SMAD4 and the TGFβ Pathway in Patients with Pancreatic Ductal Adenocarcinoma


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
Search Strategy
3. Results
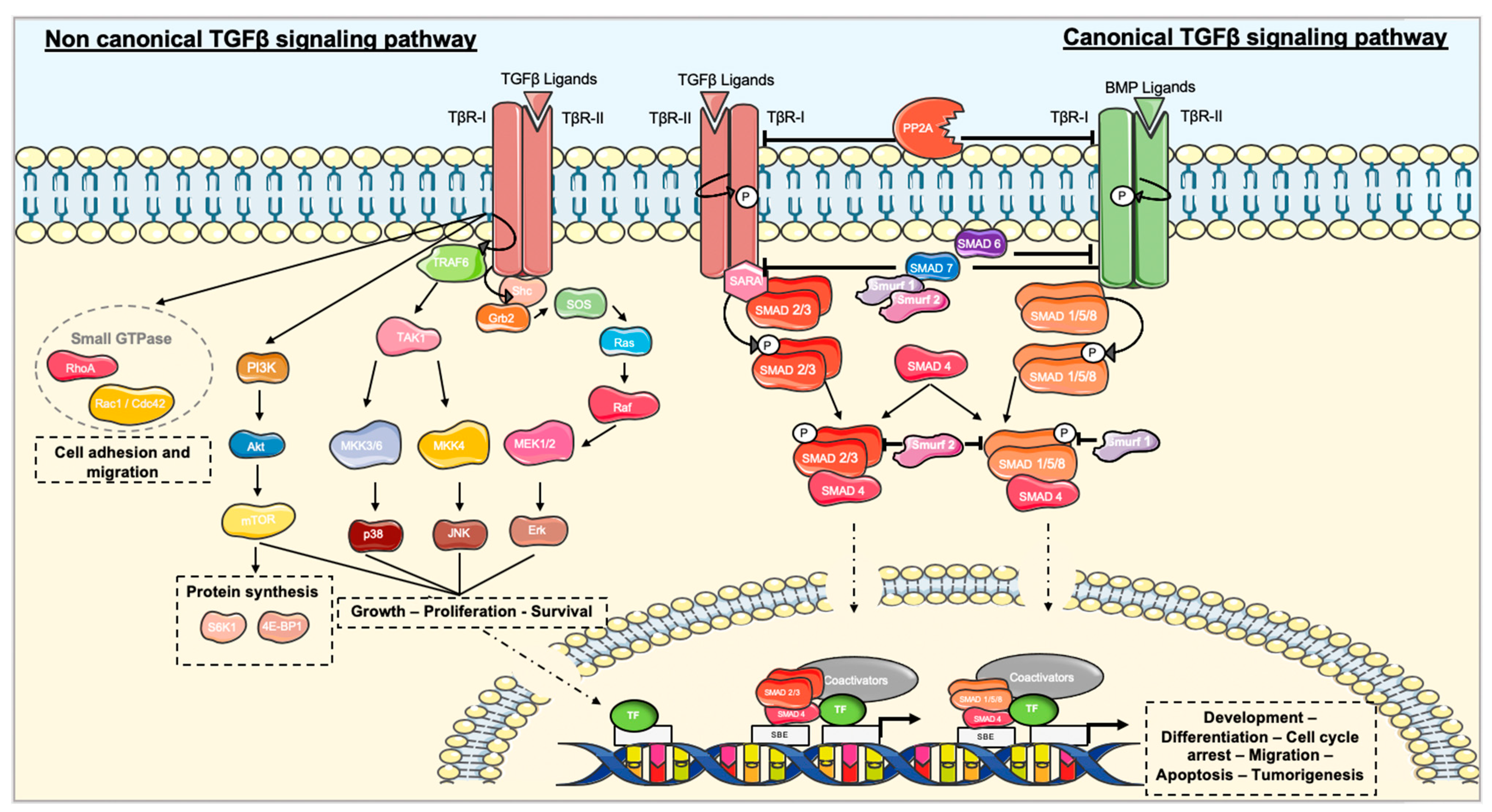
3.1. TGFβ Signaling Pathways
3.1.1. The Canonical TGFβ Signaling Pathway
3.1.2. Non-canonical TGFβ Signaling Pathway
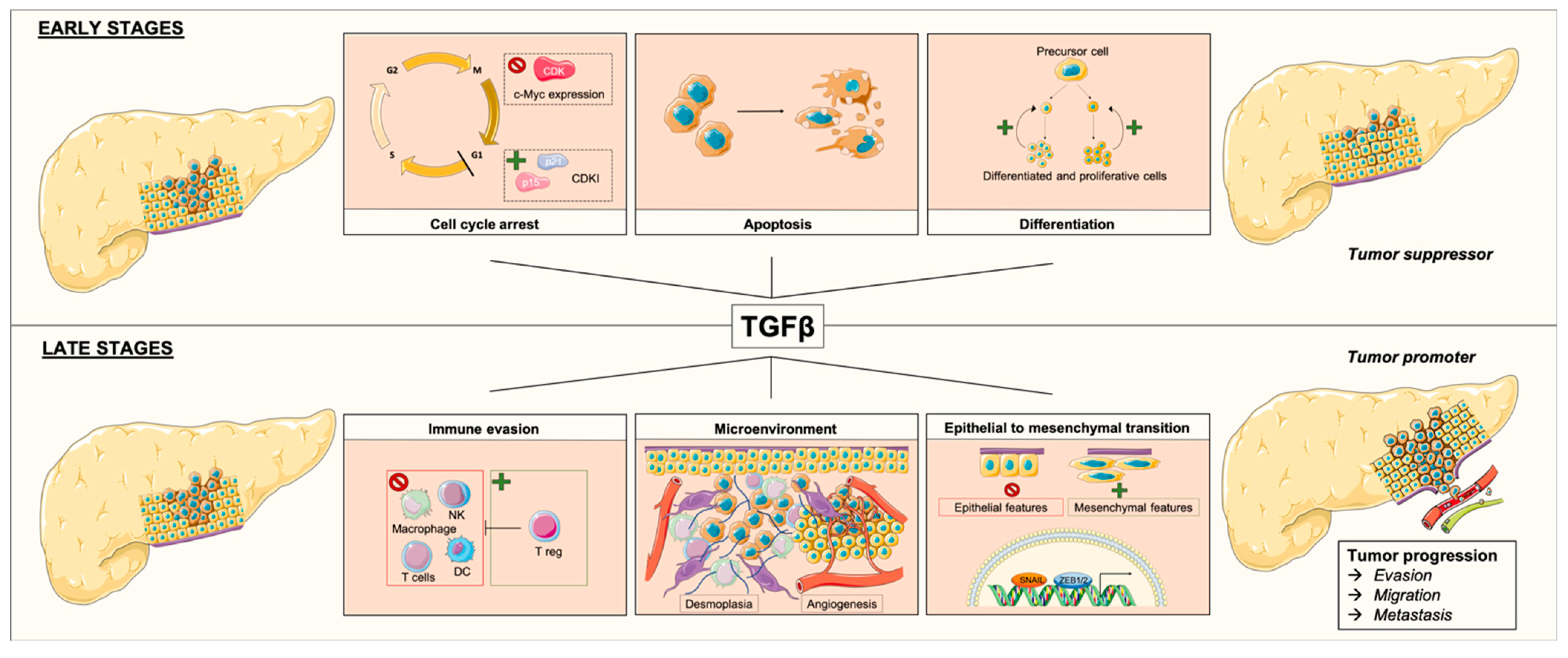
3.2. TGFβ and PDAC
3.2.1. TGFβ as a Tumor Suppressor
3.2.2. TGFβ as a Tumor Promoter
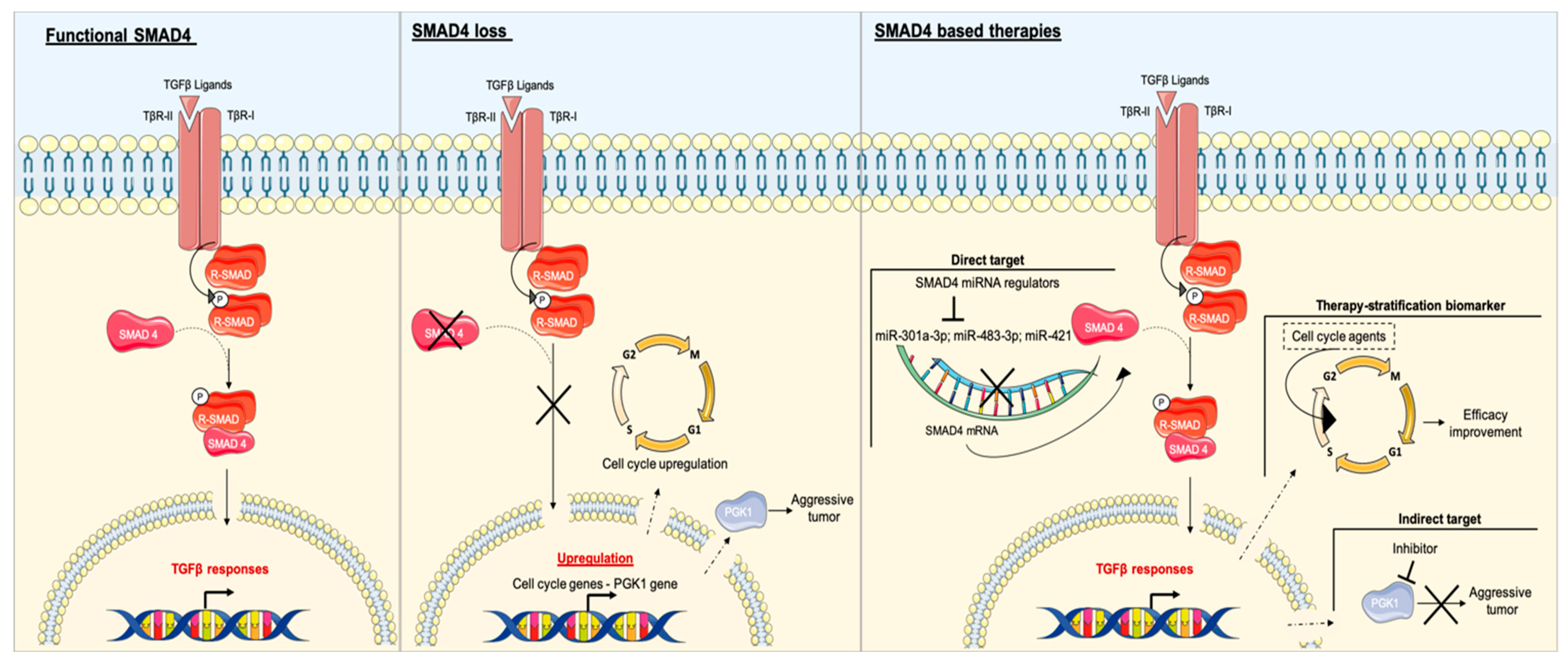
3.3. SMAD4
3.4. SMAD4 and PDAC
3.4.1. SMAD4 and EMT
3.4.2. Effect of SMAD4 Expression in Patients with PDAC
4. Discussion
SMAD4 as a Potential Target in Patients with PDAC and Impact on Other Therapies
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; Vecchia, C.L.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primer 2016, 2, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Lambert, A.; Schwarz, L.; Borbath, I.; Henry, A.; Van Laethem, J.-L.; Malka, D.; Ducreux, M.; Conroy, T. An update on treatment options for pancreatic adenocarcinoma. Ther. Adv. Med. Oncol. 2019, 11, 1758835919875568. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203. [Google Scholar] [CrossRef]
- Waddell, N.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.A.; Schutte, M.; Hoque, A.T.; Moskaluk, C.A.; da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; Hruban, R.H.; et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Souchelnytskyi, S.; Heldin, C.H. Smad regulation in TGF-beta signal transduction. J. Cell Sci. 2001, 114, 4359–4369. [Google Scholar]
- Massagué, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Neil, J.R.; Schiemann, W.P. Transforming growth factor-β and the hallmarks of cancer. Cell. Signal. 2011, 23, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Wendt, M.K.; Tian, M.; Schiemann, W.P. Deconstructing the mechanisms and consequences of TGF-β-induced EMT during cancer progression. Cell Tissue Res. 2012, 347, 85–101. [Google Scholar] [CrossRef]
- Mu, Y.; Gudey, S.K.; Landström, M. Non-Smad signaling pathways. Cell Tissue Res. 2012, 347, 11–20. [Google Scholar] [CrossRef]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Massagué, J.; Gomis, R.R. The logic of TGFbeta signaling. FEBS Lett. 2006, 580, 2811–2820. [Google Scholar] [CrossRef]
