Enhanced Clearance of Neurotoxic Misfolded Proteins by the Natural Compound Berberine and Its Derivatives

 ,
,  ,
, 

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
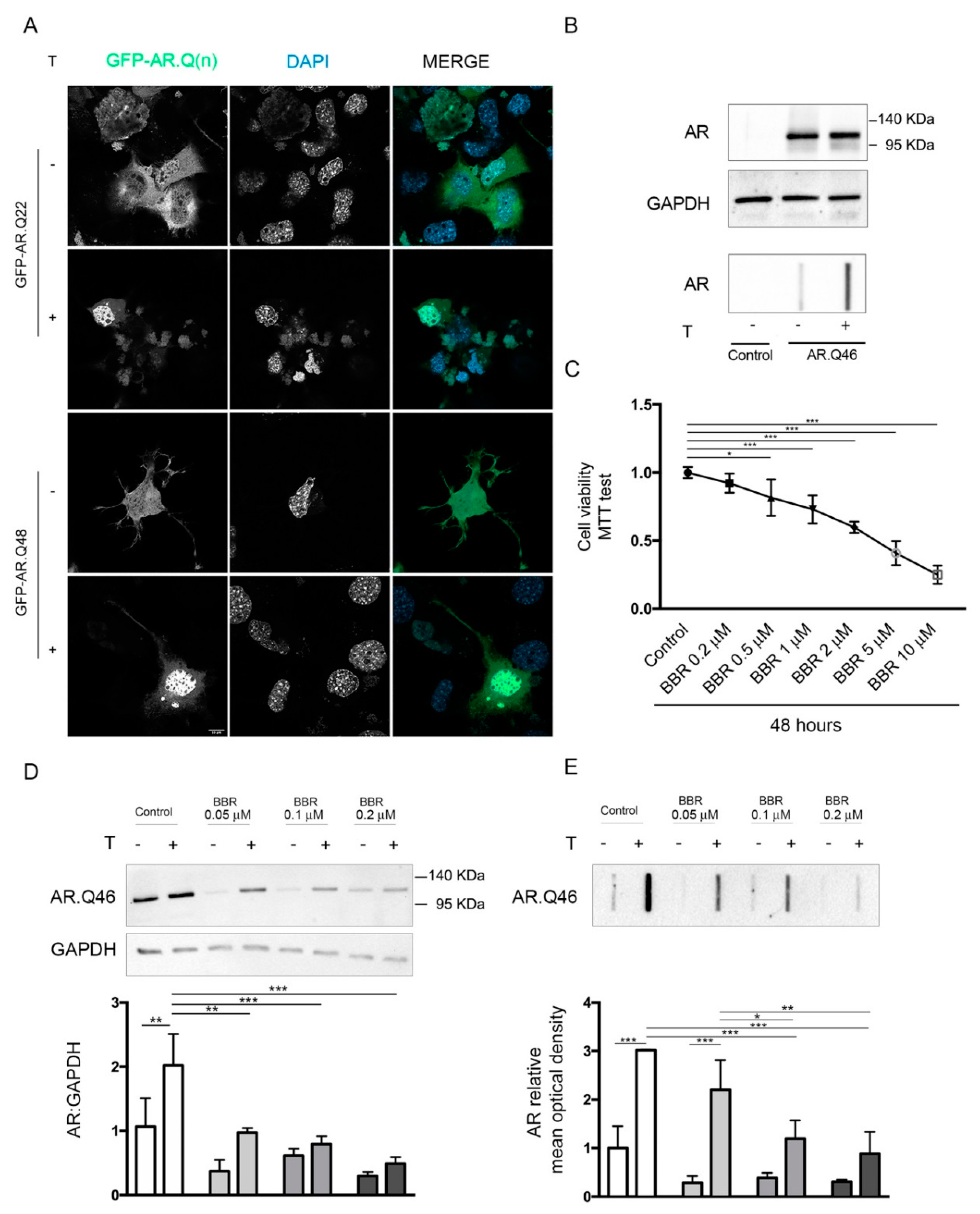
2.1. Effects of BBR on ARpolyQ Clearance
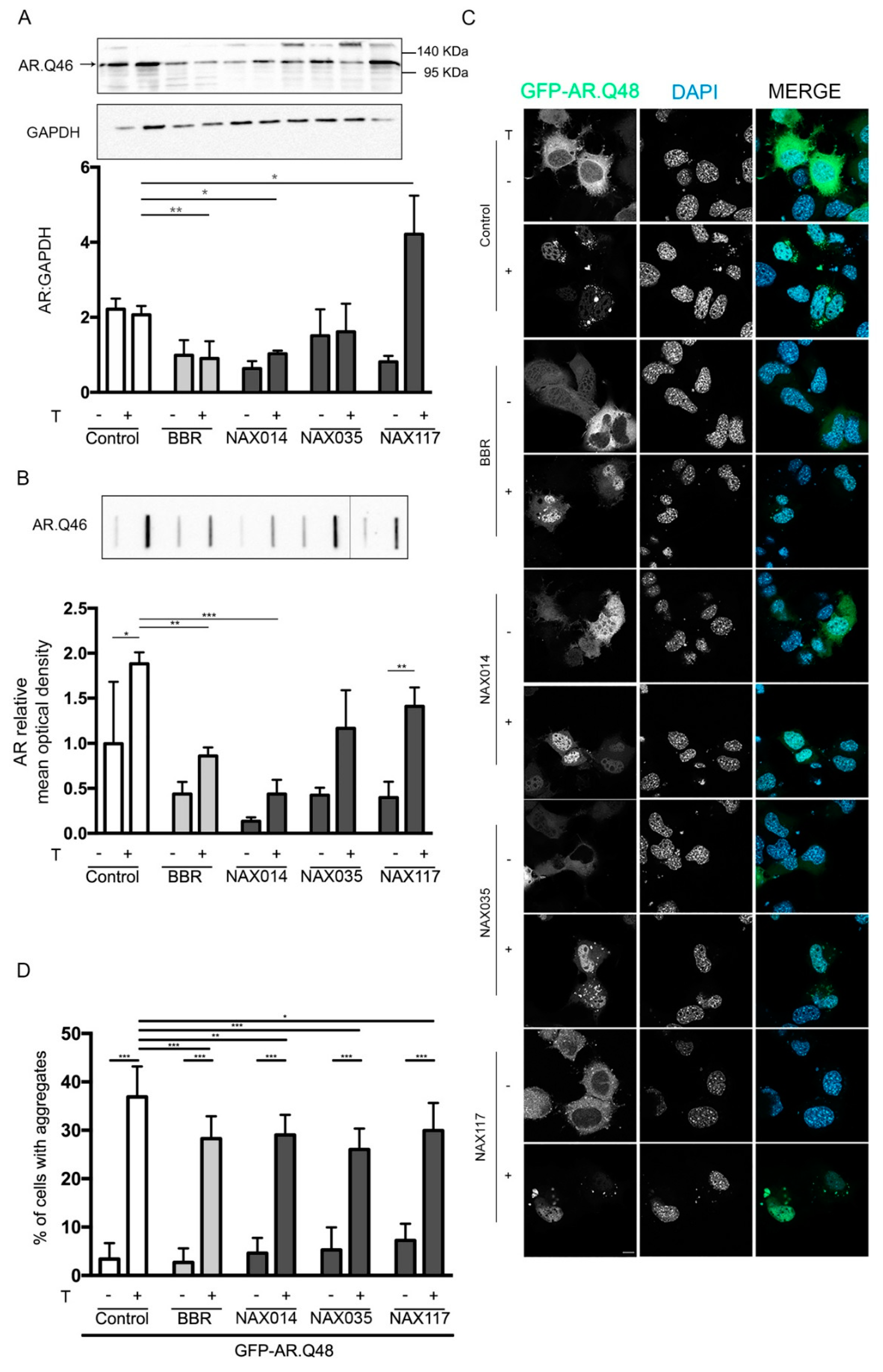
2.2. Effects of BBR-Derived Compounds on ARpolyQ Clearance
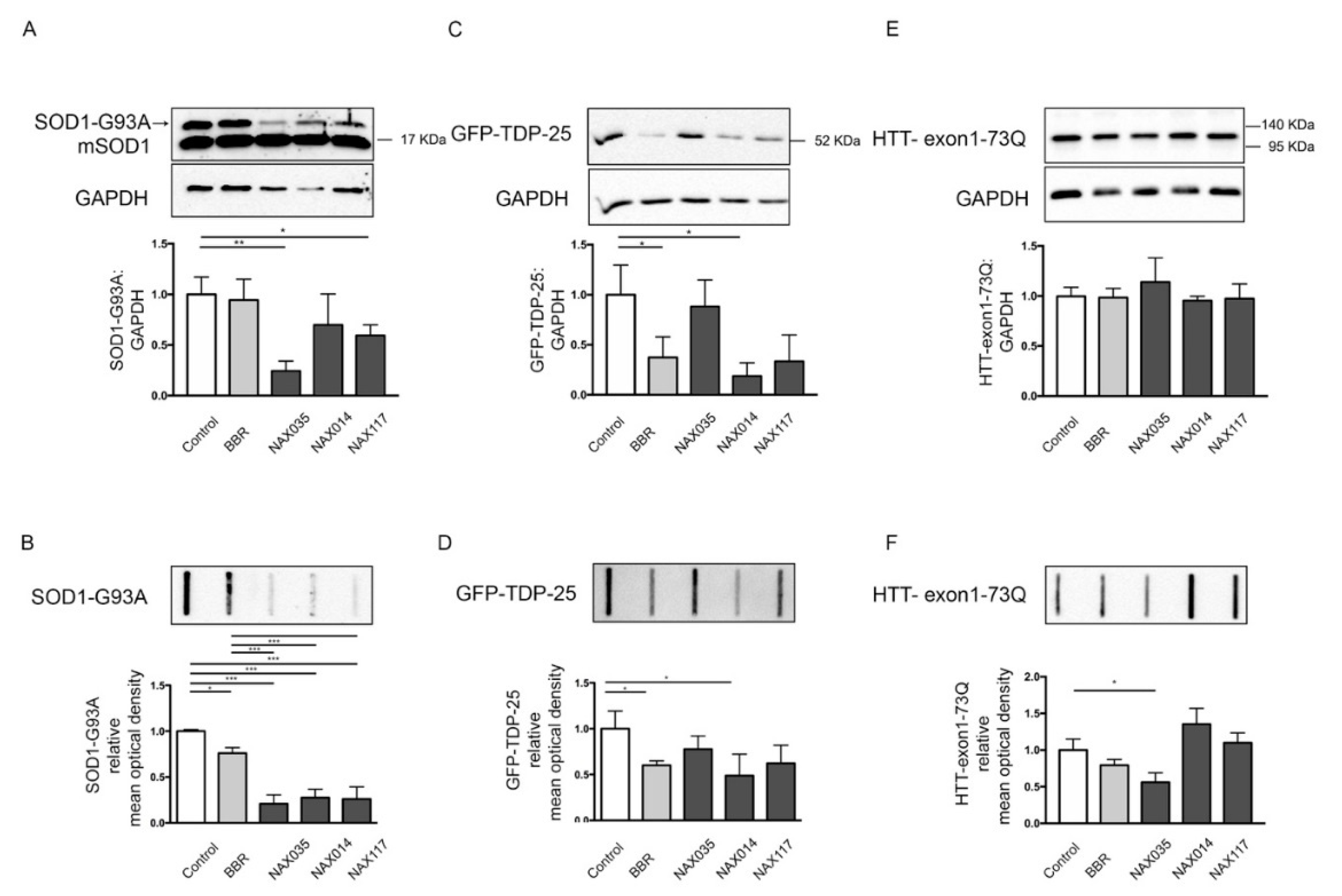
2.3. Effects of BBR and BBR-Derived Compounds on Misfolded Proteins Involved in ALS and HD
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Plasmids
4.3. Cell Cultures and Transfections
4.4. MTT Assay
4.5. Western Blot and Filter Retardation Assay
4.6. Fluorescence Microscopy Analysis
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Skowyra, M.L.; Schlesinger, P.H.; Naismith, T.V.; Hanson, P.I. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 2018, 360, eaar5078. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Retzlaff, M.; Roos, T.; Frydman, J. Cellular Strategies of Protein Quality Control. Cold Spring Harb. Perspect. Biol. 2011, 3, a004374. [Google Scholar] [CrossRef] [PubMed]
- Korolchuk, V.I.; Menzies, F.M.; Rubinsztein, D.C. A novel link between autophagy and the ubiquitin-proteasome system. Autophagy 2009, 5, 862–863. [Google Scholar] [CrossRef] [PubMed]
- Saelices, L.; Johnson, L.M.; Liang, W.Y.; Sawaya, M.R.; Cascio, D.; Ruchala, P.; Whitelegge, J.; Jiang, L.; Riek, R.; Eisenberg, D.S. Uncovering the Mechanism of Aggregation of Human Transthyretin. J. Biol. Chem. 2015, 290, 28932–28943. [Google Scholar] [CrossRef]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the Ubiquitin-Proteasome System by Protein Aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef]
- Huang, Y.; Mucke, L. Alzheimer Mechanisms and Therapeutic Strategies. Cell 2012, 148, 1204–1222. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef]
- Cox, D.; Raeburn, C.; Sui, X.; Hatters, D.M. Protein aggregation in cell biology: An aggregomics perspective of health and disease. Semin. Cell Dev. Biol. 2020, 99, 40–54. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Bergamini, E.; Brunk, U.T.; Dröge, W.; Ffrench, M.; Terman, A. Autophagy and aging: The importance of maintaining “clean” cells. Autophagy 2005, 1, 131–140. [Google Scholar] [CrossRef]
- Lee, S.; Sato, Y.; Nixon, R.A. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J. Neurosci. 2011, 31, 7817–7830. [Google Scholar] [CrossRef]
- Fan, H.-C.; Ho, L.-I.; Chi, C.-S.; Chen, S.-J.; Peng, G.-S.; Chan, T.-M.; Lin, S.-Z.; Harn, H.-J. Polyglutamine (PolyQ) Diseases: Genetics to Treatments. Cell Transplant. 2014, 23, 441–458. [Google Scholar] [CrossRef] [PubMed]
- Adegbuyiro, A.; Sedighi, F.; Pilkington, A.W.; Groover, S.; Legleiter, J. Proteins Containing Expanded Polyglutamine Tracts and Neurodegenerative Disease. Biochemistry 2017, 56, 1199–1217. [Google Scholar] [CrossRef] [PubMed]
- Fischbeck, K.H.; Lieberman, A.; Bailey, C.K.; Abel, A.; Merry, D.E. Androgen receptor mutation in Kennedy’s disease. Philos. Trans. R. Soc. B Biol. Sci. 1999, 354, 1075–1078. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kennedy, W.R.; Alter, M.; Sung, J.H. Progressive proximal spinal and bulbar muscular atrophy of late onset: A sex-linked recessive trait. Neurology 1968, 18, 671. [Google Scholar] [CrossRef]
