DNA Damage in Human Melanocytes. Pigment Cell Melanoma Res. 2009, 22, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Scherer, D.; Bermejo, J.L.; Rudnai, P.; Gurzau, E.; Koppova, K.; Hemminki, K.; Kumar, R. Mc1r Variants Associated Susceptibility to Basal Cell Carcinoma of Skin: Interaction with Host Factors and Xrcc3 Polymorphism. Int. J. Cancer 2008, 122, 1787–1793. [Google Scholar] [CrossRef] [PubMed]
- Hauser, J.E.; Kadekaro, A.L.; Kavanagh, R.J.; Wakamatsu, K.; Terzieva, S.; Schwemberger, S.; Babcock, G.; Rao, M.B.; Ito, S.; Abdel-Malek, Z.A. Melanin Content and Mc1r Function Independently Affect Uvr-Induced DNA Damage in Cultured Human Melanocytes. Pigment Cell Res. 2006, 19, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Feller, L.; Khammissa, R.A.G.; Kramer, B.; Altini, M.; Lemmer, J. Basal Cell Carcinoma, Squamous Cell Carcinoma and Melanoma of the Head and Face. Head Face Med. 2016, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Nan, H.; Kraft, P.; Hunter, D.J.; Han, J. Genetic Variants in Pigmentation Genes, Pigmentary Phenotypes, and Risk of Skin Cancer in Caucasians. Int. J. Cancer 2009, 125, 909–917. [Google Scholar] [CrossRef]
- Kanetsky, P.A.; Panossian, S.; Elder, D.E.; Guerry, D.; Ming, M.E.; Schuchter, L.; Rebbeck, T.R. Does Mc1r Genotype Convey Information About Melanoma Risk Beyond Risk Phenotypes? Cancer 2010, 116, 2416–2428. [Google Scholar]
- Williams, P.F.; Olsen, C.M.; Hayward, N.K.; Whiteman, D.C. Melanocortin 1 Receptor and Risk of Cutaneous Melanoma: A Meta-Analysis and Estimates of Population Burden. Int. J. Cancer 2011, 129, 1730–1740. [Google Scholar] [CrossRef]
- Wendt, J.; Rauscher, S.; Burgstaller-Muehlbacher, S.; Fae, I.; Fischer, G.; Pehamberger, H.; Okamoto, I. Human Determinants and the Role of Melanocortin-1 Receptor Variants in Melanoma Risk Independent of Uv Radiation Exposure. JAMA Dermatol. 2016, 152, 776–782. [Google Scholar] [CrossRef]
- Taylor, N.J.; Busam, K.J.; From, L.; Groben, P.A.; Anton-Culver, H.; Cust, A.E.; Begg, C.B.; Dwyer, T.; Gallagher, R.P.; Gruber, S.B.; et al. Inherited Variation at Mc1r and Histological Characteristics of Primary Melanoma. PLoS ONE 2015, 10, e0119920. [Google Scholar] [CrossRef]
- Robles-Espinoza, C.D.; Roberts, N.D.; Chen, S.; Leacy, F.P.; Alexandrov, L.B.; Pornputtapong, N.; Halaban, R.; Krauthammer, M.; Cui, R.; Timothy Bishop, D.; et al. Germline Mc1r Status Influences Somatic Mutation Burden in Melanoma. Nat. Commun. 2016, 7, 12064. [Google Scholar] [CrossRef] [PubMed]
- Fargnoli, M.C.; Gandini, S.; Peris, K.; Maisonneuve, P.; Raimondi, S. Mc1r Variants Increase Melanoma Risk in Families with Cdkn2a Mutations: A Meta-Analysis. Eur. J. Cancer 2010, 46, 1413–1420. [Google Scholar] [CrossRef] [PubMed]
- King, R.; Googe, P.B.; Weilbaecher, K.N.; Mihm, M.C., Jr.; Fisher, D.E. Microphthalmia Transcription Factor Expression in Cutaneous Benign, Malignant Melanocytic, and Nonmelanocytic Tumors. Am. J. Surg. Pathol. 2001, 25, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Franco-Palacios, M.; Russell, M.; Goddard, L.; Hassell, L.; Gillies, E.; Fung, K.-M. Micropthalmia Transcription Factor (Mitf) as a Diagnostic Marker for Metastatic Melanomas Negative for Other Melanoma Markers. Int. J. Clin. Exp. Pathol. 2013, 6, 1658–1664. [Google Scholar] [PubMed]
- Miettinen, M.; Fernandez, M.; Franssila, K.; Gatalica, Z.; Lasota, J.; Sarlomo-Rikala, M. Microphthalmia Transcription Factor in the Immunohistochemical Diagnosis of Metastatic Melanoma: Comparison with Four Other Melanoma Markers. Am. J. Surg. Pathol. 2001, 25, 205–211. [Google Scholar] [CrossRef]
- Carreira, S.; Goodall, J.; Denat, L.; Rodriguez, M.; Nuciforo, P.; Hoek, K.S.; Testori, A.; Larue, L.; Goding, C.R. Mitf Regulation of Dia1 Controls Melanoma Proliferation and Invasiveness. Genes Dev. 2006, 20, 3426–3439. [Google Scholar] [CrossRef]