- Shi, Y.; Massagué, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Macias, M.J.; Martin-Malpartida, P.; Massagué, J. Structural determinants of Smad function in TGF-β signaling. Trends Biochem. Sci. 2015, 40, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.-H.; Derynck, R. Specificity and versatility in tgf-beta signaling through Smads. Annu. Rev. Cell Dev. Biol. 2005, 21, 659–693. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Alarcón, C.; Sapkota, G.; Rahman, S.; Chen, P.-Y.; Goerner, N.; Macias, M.J.; Erdjument-Bromage, H.; Tempst, P.; Massagué, J. Ubiquitin Ligase Nedd4L Targets Activated Smad2/3 to Limit TGF-β Signaling. Mol. Cell 2009, 36, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. How cells read TGF-beta signals. Nat. Rev. Mol. Cell Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef]
- Lamouille, S.; Derynck, R. Cell size and invasion in TGF-beta-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J. Cell Biol. 2007, 178, 437–451. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.-H. Non-Smad TGF-beta signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef]
- Kang, J.S.; Liu, C.; Derynck, R. New regulatory mechanisms of TGF-beta receptor function. Trends Cell Biol. 2009, 19, 385–394. [Google Scholar] [CrossRef]
- Yamashita, M.; Fatyol, K.; Jin, C.; Wang, X.; Liu, Z.; Zhang, Y.E. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol. Cell 2008, 31, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Galliher, A.J.; Schiemann, W.P. Src Phosphorylates Tyr284 in TGF-β Type II Receptor and Regulates TGF-β Stimulation of p38 MAPK during Breast Cancer Cell Proliferation and Invasion. Cancer Res. 2007, 67, 3752–3758. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Derynck, R. Emergence of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin axis in transforming growth factor-β-induced epithelial-mesenchymal transition. Cells Tissues Organs 2011, 193, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Witte, D.; Lehnert, H. The role of small GTPases of the Rho/Rac family in TGF-β-induced EMT and cell motility in cancer. Dev. Dyn. 2018, 247, 451–461. [Google Scholar] [CrossRef]