- Querin, G.; Bede, P.; Marchand-Pauvert, V.; Pradat, P.-F. Biomarkers of Spinal and Bulbar Muscle Atrophy (SBMA): A Comprehensive Review. Front. Neurol. 2018, 9, 844. [Google Scholar] [CrossRef]
- Tanaka, F.; Doyu, M.; Ito, Y.; Matsumoto, M.; Mitsuma, T.; Abe, K.; Aoki, M.; Itoyama, Y.; Fischbeck, K.H.; Sobue, G. Founder effect in spinal and bulbar muscular atrophy (SBMA). Hum. Mol. Genet. 1996, 5, 1253–1257. [Google Scholar] [CrossRef]
- Grunseich, C.; Rinaldi, C.; Fischbeck, K.H. Spinal and bulbar muscular atrophy: Pathogenesis and clinical management. Oral Dis. 2013, 20, 6–9. [Google Scholar] [CrossRef]
- Dejager, S.; Bry-Gauillard, H.; Bruckert, E.; Eymard, B.; Salachas, F.; LeGuern, E.; Tardieu, S.; Chadarevian, R.; Giral, P.; Turpin, G. A Comprehensive Endocrine Description of Kennedy’s Disease Revealing Androgen Insensitivity Linked to CAG Repeat Length. J. Clin. Endocrinol. Metab. 2002, 87, 3893–3901. [Google Scholar] [CrossRef][Green Version]
- Vegeto, E.; Villa, A.; Della Torre, S.; Crippa, V.; Rusmini, P.; Cristofani, R.; Galbiati, M.; Maggi, A.; Poletti, A. The Role of Sex and Sex Hormones in Neurodegenerative Diseases. Endocr. Rev. 2019, 41, 273–319. [Google Scholar] [CrossRef]
- Davey, R.A.; Grossmann, M. Androgen Receptor Structure, Function and Biology: From Bench to Bedside. Clin. Biochem. Rev. 2016, 37, 3–15. [Google Scholar]
- Mhaouty-Kodja, S. Role of the androgen receptor in the central nervous system. Mol. Cell. Endocrinol. 2018, 465, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Dubois, V.; Laurent, M.R.; Boonen, S.; Vanderschueren, D.; Claessens, F. Androgens and skeletal muscle: Cellular and molecular action mechanisms underlying the anabolic actions. Cell. Mol. Life Sci. 2011, 69, 1651–1667. [Google Scholar] [CrossRef] [PubMed]
- Manzano, R.; Sorarù, G.; Grunseich, C.; Fratta, P.; Zuccaro, E.; Pennuto, M.; Rinaldi, C. Beyond motor neurons: Expanding the clinical spectrum in Kennedy’s disease. J. Neurol. Neurosurg. Psychiatry 2018, 89, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Poletti, A. The polyglutamine tract of androgen receptor: From functions to dysfunctions in motor neurons. Front. Neuroendocr. 2004, 25, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Piccioni, F.; Simeoni, S.; Andriola, I.; Armatura, E.; Bassanini, S.; Pozzi, P.; Poletti, A. Polyglutamine tract expansion of the androgen receptor in a motoneuronal model of spinal and bulbar muscular atrophy. Brain Res. Bull. 2001, 56, 215–220. [Google Scholar] [CrossRef]
- Simeoni, S.; Mancini, M.; Stenoien, D.L.; Marcelli, M.; Weigel, N.L.; Zanisi, M.; Martini, L.; Poletti, A. Motoneuronal cell death is not correlated with aggregate formation of androgen receptors containing an elongated polyglutamine tract. Hum. Mol. Genet. 2000, 9, 133–144. [Google Scholar] [CrossRef]