- Cheli, Y.; Giuliano, S.; Fenouille, N.; Allegra, M.; Hofman, V.; Hofman, P.; Bahadoran, P.; Lacour, J.P.; Tartare-Deckert, S.; Bertolotto, C.; et al. Hypoxia and Mitf Control Metastatic Behaviour in Mouse and Human Melanoma Cells. Oncogene 2012, 31, 2461–2470. [Google Scholar] [CrossRef]
- Bertolotto, C.; Lesueur, F.; Giuliano, S.; Strub, T.; de Lichy, M.; Bille, K.; Dessen, P.; d’Hayer, B.; Mohamdi, H.; Remenieras, A.; et al. A Sumoylation-Defective Mitf Germline Mutation Predisposes to Melanoma and Renal Carcinoma. Nature 2011, 480, 94–98. [Google Scholar] [CrossRef]
- Ciccarese, G.; Dalmasso, B.; Bruno, W.; Queirolo, P.; Pastorino, L.; Andreotti, V.; Spagnolo, F.; Tanda, E.; Ponti, G.; Massone, C.; et al. Clinical, Pathological and Dermoscopic Phenotype of Mitf P.E318k Carrier Cutaneous Melanoma Patients. J. Transl. Med. 2020, 18, 78. [Google Scholar] [CrossRef]
- Balch, C.M.; Soong, S.J.; Gershenwald, J.E.; Thompson, J.F.; Coit, D.G.; Atkins, M.B.; Ding, S.; Cochran, A.J.; Eggermont, A.M.; Flaherty, K.T.; et al. Age as a Prognostic Factor in Patients with Localized Melanoma and Regional Metastases. Ann. Surg. Oncol. 2013, 20, 3961–3968. [Google Scholar] [CrossRef]
- Ecker, B.L.; Kaur, A.; Douglass, S.M.; Webster, M.R.; Almeida, F.V.; Marino, G.E.; Sinnamon, A.J.; Neuwirth, M.G.; Alicea, G.M.; Ndoye, A.; et al. Age-Related Changes in Hapln1 Increase Lymphatic Permeability and Affect Routes of Melanoma Metastasis. Cancer Discov. 2019, 9, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Herrling, T.; Jung, K.; Fuchs, J. The Role of Melanin as Protector against Free Radicals in Skin and Its Role as Free Radical Indicator in Hair. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2008, 69, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Zmijewski, M.A.; Plonka, P.M.; Szaflarski, J.P.; Paus, R. How Uv Light Touches the Brain and Endocrine System through Skin, and Why. Endocrinology 2018, 159, 1992–2007. [Google Scholar] [CrossRef] [PubMed]
- Jimbow, K.; Lee, S.K.; King, M.G.; Hara, H.; Chen, H.; Dakour, J.; Marusyk, H. Melanin Pigments and Melanosomal Proteins as Differentiation Markers Unique to Normal and Neoplastic Melanocytes. J. Investig. Dermatol. 1993, 100, 259S–268S. [Google Scholar] [CrossRef] [PubMed]
- Brożyna, A.A.; Jóźwicki, W.; Carlson, J.A.; Slominski, A.T. Melanogenesis Affects Overall and Disease-Free Survival in Patients with Stage Iii and Iv Melanoma. Hum. Pathol. 2013, 44, 2071–2074. [Google Scholar] [CrossRef]
- Slominski, A.; Kim, T.K.; Brożyna, A.A.; Janjetovic, Z.; Brooks, D.L.P.; Schwab, L.P.; Skobowiat, C.; Jóźwicki, W.; Seagroves, T.N. The Role of Melanogenesis in Regulation of Melanoma Behavior: Melanogenesis Leads to Stimulation of Hif-1α Expression and Hif-Dependent Attendant Pathways. Arch. Biochem. Biophys. 2014, 563, 79–93. [Google Scholar] [CrossRef]
- Urbanska, K.; Romanowska-Dixon, B.; Elas, M.; Pajak, S.; Paziewski, E.; Bryk, J.; Kukielczak, B.; Slominski, A.; Zygulska-Mach, H.; Lukiewicz, S. Experimental Ruthenium Plaque Therapy of Amelanotic and Melanotic Melanomas in the Hamster Eye. Melanoma Res. 2000, 10, 26–35. [Google Scholar] [CrossRef]
- Shields, C.L.; Kaliki, S.; Cohen, M.N.; Shields, P.W.; Furuta, M.; Shields, J.A. Prognosis of Uveal Melanoma Based on Race in 8100 Patients: The 2015 Doyne Lecture. Eye (London) 2015, 29, 1027–1035. [Google Scholar] [CrossRef]
- Shields, C.L.; Kaliki, S.; Furuta, M.; Fulco, E.; Alarcon, C.; Shields, J.A. American Joint Committee on Cancer Classification of Uveal Melanoma (Anatomic Stage) Predicts Prognosis in 7731 Patients: The 2013 Zimmerman Lecture. Ophthalmology 2015, 122, 1180–1186. [Google Scholar] [CrossRef]