- Park, H.; Bang, J.-H.; Nam, A.-R.; Park, J.E.; Jin, M.H.; Bang, Y.-J.; Oh, D.-Y. The prognostic role of soluble TGF-beta and its dynamics in unresectable pancreatic cancer treated with chemotherapy. Cancer Med. 2020, 9, 43–51. [Google Scholar] [CrossRef]
- Javle, M.; Li, Y.; Tan, D.; Dong, X.; Chang, P.; Kar, S.; Li, D. Biomarkers of TGF-β signaling pathway and prognosis of pancreatic cancer. PLoS ONE 2014, 9, e85942. [Google Scholar] [CrossRef]
- Derynck, R.; Akhurst, R.J. Differentiation plasticity regulated by TGF-β family proteins in development and disease. Nat. Cell Biol. 2007, 9, 1000–1004. [Google Scholar] [CrossRef]
- Principe, D.R.; Doll, J.A.; Bauer, J.; Jung, B.; Munshi, H.G.; Bartholin, L.; Pasche, B.; Lee, C.; Grippo, P.J. TGF-β: Duality of function between tumor prevention and carcinogenesis. J. Natl. Cancer Inst. 2014, 106, djt369. [Google Scholar] [CrossRef]
- Friess, H.; Yamanaka, Y.; Büchler, M.; Ebert, M.; Beger, H.G.; Gold, L.I.; Korc, M. Enhanced expression of transforming growth factor β isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology 1993, 105, 1846–1856. [Google Scholar] [CrossRef]
- Wagner, M.; Kleeff, J.; Friess, H.; Büchler, M.W.; Korc, M. Enhanced Expression of the Type II Transforming Growth Factor-β Receptor Is Associated with Decreased Survival in Human Pancreatic Cancer. Pancreas 1999, 19, 370–376. [Google Scholar] [CrossRef]
- Alvarez, M.A.; Freitas, J.P.; Mazher Hussain, S.; Glazer, E.S. TGF-β Inhibitors in Metastatic Pancreatic Ductal Adenocarcinoma. J. Gastrointest. Cancer 2019, 50, 207–213. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Huang, Y.-H.; Chen, M.; Su, J.; Zou, Y.; Bardeesy, N.; Iacobuzio-Donahue, C.A.; Massagué, J. TGF-β Tumor Suppression through a Lethal EMT. Cell 2016, 164, 1015–1030. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.F.B.; Mortensen, M.B.; Detlefsen, S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 2016, 22, 2678–2700. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of Endothelial to Mesenchymal Transition as a Source for Carcinoma-Associated Fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef]
