Dual Deep Sequencing Improves the Accuracy of Low-Frequency Somatic Mutation Detection in Cancer Gene Panel Testing



Abstract
1. Introduction
2. Results
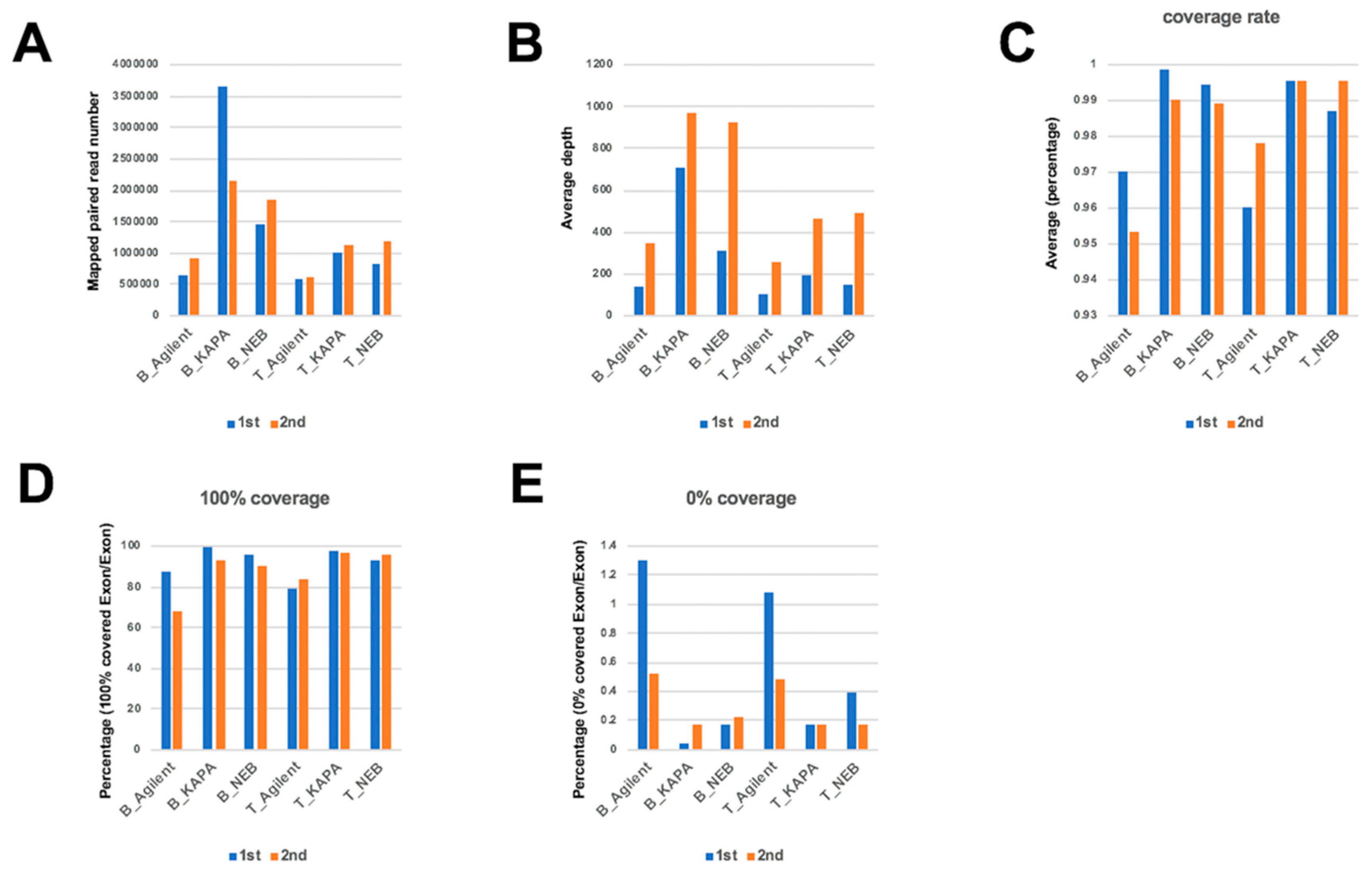
2.1. Comparison of Mapping Status between Dual Deep Sequencing
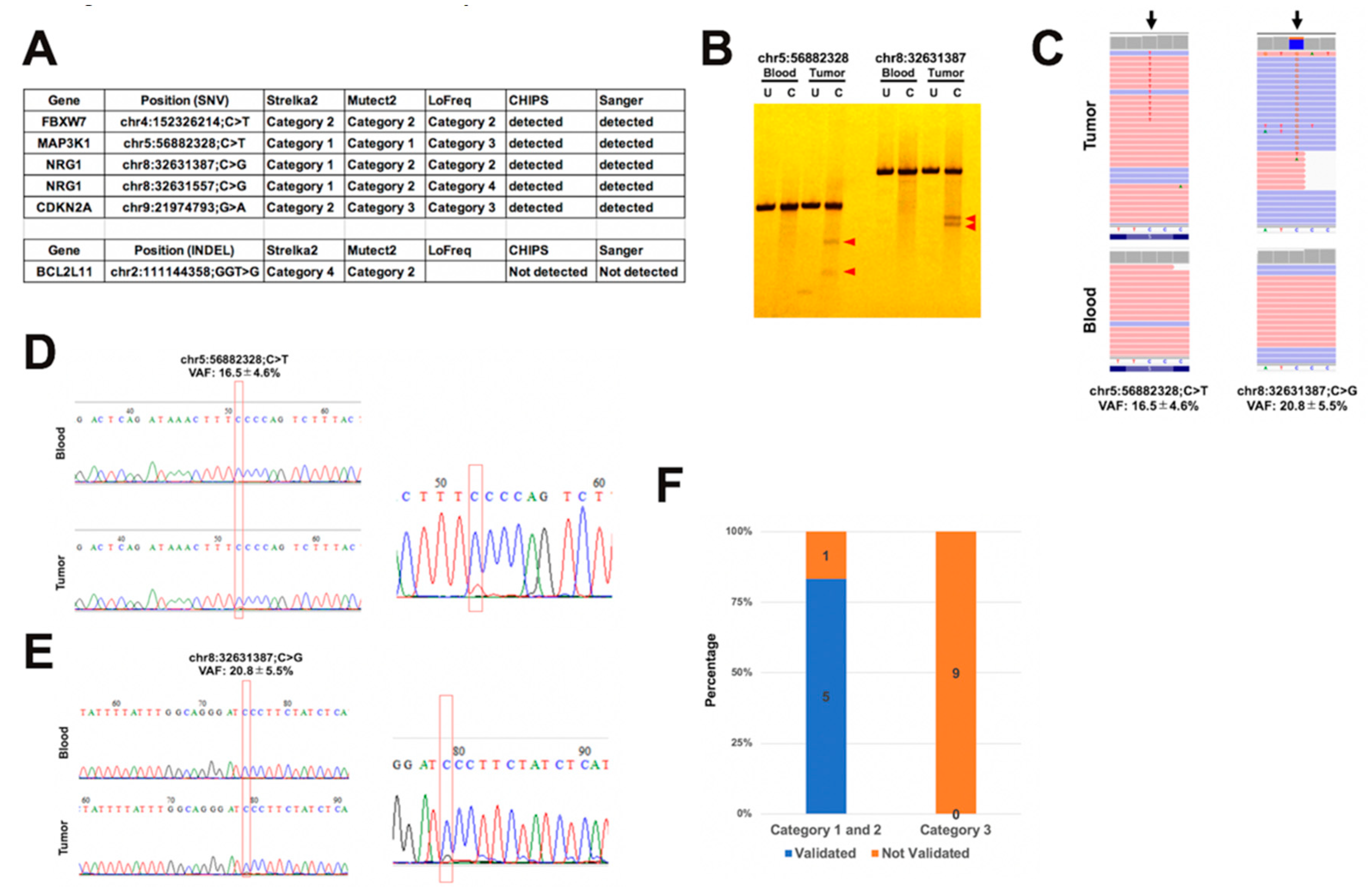
2.2. Comparison of Somatic Variants between Dual Deep Sequencing
2.3. Validation of Detected Variants by CHIPS Technology and Sanger Sequencing
2.4. Comparison of Variant Allele Frequency between Dual Deep Sequencing
2.5. Annotation of Validated Somatic Variants
2.6. Evaluation of Read Number and Different DNA Polymerases
3. Discussion
4. Materials and Methods
4.1. Patient and Sample
4.2. Genomic DNA Extraction
4.3. Library Preparation
4.4. Sequencing and Generation of FASTQ Files
4.5. Data Analysis
4.6. CHIPS and Sanger Sequencing
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gonzaga-Jauregui, C.; Lupski, J.R.; Gibbs, R.A. Human genome sequencing in health and disease. Annu. Rev. Med. 2012, 63, 35–61. [Google Scholar] [CrossRef] [PubMed]
- Gullapalli, R.R.; Desai, K.V.; Santana-Santos, L.; Kant, J.A.; Becich, M.J. Next generation sequencing in clinical medicine: Challenges and lessons for pathology and biomedical informatics. J. Pathol. Inform. 2012, 3, 40. [Google Scholar] [CrossRef] [PubMed]
- Metzker, M.L. Sequencing technologies - the next generation. Nat. Reviews. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Ghazani, A.A.; Oliver, N.M.; St Pierre, J.P.; Garofalo, A.; Rainville, I.R.; Hiller, E.; Treacy, D.J.; Rojas-Rudilla, V.; Wood, S.; Bair, E.; et al. Assigning clinical meaning to somatic and germ-line whole-exome sequencing data in a prospective cancer precision medicine study. Genet. Med. Off. J. Am. Coll. Med Genet. 2017, 19, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Sunami, K.; Ichikawa, H.; Kubo, T.; Kato, M.; Fujiwara, Y.; Shimomura, A.; Koyama, T.; Kakishima, H.; Kitami, M.; Matsushita, H.; et al. Feasibility and utility of a panel testing for 114 cancer-associated genes in a clinical setting: A hospital-based study. Cancer Sci. 2019, 110, 1480–1490. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Real, F.X. Many mosaic mutations. Curr. Oncol. (Tor. Ont.) 2013, 20, 85–87. [Google Scholar] [CrossRef]
- Aird, D.; Ross, M.G.; Chen, W.S.; Danielsson, M.; Fennell, T.; Russ, C.; Jaffe, D.B.; Nusbaum, C.; Gnirke, A. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011, 12, R18. [Google Scholar] [CrossRef]