- Thomas, N.E.; Kricker, A.; Waxweiler, W.T.; Dillon, P.M.; Busman, K.J.; From, L.; Groben, P.A.; Armstrong, B.K.; Anton-Culver, H.; Gruber, S.B.; et al. Comparison of Clinicopathologic Features and Survival of Histopathologically Amelanotic and Pigmented Melanomas: A Population-Based Study. JAMA Dermatol. 2014, 150, 1306–1314. [Google Scholar] [CrossRef]
- Sarna, M.; Krzykawska-Serda, M.; Jakubowska, M.; Zadlo, A.; Urbanska, K. Melanin Presence Inhibits Melanoma Cell Spread in Mice in a Unique Mechanical Fashion. Sci. Rep. 2019, 9, 9280. [Google Scholar] [CrossRef] [PubMed]
- Sarna, M.; Zadlo, A.; Czuba-Pelech, B.; Urbanska, K. Nanomechanical Phenotype of Melanoma Cells Depends Solely on the Amount of Endogenous Pigment in the Cells. Int. J. Mol. Sci. 2018, 19, 607. [Google Scholar] [CrossRef] [PubMed]
- Brożyna, A.A.; Jóźwicki, W.; Roszkowski, K.; Filipiak, J.; Slominski, A.T. Melanin Content in Melanoma Metastases Affects the Outcome of Radiotherapy. Oncotarget 2016, 7, 17844–17853. [Google Scholar] [CrossRef] [PubMed]
- Brozyna, A.A.; VanMiddlesworth, L.; Slominski, A.T. Inhibition of Melanogenesis as a Radiation Sensitizer for Melanoma Therapy. Int. J. Cancer 2008, 123, 1448–1456. [Google Scholar] [CrossRef]
- Slominski, A.; Zbytek, B.; Slominski, R. Inhibitors of Melanogenesis Increase Toxicity of Cyclophosphamide and Lymphocytes against Melanoma Cells. Int. J. Cancer 2009, 124, 1470–1477. [Google Scholar] [CrossRef]
- Slominski, A.; Goodman-Snitkoff, G.G. Dopa Inhibits Induced Proliferative Activity of Murine and Human Lymphocytes. Anticancer Res. 1992, 12, 753–756. [Google Scholar]
- Slominski, A.; Paus, R.; Mihm, M.C. Inhibition of Melanogenesis as an Adjuvant Strategy in the Treatment of Melanotic Melanomas: Selective Review and Hypothesis. Anticancer Res. 1998, 18, 3709–3715. [Google Scholar]
- Slominski, A.T.; Carlson, J.A. Melanoma Resistance: A Bright Future for Academicians and a Challenge for Patient Advocates. Mayo Clin. Proc. 2014, 89, 429–433. [Google Scholar] [CrossRef]
- Hofbauer, G.F.; Kamarashev, J.; Geertsen, R.; Böni, R.; Dummer, R. Tyrosinase Immunoreactivity in Formalin-Fixed, Paraffin-Embedded Primary and Metastatic Melanoma: Frequency and Distribution. J. Cutan. Pathol. 1998, 25, 204–209. [Google Scholar] [CrossRef]
- Kaufmann, O.; Koch, S.; Burghardt, J.; Audring, H.; Dietel, M. Tyrosinase, Melan-a, and Kba62 as Markers for the Immunohistochemical Identification of Metastatic Amelanotic Melanomas on Paraffin Sections. Mod. Pathol. 1998, 11, 740–746. [Google Scholar]
- Clarkson, K.S.; Sturdgess, I.C.; Molyneux, A.J. The Usefulness of Tyrosinase in the Immunohistochemical Assessment of Melanocytic Lesions: A Comparison of the Novel T311 Antibody (Anti-Tyrosinase) with S-100, Hmb45, and A103 (Anti-Melan-a). J. Clin. Pathol. 2001, 54, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Reinke, S.; Königer, P.; Herberth, G.; Audring, H.; Wang, H.; Ma, J.; Guo, Y.; Sterry, W.; Trefzer, U. Differential Expression of Mart-1, Tyrosinase, and Sm5-1 in Primary and Metastatic Melanoma. Am. J. Dermatopathol. 2005, 27, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Murtas, D.; Pilloni, L.; Diana, A.; Casula, L.; Tomei, S.; Piras, F.; Ferreli, C.; Maxia, C.; Perra, M.T. Tyrosinase and Nestin Immunohistochemical Expression in Melanocytic Nevi as a Histopathologic Pattern to Trace Melanocyte Differentiation and Nevogenesis. Histochem. Cell Biol. 2019, 151, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Bloom, M.B.; Perry-Lalley, D.; Robbins, P.F.; Li, Y.; el-Gamil, M.; Rosenberg, S.A.; Yang, J.C. Identification of Tyrosinase-Related Protein 2 as a Tumor Rejection Antigen for the B16 Melanoma. J. Exp. Med. 1997, 185, 453–459. [Google Scholar] [CrossRef]