- Zhou, Q.; Xia, S.; Guo, F.; Hu, F.; Wang, Z.; Ni, Y.; Wei, T.; Xiang, H.; Shang, D. Transforming growth factor-β in pancreatic diseases: Mechanisms and therapeutic potential. Pharmacol. Res. 2019, 142, 58–69. [Google Scholar] [CrossRef]
- Achyut, B.R.; Yang, L. Transforming Growth Factor-β in the Gastrointestinal and Hepatic Tumor Microenvironment. Gastroenterology 2011, 141, 1167–1178. [Google Scholar] [CrossRef]
- Seo, Y.D.; Pillarisetty, V.G. T-cell programming in pancreatic adenocarcinoma: A review. Cancer Gene Ther. 2017, 24, 106–113. [Google Scholar] [CrossRef]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Zhao, M.; Mishra, L.; Deng, C.-X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Demagny, H.; Araki, T.; De Robertis, E.M. The Tumor Suppressor Smad4/DPC4 Is Regulated by Phosphorylations that Integrate FGF, Wnt, and TGF-β Signaling. Cell Rep. 2014, 9, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Polireddy, K.; Chen, Q. Cancer of the Pancreas: Molecular Pathways and Current Advancement in Treatment. J. Cancer 2016, 7, 1497–1514. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Sunamura, M.; Horii, A. Molecular mechanisms of pancreatic carcinogenesis. Cancer Sci. 2006, 97, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kubiczkova, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. TGF-β—An excellent servant but a bad master. J. Transl. Med. 2012, 10, 183. [Google Scholar] [CrossRef]
- Xia, X.; Wu, W.; Huang, C.; Cen, G.; Jiang, T.; Cao, J.; Huang, K.; Qiu, Z. SMAD4 and its role in pancreatic cancer. Tumor Biol. 2015, 36, 111–119. [Google Scholar] [CrossRef]
- Demagny, H.; Robertis, E.M.D. Point mutations in the tumor suppressor Smad4/DPC4 enhance its phosphorylation by GSK3 and reversibly inactivate TGF-β signaling. Mol. Cell. Oncol. 2016, 3, e1025181. [Google Scholar] [CrossRef]
- Levy, L.; Hill, C.S. Smad4 dependency defines two classes of transforming growth factor {beta} (TGF-{beta}) target genes and distinguishes TGF-{beta}-induced epithelial-mesenchymal transition from its antiproliferative and migratory responses. Mol. Cell. Biol. 2005, 25, 8108–8125. [Google Scholar] [CrossRef]