- Dohm, J.C.; Lottaz, C.; Borodina, T.; Himmelbauer, H. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 2008, 36, e105. [Google Scholar] [CrossRef]
- Gundry, M.; Vijg, J. Direct mutation analysis by high-throughput sequencing: from germline to low-abundant, somatic variants. Mutat. Res. 2012, 729, 1–15. [Google Scholar] [CrossRef]
- Brodin, J.; Mild, M.; Hedskog, C.; Sherwood, E.; Leitner, T.; Andersson, B.; Albert, J. PCR-induced transitions are the major source of error in cleaned ultra-deep pyrosequencing data. PLoS ONE 2013, 8, e70388. [Google Scholar] [CrossRef]
- Nadeu, F.; Clot, G.; Delgado, J.; Martin-Garcia, D.; Baumann, T.; Salaverria, I.; Bea, S.; Pinyol, M.; Jares, P.; Navarro, A.; et al. Clinical impact of the subclonal architecture and mutational complexity in chronic lymphocytic leukemia. Leukemia 2018, 32, 645–653. [Google Scholar] [CrossRef]
- Potapov, V.; Ong, J.L. Examining Sources of Error in PCR by Single-Molecule Sequencing. PLoS ONE 2017, 12, e0169774. [Google Scholar] [CrossRef] [PubMed]
- Salk, J.J.; Schmitt, M.W.; Loeb, L.A. Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations. Nat. Reviews. Genet. 2018, 19, 269–285. [Google Scholar] [CrossRef] [PubMed]
- De Paz, A.M.; Cybulski, T.R.; Marblestone, A.H.; Zamft, B.M.; Church, G.M.; Boyden, E.S.; Kording, K.P.; Tyo, K.E.J. High-resolution mapping of DNA polymerase fidelity using nucleotide imbalances and next-generation sequencing. Nucleic Acids Res. 2018, 46, e78. [Google Scholar] [CrossRef] [PubMed]
- Bohannan, Z.S.; Mitrofanova, A. Calling Variants in the Clinic: Informed Variant Calling Decisions Based on Biological, Clinical, and Laboratory Variables. Comput. Struct. Biotechnol. J. 2019, 17, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Kallberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Wilm, A.; Aw, P.P.; Bertrand, D.; Yeo, G.H.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef]
- Cai, L.; Yuan, W.; Zhang, Z.; He, L.; Chou, K.C. In-depth comparison of somatic point mutation callers based on different tumor next-generation sequencing depth data. Sci. Rep. 2016, 6, 36540. [Google Scholar] [CrossRef]
- Kroigard, A.B.; Thomassen, M.; Laenkholm, A.V.; Kruse, T.A.; Larsen, M.J. Evaluation of Nine Somatic Variant Callers for Detection of Somatic Mutations in Exome and Targeted Deep Sequencing Data. PLoS ONE 2016, 11, e0151664. [Google Scholar] [CrossRef]