- Rusmini, P.; Sau, D.; Crippa, V.; Palazzolo, I.; Simonini, F.; Onesto, E.; Martini, L.; Poletti, A. Aggregation and proteasome: The case of elongated polyglutamine aggregation in spinal and bulbar muscular atrophy. Neurobiol. Aging 2007, 28, 1099–1111. [Google Scholar] [CrossRef]
- Palazzolo, I.; Gliozzi, A.; Rusmini, P.; Sau, D.; Crippa, V.; Simonini, F.; Onesto, E.; Bolzoni, E.; Poletti, A. The role of the polyglutamine tract in androgen receptor. J. Steroid Biochem. Mol. Biol. 2008, 108, 245–253. [Google Scholar] [CrossRef]
- Podvin, S.; Reardon, H.; Yin, K.; Mosier, C.; Hook, V. Multiple clinical features of Huntington’s disease correlate with mutant HTT gene CAG repeat lengths and neurodegeneration. J. Neurol. 2018, 266, 551–564. [Google Scholar] [CrossRef]
- Reiner, A.; Dragatsis, I.; Zeitlin, S.O.; Goldowitz, D. Wild-Type Huntingtin Plays a Role in Brain Development and Neuronal Survival. Mol. Neurobiol. 2003, 28, 259–276. [Google Scholar] [CrossRef]
- Rui, Y.-N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, N.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262–275. [Google Scholar] [CrossRef] [PubMed]
- White, J.; Anderson, E.; Zimmerman, K.; Zheng, K.H.; Rouhani, R.; Gunawardena, S. Huntingtin differentially regulates the axonal transport of a sub-set of Rab-containing vesicles in vivo. Hum. Mol. Genet. 2015, 24, 7182–7195. [Google Scholar] [CrossRef] [PubMed]
- Schulte, J.; Littleton, J.T. Relevance to Huntington’ s Disease Pathology. Curr. Trends Neurol. 2011, 5, 65–78. [Google Scholar] [PubMed]
- Rusmini, P.; Cortese, K.; Crippa, V.; Cristofani, R.; Cicardi, M.E.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Meroni, M.; Messi, E.; et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy 2018, 15, 631–651. [Google Scholar] [CrossRef] [PubMed]
- Cristofani, R.; Crippa, V.; Rusmini, P.; Cicardi, M.E.; Meroni, M.; Licata, N.V.; Sala, G.; Giorgetti, E.; Grunseich, C.; Galbiati, M.; et al. Inhibition of retrograde transport modulates misfolded protein accumulation and clearance in motoneuron diseases. Autophagy 2017, 13, 1280–1303. [Google Scholar] [CrossRef]
- Klickovic, U.; Zampedri, L.; Sinclair, C.D.; Wastling, S.J.; Trimmel, K.; Howard, R.S.; Malaspina, A.; Sharma, N.; Sidle, K.; Emira, A.; et al. Skeletal muscle MRI differentiates SBMA and ALS and correlates with disease severity. Neurology 2019, 93, e895–e907. [Google Scholar] [CrossRef]
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.-M.; Le Masson, G. Masson Genetics of amyotrophic lateral sclerosis: A review. J. Neurol. Sci. 2019, 399, 217–226. [Google Scholar] [CrossRef]
- Sabatelli, M.; Conte, A.; Zollino, M. Clinical and genetic heterogeneity of amyotrophic lateral sclerosis. Clin. Genet. 2013, 83, 408–416. [Google Scholar] [CrossRef]
- Pansarasa, O.; Bordoni, M.; Diamanti, L.; Sproviero, D.; Gagliardi, S.; Cereda, C. SOD1 in Amyotrophic Lateral Sclerosis: “Ambivalent” Behavior Connected to the Disease. Int. J. Mol. Sci. 2018, 19, 1345. [Google Scholar] [CrossRef]
- Berning, B.; Walker, A.K. The Pathobiology of TDP-43 C-Terminal Fragments in ALS and FTLD. Front. Mol. Neurosci. 2019, 13, 335. [Google Scholar] [CrossRef]