- Jazag, A.; Ijichi, H.; Kanai, F.; Imamura, T.; Guleng, B.; Ohta, M.; Imamura, J.; Tanaka, Y.; Tateishi, K.; Ikenoue, T.; et al. Smad4 silencing in pancreatic cancer cell lines using stable RNA interference and gene expression profiles induced by transforming growth factor-β. Oncogene 2005, 24, 662–671. [Google Scholar] [CrossRef][Green Version]
- Ahmed, S.; Bradshaw, A.-D.; Gera, S.; Dewan, M.Z.; Xu, R. The TGF-β/Smad4 Signaling Pathway in Pancreatic Carcinogenesis and Its Clinical Significance. J. Clin. Med. 2017, 6. [Google Scholar] [CrossRef]
- Shichi, Y.; Sasaki, N.; Michishita, M.; Hasegawa, F.; Matsuda, Y.; Arai, T.; Gomi, F.; Aida, J.; Takubo, K.; Toyoda, M.; et al. Enhanced morphological and functional differences of pancreatic cancer with epithelial or mesenchymal characteristics in 3D culture. Sci. Rep. 2019, 9, 10871. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Fujii, T.; Shimoyama, Y.; Kanda, M.; Nakayama, G.; Sugimoto, H.; Koike, M.; Nomoto, S.; Fujiwara, M.; Nakao, A.; et al. Expression Predicts Local Spread and Treatment Failure in Resected Pancreatic Cancer. Pancreas 2015, 44, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-W.; Hsiao, P.-J.; Weng, C.-C.; Kuo, K.-K.; Kuo, T.-L.; Wu, D.-C.; Hung, W.-C.; Cheng, K.-H. SMAD4 Loss triggers the phenotypic changes of pancreatic ductal adenocarcinoma cells. BMC Cancer 2014, 14, 181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cao, J.; Pei, Y.; Zhang, J.; Wang, Q. Smad4 inhibits cell migration via suppression of JNK activity in human pancreatic carcinoma PANC-1 cells. Oncol. Lett. 2016, 11, 3465–3470. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.-H. Mechanisms of TGFβ-Induced Epithelial–Mesenchymal Transition. J. Clin. Med. 2016, 5, 63. [Google Scholar] [CrossRef]
- Shi, S.; Ji, S.; Qin, Y.; Xu, J.; Zhang, B.; Xu, W.; Liu, J.; Long, J.; Liu, C.; Liu, L.; et al. Metabolic tumor burden is associated with major oncogenomic alterations and serum tumor markers in patients with resected pancreatic cancer. Cancer Lett. 2015, 360, 227–233. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Zhan, S.; Zhang, L.; Zhang, S.; Tang, Q.; Li, M.; Tan, Z.; Liu, S.; Xing, X. SMAD4 Y353C promotes the progression of PDAC. BMC Cancer 2019, 19, 1037. [Google Scholar] [CrossRef]