- Kameyama, K.; Sakai, C.; Kuge, S.; Nishiyama, S.; Tomita, Y.; Ito, S.; Wakamatsu, K.; Hearing, V.J. The Expression of Tyrosinase, Tyrosinase-Related Proteins 1 and 2 (Trp1 and Trp2), the Silver Protein, and a Melanogenic Inhibitor in Human Melanoma Cells of Differing Melanogenic Activities. Pigment Cell Res. 1995, 8, 97–104. [Google Scholar] [CrossRef]
- Journe, F.; Id Boufker, H.; Van Kempen, L.; Galibert, M.D.; Wiedig, M.; Salès, F.; Theunis, A.; Nonclercq, D.; Frau, A.; Laurent, G.; et al. Tyrp1 Mrna Expression in Melanoma Metastases Correlates with Clinical Outcome. Br. J. Cancer 2011, 105, 1726–1732. [Google Scholar] [CrossRef]
- Bolander, A.; Agnarsdóttir, M.; Strömberg, S.; Ponten, F.; Hesselius, P.; Uhlen, M.; Bergqvist, M. The Protein Expression of Trp-1 and Galectin-1 in Cutaneous Malignant Melanomas. Cancer Genomics Proteomics 2008, 5, 293–300. [Google Scholar]
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.J.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Prognostic and Clinicopathologic Associations of Oncogenic Braf in Metastatic Melanoma. J. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kim, S.N.; Hahn, H.J.; Lee, Y.W.; Choe, Y.B.; Ahn, K.J. Metaanalysis of Braf Mutations and Clinicopathologic Characteristics in Primary Melanoma. J. Am. Acad. Dermatol. 2015, 72, 1036–1046.e2. [Google Scholar] [CrossRef]
- Menzies, A.M.; Haydu, L.E.; Visintin, L.; Carlino, M.S.; Howle, J.R.; Thompson, J.F.; Kefford, R.F.; Scolyer, R.A.; Long, G.V. Distinguishing Clinicopathologic Features of Patients with V600e and V600k Braf-Mutant Metastatic Melanoma. Clin. Cancer Res. 2012, 18, 3242–3249. [Google Scholar] [CrossRef]
- Colebatch, A.J.; Ferguson, P.; Newell, F.; Kazakoff, S.H.; Witkowski, T.; Dobrovic, A.; Johansson, P.A.; Saw, R.P.M.; Stretch, J.R.; McArthur, G.A.; et al. Molecular Genomic Profiling of Melanocytic nevi. J. Investig. Dermatol. 2019, 139, 1762–1768. [Google Scholar] [CrossRef] [PubMed]
- Brash, D.E. Uv Signature Mutations. Photochem. Photobiol. 2015, 91, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.; Curtin, J.A.; Pinkel, D.; Bastian, B.C. Congenital Melanocytic Nevi Frequently Harbor Nras Mutations but No Braf Mutations. J. Investig. Dermatol. 2007, 127, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, J.L.; Fridlyand, J.; Patel, H.; Jain, A.N.; Busam, K.; Kageshita, T.; Ono, T.; Albertson, D.G.; Pinkel, D.; Bastian, B.C. Determinants of Braf Mutations in Primary Melanomas. J. Natl. Cancer Inst. 2003, 95, 1878–1890. [Google Scholar] [CrossRef]
- Melamed, R.D.; Aydin, I.T.; Rajan, G.S.; Phelps, R.; Silvers, D.N.; Emmett, K.J.; Brunner, G.; Rabadan, R.; Celebi, J.T. Genomic Characterization of Dysplastic Nevi Unveils Implications for Diagnosis Of melanoma. J. Investig. Dermatol. 2017, 137, 905–909. [Google Scholar] [CrossRef]
- Akslen, L.A.; Angelini, S.; Straume, O.; Bachmann, I.M.; Molven, A.; Hemminki, K.; Kumar, R. Braf and Nras Mutations Are Frequent in Nodular Melanoma but Are Not Associated with Tumor Cell Proliferation or Patient Survival. J. Investig. Dermatol. 2005, 125, 312–317. [Google Scholar] [CrossRef]
- Abd Elmageed, Z.Y.; Moore, R.F.; Tsumagari, K.; Lee, M.M.; Sholl, A.B.; Friedlander, P.; Al-Qurayshi, Z.; Hassan, M.; Wang, A.R.; Boulares, H.A.; et al. Prognostic Role of Braf(V600e) Cellular Localization in Melanoma. J. Am. Coll. Surg. 2018, 226, 526–537. [Google Scholar] [CrossRef]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.-K.; Attar, N.; Sazegar, H.; et al. Melanomas Acquire Resistance to B-Raf(V600e) Inhibition by Rtk or N-Ras Upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef]