- Winklhofer, K.F.; Tatzelt, J.; Haass, C. The two faces of protein misfolding: Gain- and loss-of-function in neurodegenerative diseases. EMBO J. 2008, 27, 336–349. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Matsuda, N. Proteostasis and neurodegeneration: The roles of proteasomal degradation and autophagy. Biochim. Biophys. Acta (BBA) Bioenerg. 2014, 1843, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Giorgetti, E.; Rusmini, P.; Crippa, V.; Cristofani, R.; Boncoraglio, A.; Cicardi, M.E.; Galbiati, M.; Poletti, A. Synergic prodegradative activity of Bicalutamide and trehalose on the mutant androgen receptor responsible for spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2014, 24, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Ting, H.-C.; Chang, C.-Y.; Lu, K.Y.; Chuang, H.-M.; Tsai, S.-F.; Huang, M.-H.; Liu, C.-A.; Lin, S.-Z.; Harn, H.-J. Targeting Cellular Stress Mechanisms and Metabolic Homeostasis by Chinese Herbal Drugs for Neuroprotection. Molecules 2018, 23, 259. [Google Scholar] [CrossRef]
- Rad, S.Z.K.; Rameshrad, M.; Hosseinzadeh, H. Toxicology effects of Berberis vulgaris (barberry) and its active constituent, berberine: A review. Iran. J. Basic Med. Sci. 2017, 20, 516–529. [Google Scholar]
- Ortiz, L.M.G.; Lombardi, P.; Tillhon, M.; Scovassi, A.I. Berberine, an Epiphany Against Cancer. Molecules 2014, 19, 12349–12367. [Google Scholar] [CrossRef]
- Zou, K.; Li, Z.; Zhang, Y.; Zhang, H.-Y.; Li, B.; Zhu, W.-L.; Shi, J.; Jia, Q.; Li, Y.-M. Advances in the study of berberine and its derivatives: A focus on anti-inflammatory and anti-tumor effects in the digestive system. Acta Pharmacol. Sin. 2016, 38, 157–167. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.; Zou, D.; Liu, W.; Yang, J.; Zhu, N.; Huo, L.; Wang, M.; Hong, J.; Wu, P.; et al. Treatment of Type 2 Diabetes and Dyslipidemia with the Natural Plant Alkaloid Berberine. J. Clin. Endocrinol. Metab. 2008, 93, 2559–2565. [Google Scholar] [CrossRef]
- Qing, Y.; Dong, X.; Hongli, L.; Yanhui, L. Berberine promoted myocardial protection of postoperative patients through regulating myocardial autophagy. Biomed. Pharmacother. 2018, 105, 1050–1053. [Google Scholar] [CrossRef]
- Cazzaniga, M.; Bonanni, B. Relationship Between Metabolic Disorders and Breast Cancer Incidence and Outcomes. Is There a Preventive and Therapeutic Role for Berberine? Anticancer. Res. 2018, 38, 4393–4402. [Google Scholar] [CrossRef]
- Imenshahidi, M.; Hosseinzadeh, H. Berberis Vulgarisand Berberine: An Update Review. Phytotherapy Res. 2016, 30, 1745–1764. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Wang, C.; He, W.; Chen, Y. Berberine Alleviates Amyloid-Beta Pathology in the Brain of APP/PS1 Transgenic Mice via Inhibiting β/γ-Secretases Activity and Enhancing α-Secretases. Curr. Alzheimer Res. 2018, 15, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhao, C.; Cao, G.; Guo, L.; Zhang, S.; Liang, Y.; Qin, C.; Su, P.; Li, H.; Zhang, W. Berberine modulates amyloid-β peptide generation by activating AMP-activated protein kinase. Neuropharmacology 2017, 125, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-F.; Lee, Y.