- Blackford, A.; Serrano, O.K.; Wolfgang, C.L.; Parmigiani, G.; Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Eshleman, J.R.; et al. SMAD4 Gene Mutations Are Associated with Poor Prognosis in Pancreatic Cancer. Clin. Cancer Res. 2009, 15, 4674–4679. [Google Scholar] [CrossRef]
- Oshima, M.; Okano, K.; Muraki, S.; Haba, R.; Maeba, T.; Suzuki, Y.; Yachida, S. Immunohistochemically detected expression of 3 major genes (CDKN2A/p16, TP53, and SMAD4/DPC4) strongly predicts survival in patients with resectable pancreatic cancer. Ann. Surg. 2013, 258, 336–346. [Google Scholar] [CrossRef]
- Ormanns, S.; Haas, M.; Remold, A.; Kruger, S.; Holdenrieder, S.; Kirchner, T.; Heinemann, V.; Boeck, S. The Impact of SMAD4 Loss on Outcome in Patients with Advanced Pancreatic Cancer Treated with Systemic Chemotherapy. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Hsieh, Y.-Y.; Liu, T.-P.; Chou, C.-J.; Chen, H.-Y.; Lee, K.-H.; Yang, P.-M. Integration of Bioinformatics Resources Reveals the Therapeutic Benefits of Gemcitabine and Cell Cycle Intervention in SMAD4-Deleted Pancreatic Ductal Adenocarcinoma. Genes 2019, 10, 766. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.M.; Tang, L.H.; Klimstra, D.S.; Liu, W.; Linkov, I.; Brennan, M.F.; DʼAngelica, M.I.; DeMatteo, R.P.; Fong, Y.; Jarnagin, W.R.; et al. Failure patterns in resected pancreas adenocarcinoma: Lack of predicted benefit to SMAD4 expression. Ann. Surg. 2013, 258, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Brosnan, J.A.; Blackford, A.L.; Sur, S.; Hruban, R.H.; Kinzler, K.W.; Vogelstein, B.; Maitra, A.; Diaz, L.A.; Iacobuzio-Donahue, C.A.; et al. Genetically Defined Subsets of Human Pancreatic Cancer Show Unique In Vitro Chemosensitivity. Clin. Cancer Res. 2012, 18, 6519–6530. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Han, H.; Von Hoff, D.D. Identification of an Agent Selectively Targeting DPC4 (Deleted in Pancreatic Cancer Locus 4)–Deficient Pancreatic Cancer Cells. Cancer Res. 2006, 66, 9722–9730. [Google Scholar] [CrossRef]
- Wang, H.; Stephens, B.; Von Hoff, D.D.; Han, H. Identification and Characterization of a Novel Anticancer Agent With Selectivity Against Deleted in Pancreatic Cancer Locus 4 (DPC4)-Deficient Pancreatic and Colon Cancer Cells. Pancreas 2009, 38, 551–557. [Google Scholar] [CrossRef]