- Amann, V.C.; Ramelyte, E.; Thurneysen, S.; Pitocco, R.; Bentele-Jaberg, N.; Goldinger, S.M.; Dummer, R.; Mangana, J. Developments in Targeted Therapy in Melanoma. Eur. J. Surg. Oncol. 2017, 43, 581–593. [Google Scholar] [CrossRef]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroyakovskiy, D.; Dummer, R.; Grange, F.; Mortier, L.; Chiarion-Sileni, V.; et al. Three-Year Estimate of Overall Survival in Combi-V, a Randomized Phase 3 Study Evaluating First-Line Dabrafenib (D) + Trametinib (T) in Patients (Pts) with Unresectable or Metastatic Braf V600e/K–Mutant Cutaneous Melanoma. Ann. Oncol. 2016, 27 (Suppl. 6). [Google Scholar] [CrossRef]
- Long, G.V.; Flaherty, K.T.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; et al. Dabrafenib Plus Trametinib Versus Dabrafenib Monotherapy in Patients with Metastatic Braf V600e/K-Mutant Melanoma: Long-Term Survival and Safety Analysis of a Phase 3 Study. Ann. Oncol. 2017, 28, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Weber, J.S.; Infante, J.R.; Kim, K.B.; Daud, A.; Gonzalez, R.; Sosman, J.A.; Hamid, O.; Schuchter, L.; Cebon, J.; et al. Overall Survival and Durable Responses in Patients with Braf V600-Mutant Metastatic Melanoma Receiving Dabrafenib Combined with Trametinib. J. Clin. Oncol. 2016, 34, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Pavlick, A.C.; Fecher, L.; Ascierto, P.A.; Sullivan, R.J. Frontline Therapy for Braf-Mutated Metastatic Melanoma: How Do You Choose, and Is There One Correct Answer? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.A.; Cullinane, C.; Kirby, L.; Abuhammad, S.; Lelliott, E.J.; Waldeck, K.; Young, R.J.; Brajanovski, N.; Cameron, D.P.; Walker, R.; et al. Palbociclib Synergizes with Braf and Mek Inhibitors in Treatment Naive Melanoma but Not after the Development of Braf Inhibitor Resistance. Int. J. Cancer 2018, 142, 2139–2152. [Google Scholar] [CrossRef]
- Misek, S.A.; Appleton, K.M.; Dexheimer, T.S.; Lisabeth, E.M.; Lo, R.S.; Larsen, S.D.; Gallo, K.A.; Neubig, R.R. Rho-Mediated Signaling Promotes Braf Inhibitor Resistance in De-Differentiated Melanoma Cells. Oncogene 2020, 39, 1466–1483. [Google Scholar] [CrossRef]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. Raf Inhibitors Prime Wild-Type Raf to Activate the Mapk Pathway and Enhance Growth. Nature 2010, 464, 431–435. [Google Scholar] [CrossRef]
- Su, F.; Viros, A.; Milagre, C.; Trunzer, K.; Bollag, G.; Spleiss, O.; Reis-Filho, J.S.; Kong, X.; Koya, R.C.; Flaherty, K.T.; et al. Ras Mutations in Cutaneous Squamous-Cell Carcinomas in Patients Treated with Braf Inhibitors. N. Engl. J. Med. 2012, 366, 207–215. [Google Scholar] [CrossRef]
- Göppner, D.; Müller, J.; Krüger, S.; Franke, I.; Gollnick, H.; Quist, S.R. High Incidence of Naevi-Associated Braf Wild-Type Melanoma and Dysplastic Naevi under Treatment with the Class I Braf Inhibitor Vemurafenib. Acta Derm. Venereol. 2014, 94, 517–520. [Google Scholar] [CrossRef]
- Shimizu, K.; Goldfarb, M.; Perucho, M.; Wigler, M. Isolation and Preliminary Characterization of the Transforming Gene of a Human Neuroblastoma Cell Line. Proc. Natl. Acad. Sci. USA 1983, 80, 383–387. [Google Scholar] [CrossRef]
- Hall, A.; Marshall, C.J.; Spurr, N.K.; Weiss, R.A. Identification of Transforming Gene in Two Human Sarcoma Cell Lines as a New Member of the Ras Gene Family Located on Chromosome 1. Nature 1983, 303, 396–400. [Google Scholar] [CrossRef]
- Chiappetta, C.; Proietti, I.; Soccodato, V.; Puggioni, C.; Zaralli, R.; Pacini, L.; Porta, N.; Skroza, N.; Petrozza, V.; Potenza, C.; et al. Braf and Nras Mutations Are Heterogeneous and Not Mutually Exclusive in Nodular Melanoma. Appl. Immunohistochem. Mol. Morphol. 2015, 23, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Van’t Veer, L.J.; Burgering, B.M.; Versteeg, R.; Boot, A.J.; Ruiter, D.J.; Osanto, S.; Schrier, P.I.; Bos, J.L. N-Ras Mutations in Human Cutaneous Melanoma from Sun-Exposed Body Sites. Mol. Cell. Biol. 1989, 9, 3114–3116. [Google Scholar] [CrossRef] [PubMed]