-C.; Lee, K.-H.; Lin, H.-C.; Chen, C.-L.; Shen, C.-K.J.; Huang, C.-C. Therapeutic effect of berberine on TDP-43-related pathogenesis in FTLD and ALS. J. Biomed. Sci. 2016, 23, 72. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Wei, W.; Gaertig, M.A.; Li, S.; Li, X.-J. Therapeutic Effect of Berberine on Huntington’s Disease Transgenic Mouse Model. PLoS ONE 2015, 10, e0134142. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Cho, K.-H.; Shin, M.; Lee, J.-M.; Cho, H.-S.; Kim, C.-J.; Shin, D.-H.; Yang, H.J. Berberine prevents nigrostriatal dopaminergic neuronal loss and suppresses hippocampal apoptosis in mice with Parkinson’s disease. Int. J. Mol. Med. 2014, 33, 870–878. [Google Scholar] [CrossRef]
- Fan, D.; Liu, L.; Wu, Z.; Cao, M. Combating Neurodegenerative Diseases with the Plant Alkaloid Berberine: Molecular Mechanisms and Therapeutic Potential. Curr. Neuropharmacol. 2019, 17, 563–579. [Google Scholar] [CrossRef]
- Cai, Z.; Wang, C.; Yang, W. Role of berberine in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2016, 12, 2509–2520. [Google Scholar] [CrossRef]
- Haghani, M.; Shabani, M.; Tondar, M. The Therapeutic Potential of Berberine against the Altered Intrinsic Properties of the CA1 Neurons Induced by Aβ Neurotoxicity. Eur. J. Pharmacol. 2015, 758, 82–88. [Google Scholar] [CrossRef]
- Huang, M.; Jiang, X.; Liang, Y.; Liu, Q.; Chen, S.; Guo, Y. Berberine improves cognitive impairment by promoting autophagic clearance and inhibiting production of β-amyloid in APP/tau/PS1 mouse model of Alzheimer’s disease. Exp. Gerontol. 2017, 91, 25–33. [Google Scholar] [CrossRef]
- Zhang, Q.; Bian, H.; Guo, L.; Zhu, H. Pharmacologic preconditioning with berberine attenuating ischemia-induced apoptosis and promoting autophagy in neuron. Am. J. Transl. Res. 2016, 8, 1197–1207. [Google Scholar] [PubMed]
- Hsu, Y.-Y.; Chen, C.-S.; Wu, S.-N.; Jong, Y.-J.; Lo, Y.-C. Berberine activates Nrf2 nuclear translocation and protects against oxidative damage via a phosphatidylinositol 3-kinase/Akt-dependent mechanism in NSC34 motor neuron-like cells. Eur. J. Pharm. Sci. 2012, 46, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Spinozzi, S.; Colliva, C.; Camborata, C.; Roberti, M.; Ianni, C.; Neri, F.; Calvarese, C.; Lisotti, A.; Mazzella, G.; Roda, A. Berberine and Its Metabolites: Relationship between Physicochemical Properties and Plasma Levels after Administration to Human Subjects. J. Nat. Prod. 2014, 77, 766–772. [Google Scholar] [CrossRef]
- Zuo, F.; Nakamura, N.; Akao, T.; Hattori, M. Pharmacokinetics of Berberine and Its Main Metabolites in Conventional and Pseudo Germ-Free Rats Determined by Liquid Chromatography/Ion Trap Mass Spectrometry. Drug Metab. Dispos. 2006, 34, 2064–2072. [Google Scholar] [CrossRef]
- Tan, X.-S.; Ma, J.-Y.; Feng, R.; Ma, C.; Chen, W.-J.; Sun, Y.-P.; Fu, J.; Huang, M.; He, C.-Y.; Shou, J.-W.; et al. Tissue Distribution of Berberine and Its Metabolites after Oral Administration in Rats. PLoS ONE 2013, 8, e77969. [Google Scholar] [CrossRef] [PubMed]