- Alioto, T.S.; Buchhalter, I.; Derdak, S.; Hutter, B.; Eldridge, M.D.; Hovig, E.; Heisler, L.E.; Beck, T.A.; Simpson, J.T.; Tonon, L.; et al. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nat. Commun. 2015, 6, 10001. [Google Scholar] [CrossRef] [PubMed]
- Sandmann, S.; de Graaf, A.O.; Karimi, M.; van der Reijden, B.A.; Hellstrom-Lindberg, E.; Jansen, J.H.; Dugas, M. Evaluating Variant Calling Tools for Non-Matched Next-Generation Sequencing Data. Sci. Rep. 2017, 7, 43169. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.H.; Kim, I.J.; Wu, D.; Climent, J.; Kang, H.C.; DelRosario, R.; Balmain, A. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Sci. (New Yorkn.Y.) 2008, 321, 1499–1502. [Google Scholar] [CrossRef] [PubMed]
- Beije, N.; Helmijr, J.C.; Weerts, M.J.A.; Beaufort, C.M.; Wiggin, M.; Marziali, A.; Verhoef, C.; Sleijfer, S.; Jansen, M.; Martens, J.W.M. Somatic mutation detection using various targeted detection assays in paired samples of circulating tumor DNA, primary tumor and metastases from patients undergoing resection of colorectal liver metastases. Mol. Oncol. 2016, 10, 1575–1584. [Google Scholar] [CrossRef]
- Teer, J.K.; Zhang, Y.; Chen, L.; Welsh, E.A.; Cress, W.D.; Eschrich, S.A.; Berglund, A.E. Evaluating somatic tumor mutation detection without matched normal samples. Hum. Genom. 2017, 11, 22. [Google Scholar] [CrossRef]
- Wang, Q.; Jia, P.; Li, F.; Chen, H.; Ji, H.; Hucks, D.; Dahlman, K.B.; Pao, W.; Zhao, Z. Detecting somatic point mutations in cancer genome sequencing data: a comparison of mutation callers. Genome Med. 2013, 5, 91. [Google Scholar] [CrossRef]
- Filges, S.; Yamada, E.; Stahlberg, A.; Godfrey, T.E. Impact of Polymerase Fidelity on Background Error Rates in Next-Generation Sequencing with Unique Molecular Identifiers/Barcodes. Sci. Rep. 2019, 9, 3503. [Google Scholar] [CrossRef]
- Zhu, X.J.; Sun, S.; Xie, B.; Hu, X.; Zhang, Z.; Qiu, M.; Dai, Z.M. Guanine-rich sequences inhibit proofreading DNA polymerases. Sci. Rep. 2016, 6, 28769. [Google Scholar] [CrossRef]
- Spencer, D.H.; Tyagi, M.; Vallania, F.; Bredemeyer, A.J.; Pfeifer, J.D.; Mitra, R.D.; Duncavage, E.J. Performance of common analysis methods for detecting low-frequency single nucleotide variants in targeted next-generation sequence data. J. Mol. Diagn. Jmd. 2014, 16, 75–88. [Google Scholar] [CrossRef]