- Thomas, N.E.; Edmiston, S.N.; Alexander, A.; Groben, P.A.; Parrish, E.; Kricker, A.; Armstrong, B.K.; Anton-Culver, H.; Gruber, S.B.; From, L.; et al. Association between Nras and Braf Mutational Status and Melanoma-Specific Survival among Patients with Higher-Risk Primary Melanoma. JAMA Oncol. 2015, 1, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Ellerhorst, J.A.; Greene, V.R.; Ekmekcioglu, S.; Warneke, C.L.; Johnson, M.M.; Cooke, C.P.; Wang, L.E.; Prieto, V.G.; Gershenwald, J.E.; Wei, Q.; et al. Clinical Correlates of Nras and Braf Mutations in Primary Human Melanoma. Clin. Cancer Res. 2011, 17, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Charbel, C.; Fontaine, R.H.; Malouf, G.G.; Picard, A.; Kadlub, N.; El-Murr, N.; How-Kit, A.; Su, X.; Coulomb-L’Hermine, A.; Tost, J.; et al. Nras Mutation Is the Sole Recurrent Somatic Mutation in Large Congenital Melanocytic Nevi. J. Investig. Dermatol. 2014, 134, 1067–1074. [Google Scholar] [CrossRef]
- Hancock, J.F.; Magee, A.I.; Childs, J.E.; Marshall, C.J. All Ras Proteins Are Polyisoprenylated but Only Some Are Palmitoylated. Cell 1989, 57, 1167–1177. [Google Scholar] [CrossRef]
- Boespflug, A.; Caramel, J.; Dalle, S.; Thomas, L. Treatment of Nras-Mutated Advanced or Metastatic Melanoma: Rationale, Current Trials and Evidence to Date. Ther. Adv. Med. Oncol. 2017, 9, 481–492. [Google Scholar] [CrossRef]
- Margolin, K.A.; Moon, J.; Flaherty, L.E.; Lao, C.D.; Akerley, W.L., 3rd; Othus, M.; Sosman, J.A.; Kirkwood, J.M.; Sondak, V.K. Randomized Phase Ii Trial of Sorafenib with Temsirolimus or Tipifarnib in Untreated Metastatic Melanoma (S0438). Clin. Cancer Res. 2012, 18, 1129–1137. [Google Scholar] [CrossRef]
- Whyte, D.B.; Kirschmeier, P.; Hockenberry, T.N.; Nunez-Oliva, I.; James, L.; Catino, J.J.; Bishop, W.R.; Pai, J.K. K- and N-Ras Are Geranylgeranylated in Cells Treated with Farnesyl Protein Transferase Inhibitors. J. Biol. Chem. 1997, 272, 14459–14464. [Google Scholar] [CrossRef]
- Dummer, R.; Schadendorf, D.; Ascierto, P.A.; Arance, A.; Dutriaux, C.; Di Giacomo, A.M.; Rutkowski, P.; Del Vecchio, M.; Gutzmer, R.; Mandala, M.; et al. Binimetinib Versus Dacarbazine in Patients with Advanced Nras-Mutant Melanoma (Nemo): A Multicentre, Open-Label, Randomised, Phase 3 Trial. Lancet Oncol. 2017, 18, 435–445. [Google Scholar] [CrossRef]
- Posch, C.; Moslehi, H.; Feeney, L.; Green, G.A.; Ebaee, A.; Feichtenschlager, V.; Chong, K.; Peng, L.; Dimon, M.T.; Phillips, T.; et al. Combined Targeting of Mek and Pi3k/Mtor Effector Pathways Is Necessary to Effectively Inhibit Nras Mutant Melanoma in Vitro and in Vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 4015–4020. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.; Nonami, A.; Chen, Z.; Liu, F.; Zhang, J.; Sattler, M.; Nelson, E.; Cowens, K.; Christie, A.L.; Mitsiades, C.; et al. Identification of Wee1 as a Novel Therapeutic Target for Mutant Ras-Driven Acute Leukemia and Other Malignancies. Leukemia 2015, 29, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, N.; Kikuchi, K.; Yagyu, S.; Miyachi, M.; Iehara, T.; Tajiri, T.; Sakai, T.; Hosoi, H. Mutations in the Ras Pathway as Potential Precision Medicine Targets in Treatment of Rhabdomyosarcoma. Biochem. Biophys. Res. Commun. 2019, 512, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Vu, H.L.; Aplin, A.E. Targeting Mutant Nras Signaling Pathways in Melanoma. Pharmacol. Res. 2016, 107, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Duensing, A.; Medeiros, F.; McConarty, B.; Joseph, N.E.; Panigrahy, D.; Singer, S.; Fletcher, C.D.; Demetri, G.D.; Fletcher, J.A. Mechanisms of Oncogenic Kit Signal Transduction in Primary Gastrointestinal Stromal Tumors (Gists). Oncogene 2004, 23, 3999–4006. [Google Scholar] [CrossRef] [PubMed]