- Kheir, M.; Wang, Y.; Hua, L.; Hu, J.; Li, L.; Lei, F.; Du, L.-J. Acute toxicity of berberine and its correlation with the blood concentration in mice. Food Chem. Toxicol. 2010, 48, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hao, H.; Xie, H.-G.; Lai, L.; Wang, Q.; Liu, C.; Wang, G. Extensive Intestinal First-Pass Elimination and Predominant Hepatic Distribution of Berberine Explain Its Low Plasma Levels in Rats. Drug Metab. Dispos. 2010, 38, 1779–1784. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-S.; Zheng, Y.-R.; Zhang, Y.-F.; Long, X.-Y. Research progress on berberine with a special focus on its oral bioavailability. Fitoterapia 2016, 109, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Waters, M.L. Aromatic interactions in model systems. Curr. Opin. Chem. Biol. 2002, 6, 736–741. [Google Scholar] [CrossRef]
- Riley, K.E.; Hobza, P. On the Importance and Origin of Aromatic Interactions in Chemistry and Biodisciplines. Accounts Chem. Res. 2012, 46, 927–936. [Google Scholar] [CrossRef]
- Akula, S.; Candido, S.; Libra, M.; Abrams, S.L.; Steelman, L.S.; Lertpiriyapong, K.; Ramazzotti, G.; Ratti, S.; Follo, M.Y.; Martelli, A.M.; et al. Abilities of berberine and chemically modified berberines to interact with metformin and inhibit proliferation of pancreatic cancer cells. Adv. Biol. Regul. 2019, 73, 100633. [Google Scholar] [CrossRef]
- Pierpaoli, E.; Damiani, E.; Orlando, F.; Lucarini, G.; Bartozzi, B.; Lombardi, P.; Salvatore, C.; Geroni, C.; Donati, A.; Provinciali, M. Antiangiogenic and antitumor activities of berberine derivative NAX014 compound in a transgenic murine model of HER2/neu-positive mammary carcinoma. Carcinogenesis 2015, 36, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Abrams, S.L.; Follo, M.Y.; Steelman, L.S.; Lertpiriyapong, K.; Cocco, L.; Ratti, S.; Martelli, A.M.; Candido, S.; Libra, M.; Murata, R.M.; et al. Abilities of berberine and chemically modified berberines to inhibit proliferation of pancreatic cancer cells. Adv. Biol. Regul. 2019, 71, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Pierpaoli, E.; Fiorillo, G.; Lombardi, P.; Salvatore, C.; Geroni, C.; Piacenza, F.; Provinciali, M. Antitumor activity of NAX060: A novel semisynthetic berberine derivative in breast cancer cells. BioFactors 2018, 44, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Marcelli, M.; Stenoien, D.L.; Szafran, A.T.; Simeoni, S.; Agoulnik, I.U.; Weigel, N.L.; Moran, T.; Mikic, I.; Price, J.H.; Mancini, M.A. Quantifying effects of ligands on androgen receptor nuclear translocation, intranuclear dynamics, and solubility. J. Cell. Biochem. 2006, 98, 770–788. [Google Scholar] [CrossRef] [PubMed]
- Szafran, A.T.; Szwarc, M.; Marcelli, M.; Mancini, M. Androgen Receptor Functional Analyses by High Throughput Imaging: Determination of Ligand, Cell Cycle, and Mutation-Specific Effects. PLoS ONE 2008, 3, e3605. [Google Scholar] [CrossRef]
- Rusmini, P.; Crippa, V.; Giorgetti, E.; Boncoraglio, A.; Cristofani, R.; Carra, S.; Poletti, A. Clearance of the mutant androgen receptor in motoneuronal models of spinal and bulbar muscular atrophy. Neurobiol. Aging 2013, 34, 2585–2603. [Google Scholar] [CrossRef]