- McInerney, P.; Adams, P.; Hadi, M.Z. Error Rate Comparison during Polymerase Chain Reaction by DNA Polymerase. Mol. Biol. Int. 2014, 2014, 287430. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinform. (Oxf. Engl.) 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- El-Ghazaly, M.A.; Fadel, N.A.; Abdel-Naby, D.H.; Abd El-Rehim, H.A.; Zaki, H.F.; Kenawy, S.A. Potential anti-inflammatory action of resveratrol and piperine in adjuvant-induced arthritis: Effect on pro-inflammatory cytokines and oxidative stress biomarkers. Egypt. Rheumatol. 2019. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinform. (Oxf. Engl.) 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, V.; Danecek, P.; Scally, A.; Xue, Y.; Tyler-Smith, C.; Durbin, R. BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinform. (Oxf. Engl.) 2016, 32, 1749–1751. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinform. (Oxf. Engl.) 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–d947. [Google Scholar] [CrossRef]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef]
- Liu, F.-C.; Tsai, Y.-F.; Tsai, H.-I.; Yu, H.-P. Anti-Inflammatory and Organ-Protective Effects of Resveratrol in Trauma-Hemorrhagic Injury. Mediat. Inflamm. 2015, 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Niida, Y.; Kuroda, M.; Mitani, Y.; Okumura, A.; Yokoi, A. Applying and testing the conveniently optimized enzyme mismatch cleavage method to clinical DNA diagnosis. Mol. Genet. Metab. 2012, 107, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Niida, Y.; Ozaki, M.; Inoue, M.; Takase, E.; Kuroda, M.; Mitani, Y.; Okumura, A.; Yokoi, A.; Fujita, S.; Yamada, K. CHIPS for genetic testing to improve a regional clinical genetic service. Clin. Genet. 2015, 88, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Niida, Y. Development of a simple and highly sensitive mutation screening system by enzyme mismatch cleavage with optimized conditions for standard laboratories. Electrophoresis 2008, 29, 1473–1483. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Position (hg38) | DNA | Mutation Type | Protein | dbSNP | ClinVar | COSMIC |
|---|---|---|---|---|---|---|---|
| FBXW7 | chr4:152326214 | NM_033632.3:c.1436G > A | missense | NP_361014.1:p.Arg479Gln | rs866987936 | 376419 | COSM1154291; COSM22974; COSM447498; COSM447499; COSM6847976: COSM94297 |