- Slipicevic, A.; Herlyn, M. Kit in Melanoma: Many Shades of Gray. J. Investig. Dermatol. 2015, 135, 337–338. [Google Scholar] [CrossRef]
- Ma, X.; Wu, Y.; Zhang, T.; Song, H.; Jv, H.; Guo, W.; Ren, G. The Clinical Significance of C-Kit Mutations in Metastatic Oral Mucosal Melanoma in China. Oncotarget 2017, 8, 82661–82673. [Google Scholar] [CrossRef]
- Guo, J.; Si, L.; Kong, Y.; Flaherty, K.T.; Xu, X.; Zhu, Y.; Corless, C.L.; Li, L.; Li, H.; Sheng, X.; et al. Phase Ii, Open-Label, Single-Arm Trial of Imatinib Mesylate in Patients with Metastatic Melanoma Harboring C-Kit Mutation or Amplification. J. Clin. Oncol. 2011, 29, 2904–2909. [Google Scholar] [CrossRef]
- O’Day, S.J.; Hamid, O.; Urba, W.J. Targeting Cytotoxic T-Lymphocyte Antigen-4 (Ctla-4): A Novel Strategy for the Treatment of Melanoma and Other Malignancies. Cancer 2007, 110, 2614–2627. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the Pd-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef]
- Zitvogel, L.; Kroemer, G. Targeting Pd-1/Pd-L1 Interactions for Cancer Immunotherapy. Oncoimmunology 2012, 1, 1223–1225. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab Plus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Cardona, D.M.; Taube, J.M.; Anders, R.A.; Taylor, C.R.; Wolchok, J.D.; Callahan, M.K.; Curran, M.A.; Lesokhin, A.M.; Grosso, J.F.; et al. Peripheral and Tumor Immune Correlates in Patients with Advanced Melanoma Treated with Nivolumab (Anti-Pd-1, Bms-936558, Ono-4538) Monotherapy or in Combination with Ipilimumab. J. Transl. Med. 2014, 12, O8. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in Previously Untreated Melanoma without Braf Mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; Kudchadkar, R.R.; Yu, B.; Gallenstein, D.; Horak, C.E.; Inzunza, H.D.; Zhao, X.; Martinez, A.J.; Wang, W.; Gibney, G.; et al. Safety, Efficacy, and Biomarkers of Nivolumab with Vaccine in Ipilimumab-Refractory or -Naive Melanoma. J. Clin. Oncol. 2013, 31, 4311–4318. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal Pd-L1 Contributes to Immunosuppression and Is Associated with Anti-Pd-1 Response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Del Re, M.; Marconcini, R.; Pasquini, G.; Rofi, E.; Vivaldi, C.; Bloise, F.; Restante, G.; Arrigoni, E.; Caparello, C.; Bianco, M.G.; et al. Pd-L1 Mrna Expression in Plasma-Derived Exosomes Is Associated with Response to Anti-Pd-1 Antibodies in Melanoma and Nsclc. Br. J. Cancer 2018, 118, 820–824. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-Driven Biomarkers to Guide Immune Checkpoint Blockade in Cancer Therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Kleffel, S.; Posch, C.; Barthel, S.R.; Mueller, H.; Schlapbach, C.; Guenova, E.; Elco, C.P.; Lee, N.; Juneja, V.R.; Zhan, Q.; et al. Melanoma Cell-Intrinsic Pd-1 Receptor Functions Promote Tumor Growth. Cell 2015, 162, 1242–1256. [Google Scholar] [CrossRef]
- Zhang, B.; Dang, J.; Ba, D.; Wang, C.; Han, J.; Zheng, F. Potential Function of Ctla-4 in the Tumourigenic Capacity of Melanoma Stem Cells. Oncol. Lett. 2018, 16, 6163–6170. [Google Scholar] [CrossRef]
- Pistillo, M.P.; Fontana, V.; Morabito, A.; Dozin, B.; Laurent, S.; Carosio, R.; Banelli, B.; Ferrero, F.; Spano, L.; Tanda, E.; et al. Soluble Ctla-4 as a Favorable Predictive Biomarker in Metastatic Melanoma Patients Treated with Ipilimumab: An Italian Melanoma Intergroup Study. Cancer Immunol. Immunother. 2019, 68, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Petrie, T.; Samatham, R.; Witkowski, A.M.; Esteva, A.; Leachman, S.A. Melanoma Early Detection: Big Data, Bigger Picture. J. Investig. Dermatol. 2019, 139, 25–30. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Gene | Incidence | Hallmarks of Tumors |
|---|---|---|
| Cutaneous Melanoma | ||
| BRAF | 40–60% | Superficial spreading subtype; younger patients; non-CSD skin [14,15,16] |