- Landles, C.; Sathasivam, K.; Weiss, A.; Woodman, B.; Moffitt, H.; Finkbeiner, S.; Sun, B.; Gafni, J.; Ellerby, L.M.; Trottier, Y.; et al. Proteolysis of Mutant Huntingtin Produces an Exon 1 Fragment That Accumulates as an Aggregated Protein in Neuronal Nuclei in Huntington Disease. J. Biol. Chem. 2010, 285, 8808–8823. [Google Scholar] [CrossRef]
- Neueder, A.; Landles, C.; Ghosh, R.; Howland, D.; Myers, R.; Faull, R.L.M.; Tabrizi, S.J.; Bates, G. The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington’s disease patients. Sci. Rep. 2017, 7, 1307. [Google Scholar] [CrossRef]
- Li, J.; Cao, B.; Liu, X.; Fu, X.; Xiong, Z.; Chen, L.; Sartor, O.; Dong, Y.; Zhang, H. Berberine suppresses androgen receptor signaling in prostate cancer. Mol. Cancer Ther. 2011, 10, 1346–1356. [Google Scholar] [CrossRef]
- Sierra, H.; Cordova, M.; Chen, C.-S.J.; Rajadhyaksha, M. Confocal imaging-guided laser ablation of basal cell carcinomas: An ex vivo study. J. Investig. Dermatol. 2014, 135, 612–615. [Google Scholar] [CrossRef]
- Waza, M.; Adachi, H.; Katsuno, M.; Minamiyama, M.; Tanaka, F.; Sobue, G. Alleviating Neurodegeneration by an Anticancer Agent: An Hsp90 Inhibitor (17-AAG). Ann. N. Y. Acad. Sci. 2006, 1086, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Rusmini, P.; Simonini, F.; Crippa, V.; Bolzoni, E.; Onesto, E.; Cagnin, M.; Sau, D.; Ferri, N.; Poletti, A. 17-AAG increases autophagic removal of mutant androgen receptor in spinal and bulbar muscular atrophy. Neurobiol. Dis. 2011, 41, 83–95. [Google Scholar] [CrossRef]
- Adachi, H.; Waza, M.; Tokui, K.; Katsuno, M.; Minamiyama, M.; Tanaka, F.; Doyu, M.; Sobue, G. CHIP Overexpression Reduces Mutant Androgen Receptor Protein and Ameliorates Phenotypes of the Spinal and Bulbar Muscular Atrophy Transgenic Mouse Model. J. Neurosci. 2007, 27, 5115–5126. [Google Scholar] [CrossRef]
- Pratt, W.B.; Gestwicki, J.E.; Osawa, Y.; Lieberman, A.P. Targeting Hsp90/Hsp70-based protein quality control for treatment of adult onset neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 2014, 55, 353–371. [Google Scholar] [CrossRef]
- Thomas, M.; Harrell, J.M.; Morishima, Y.; Peng, H.-M.; Pratt, W.B.; Lieberman, A.P. Pharmacologic and genetic inhibition of hsp90-dependent trafficking reduces aggregation and promotes degradation of the expanded glutamine androgen receptor without stress protein induction. Hum. Mol. Genet. 2006, 15, 1876–1883. [Google Scholar] [CrossRef] [PubMed]
- Montie, H.L.; Cho, M.S.; Holder, L.; Liu, Y.; Tsvetkov, A.S.; Finkbeiner, S.; Merry, D.E. Cytoplasmic retention of polyglutamine-expanded androgen receptor ameliorates disease via autophagy in a mouse model of spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2009, 18, 1937–1950. [Google Scholar] [CrossRef]
- Nedelsky, N.B.; Pennuto, M.; Smith, R.B.; Palazzolo, I.; Moore, J.; Nie, Z.; Neale, G.; Taylor, J.P. Native Functions of the Androgen Receptor Are Essential to Pathogenesis in a Drosophila Model of Spinobulbar Muscular Atrophy. Neuron 2010, 67, 936–952. [Google Scholar] [CrossRef]