- Liang, C.; Qin, Y.; Zhang, B.; Ji, S.; Shi, S.; Xu, W.; Liu, J.; Xiang, J.; Liang, D.; Hu, Q.; et al. Metabolic plasticity in heterogeneous pancreatic ductal adenocarcinoma. Biochim. Biophys. Acta 2016, 1866, 177–188. [Google Scholar] [CrossRef]
- Liang, C.; Shi, S.; Qin, Y.; Meng, Q.; Hua, J.; Hu, Q.; Ji, S.; Zhang, B.; Xu, J.; Yu, X.-J. Localisation of PGK1 determines metabolic phenotype to balance metastasis and proliferation in patients with SMAD4-negative pancreatic cancer. Gut 2020, 69, 888–900. [Google Scholar] [CrossRef]
- Hao, J.; Zhang, S.; Zhou, Y.; Liu, C.; Hu, X.; Shao, C. MicroRNA 421 suppresses DPC4/Smad4 in pancreatic cancer. Biochem. Biophys. Res. Commun. 2011, 406, 552–557. [Google Scholar] [CrossRef]
- Hao, J.; Zhang, S.; Zhou, Y.; Hu, X.; Shao, C. MicroRNA 483-3p suppresses the expression of DPC4/Smad4 in pancreatic cancer. FEBS Lett. 2011, 585, 207–213. [Google Scholar] [CrossRef]
- Xia, X.; Zhang, K.; Cen, G.; Jiang, T.; Cao, J.; Huang, K.; Huang, C.; Zhao, Q.; Qiu, Z. MicroRNA-301a-3p promotes pancreatic cancer progression via negative regulation of SMAD4. Oncotarget 2015, 6. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dardare, J.; Witz, A.; Merlin, J.-L.; Gilson, P.; Harlé, A. SMAD4 and the TGFβ Pathway in Patients with Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 3534. https://doi.org/10.3390/ijms21103534
Dardare J, Witz A, Merlin J-L, Gilson P, Harlé A. SMAD4 and the TGFβ Pathway in Patients with Pancreatic Ductal Adenocarcinoma. International Journal of Molecular Sciences. 2020; 21(10):3534. https://doi.org/10.3390/ijms21103534
Chicago/Turabian StyleDardare, Julie, Andréa Witz, Jean-Louis Merlin, Pauline Gilson, and Alexandre Harlé. 2020. "SMAD4 and the TGFβ Pathway in Patients with Pancreatic Ductal Adenocarcinoma" International Journal of Molecular Sciences 21, no. 10: 3534. https://doi.org/10.3390/ijms21103534
APA StyleDardare, J., Witz, A., Merlin, J.-L., Gilson, P., & Harlé, A. (2020). SMAD4 and the TGFβ Pathway in Patients with Pancreatic Ductal Adenocarcinoma. International Journal of Molecular Sciences, 21(10), 3534. https://doi.org/10.3390/ijms21103534