| NRAS | 15–30% | Nodular subtype; CSD skin [17,18] |
| KIT | 1–2% | Mucosal and acral types; CSD skin [16,19] |
| CDKN2A | 25–40% (familial) | Superficial spreading subtype; dysplastic nevus syndrome [20,21,22] |
| VDR | Unknown | Inverse correlation with tumor progression and mitotic rates [23,24] |
| MC1R | Variants in up to 60% | Fair skin, red hair phenotype; presentation on arms [25,26] |
| MITF | 1–2% (familial) | Direct correlation with survival and negative lymph node status [27,28] |
| Uveal Melanoma | ||
| GNAQ/GNA11 | 80–90% | Present in most cases of uveal melanoma; rarely cutaneous melanoma; benign blue and uveal nevi [29] |
| BAP1 | 8–50% (familial) | High metastatic risk; BAPoma (atypical spitzoid tumor) [30,31,32] |
| SF3B1 | 10–21% | Intermediate metastatic risk; younger patients [33,34,35,36] |
| EIF1AX | 13–21% | Low metastatic risk; good prognosis [33,34,36,37] |
| Marker | Activity | Targeted Therapy |
|---|---|---|
| Cutaneous Melanoma | ||
| B-raf | Protein kinase along the MAPK pathway; most common mutation | Vemurafenib, dabrafenib, encorafenib [14,38,39,40] |
| N-ras | GTPase with signal transduction along the MAPK and PI3K pathways | Phase II trials of FTIs, lonafarnib and tipifarnib (NCT00060125 and NCT00281957) [15,41,42,43] |
| c-Kit | Growth factor-binding RTK; first signal along the MAPK and PI3K pathways | Phase II trials of imatinib and nilotinib; phase II trial of regorafenib (NCT02501551) [15,44,45,46,47] |
| CDKN2A | Encodes p16 and p14ARF to regulate cell cycle and apoptosis | Phase II trial of CDK inhibitor, flavopiridol (NCT00005971) [48,49,50,51] |
| VDR | Binds active vitamin D to mediate various downstream functions | Phase II trial of high-dose vitamin D (ACTRN12609000351213); phase III trial of vitamin D supplementation (NCT01748448) [52,53,54,55,56] |
| MC1R | Binds MSH and ACTH to regulate melanogenesis and skin pigmentation | None [25] |
| MITF | Regulates melanocyte development, differentiation, and function | None [57,58,59] |
| Melanin | Pigment that scavenges free radicals | None [60,61,62] |
| TYR/TRP1/TRP2 | Proteins related to melanin synthesis | None [63,64] |
| HAPLN1 | ECM component associated with age-related loss | None [65] |
| CTLA4/PD-1/PD-L1 | Downregulates the T cell immune response | Ipilimumab, pembrolizumab, and nivolumab [66,67,68,69] |
| Uveal Melanoma | ||
| GNAQ/GNA11 | G protein alpha subunits involved in the MAPK and PI3K pathways | None [70,71,72] |
| BAP1 | Deubiquitinase involved in cell cycle progression | Phase II trial of PARP inhibitor, niraparib (NCT03207347) [73,74] |
| SF3B1 | Splicing factor subunit | None [34,75] |
| EIF1AX | Eukaryotic translation initiation factor that stabilizes ribosome | None [34,76] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, K.; Oak, A.S.W.; Slominski, R.M.; Brożyna, A.A.; Slominski, A.T. Current Molecular Markers of Melanoma and Treatment Targets. Int. J. Mol. Sci. 2020, 21, 3535. https://doi.org/10.3390/ijms21103535
Yang K, Oak ASW, Slominski RM, Brożyna AA, Slominski AT. Current Molecular Markers of Melanoma and Treatment Targets. International Journal of Molecular Sciences. 2020; 21(10):3535. https://doi.org/10.3390/ijms21103535
Chicago/Turabian StyleYang, Kevin, Allen S.W. Oak, Radomir M. Slominski, Anna A. Brożyna, and Andrzej T. Slominski. 2020. "Current Molecular Markers of Melanoma and Treatment Targets" International Journal of Molecular Sciences 21, no. 10: 3535. https://doi.org/10.3390/ijms21103535
APA StyleYang, K., Oak, A. S. W., Slominski, R. M., Brożyna, A. A., & Slominski, A. T. (2020). Current Molecular Markers of Melanoma and Treatment Targets. International Journal of Molecular Sciences, 21(10), 3535. https://doi.org/10.3390/ijms21103535