| MAP3K1 | chr5:56882328 | NM005921.1:c.3138C > T | missense | NP_005912.1:p.Ser1043Phe | - | - | COSM6889390 |
| NRG1 | chr8:32631387 | NM_13960.4:c.502+14502C > G | intron variant | - | - | - | - |
| NRG1 | chr8:32631557 | NM_13960.4:c.502+14672C > G | intron variant | - | - | - | - |
| CDKN2A | chr9:21974793 | NM_058197.4:c.35C > T | missense | NP_478104.2:p.Ser12Leu | rs141798398 | 236988 | COSM6985693; COSM6985694; COSM6985695 |
| Gene | Position | Mutation type | Protein | SIFT | Polyphen2_HDIV | Polyphen2_HVAR | LRT | Mutation Taster | MutationAssessor | FATHMM | PROVEAN | MetaSVM | MetaLR | fathmm-MKL_coding |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FBXW7 | chr4:152326214;C > T | missense | p.Arg479Gln | Damaging | Probably damaging | Probably damaging | Deleterious | Disease causing | Low | Tolerated | Deleterious | Tolerated | Tolerated | Deleterious |
| MAP3K1 | chr5:56882328;C > T | missense | p.Ser1043Phe | Damaging | Probably damaging | Probably damaging | Deleterious | Disease causing | Medium | Tolerated | Neutral | Deleterious | Deleterious | Deleterious |
| CDKN2A | chr9:21974793;G > A | missense | p.Ser12Leu | Tolerated | Benign | Benign | . | Polymorphism | Neutral | Tolerated | Neutral | Tolerated | Tolerated | Neutral |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ura, H.; Togi, S.; Niida, Y. Dual Deep Sequencing Improves the Accuracy of Low-Frequency Somatic Mutation Detection in Cancer Gene Panel Testing. Int. J. Mol. Sci. 2020, 21, 3530. https://doi.org/10.3390/ijms21103530
Ura H, Togi S, Niida Y. Dual Deep Sequencing Improves the Accuracy of Low-Frequency Somatic Mutation Detection in Cancer Gene Panel Testing. International Journal of Molecular Sciences. 2020; 21(10):3530. https://doi.org/10.3390/ijms21103530
Chicago/Turabian StyleUra, Hiroki, Sumihito Togi, and Yo Niida. 2020. "Dual Deep Sequencing Improves the Accuracy of Low-Frequency Somatic Mutation Detection in Cancer Gene Panel Testing" International Journal of Molecular Sciences 21, no. 10: 3530. https://doi.org/10.3390/ijms21103530
APA StyleUra, H., Togi, S., & Niida, Y. (2020). Dual Deep Sequencing Improves the Accuracy of Low-Frequency Somatic Mutation Detection in Cancer Gene Panel Testing. International Journal of Molecular Sciences, 21(10), 3530. https://doi.org/10.3390/ijms21103530