- Takeyama, K.-I.; Ito, S.; Yamamoto, A.; Tanimoto, H.; Furutani, T.; Kanuka, H.; Miura, M.; Tabata, T.; Kato, S. Androgen-dependent neurodegeneration by polyglutamine-expanded human androgen receptor in Drosophila. Neuron 2002, 35, 855–864. [Google Scholar] [CrossRef]
- Walcott, J.L.; Merry, D.E. Ligand Promotes Intranuclear Inclusions in a Novel Cell Model of Spinal and Bulbar Muscular Atrophy. J. Biol. Chem. 2002, 277, 50855–50859. [Google Scholar] [CrossRef]
- Li, M.; Miwa, S.; Kobayashi, Y.; Merry, D.E.; Yamamoto, M.; Tanaka, F.; Doyu, M.; Hashizum, Y.; Fischbeck, K.H.; Sobue, G. Nuclear inclusions of the androgen receptor protein in spinal and bulbar muscular atrophy. Ann. Neurol. 1998, 44, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Parodi, S.; Pennuto, M. Neurotoxic effects of androgens in spinal and bulbar muscular atrophy. Front. Neuroendocr. 2011, 32, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Thirupurasundari, C.J.; Padmini, R.; Devaraj, S.N. Effect of berberine on the antioxidant status, ultrastructural modifications and protein bound carbohydrates in azoxymethane-induced colon cancer in rats. Chem. Interactions 2009, 177, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Li, H.; Singh, A.B.; Cao, A.; Liu, J. Inhibition ofPCSK9Transcription by Berberine Involves Down-regulation of Hepatic HNF1α Protein Expression through the Ubiquitin-Proteasome Degradation Pathway. J. Biol. Chem. 2014, 290, 4047–4058. [Google Scholar] [CrossRef]
- Wang, N.; Wang, X.; Tan, H.-Y.; Li, S.; Tsang, C.M.; Tsao, S.-W.; Feng, Y. Berberine Suppresses Cyclin D1 Expression through Proteasomal Degradation in Human Hepatoma Cells. Int. J. Mol. Sci. 2016, 17, 1899. [Google Scholar] [CrossRef]
- Li, A.; Liu, Q.; Li, Q.; Liu, B.; Yang, Y.; Zhang, N. Berberine Reduces Pyruvate-driven Hepatic Glucose Production by Limiting Mitochondrial Import of Pyruvate through Mitochondrial Pyruvate Carrier 1. EBioMedicine 2018, 34, 243–255. [Google Scholar] [CrossRef]
- Ortiz, L.M.G.; Tillhon, M.; Parks, M.; Dutto, I.; Prosperi, E.; Savio, M.; Arcamone, A.G.; Buzzetti, F.; Lombardi, P.; Scovassi, A.I. Multiple Effects of Berberine Derivatives on Colon Cancer Cells. BioMed Res. Int. 2014, 2014, 924585. [Google Scholar] [CrossRef]
- Stenoien, D.L.; Cummings, C.J.; Adams, H.P.; Mancini, M.A.; Patel, K.; DeMartino, G.N.; Marcelli, M.; Weigel, N.L. Polyglutamine-Expanded Androgen Receptors Form Aggregates That Sequester Heat Shock Proteins, Proteasome Components and SRC-1, and Are Suppressed by the HDJ-2 Chaperone. Hum. Mol. Genet. 1999, 8, 731–741. [Google Scholar] [CrossRef]
- Cashman, N.R.; Durham, H.D.; Blusztajn, J.K.; Oda, K.; Tabira, T.; Shaw, I.T.; Dahrouge, S.; Antel, J.P. Neuroblastoma × spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev. Dyn. 1992, 194, 209–221. [Google Scholar] [CrossRef]
- Piccioni, F.; Pinton, P.; Simeoni, S.; Pozzi, P.; Fascio, U.; Vismara, G.; Martini, L.; Rizzuto, R.; Poletti, A. Androgen receptor with elongated polyglutamine tract forms aggregates that alter axonal trafficking and mitochondrial distribution in motoneuronal processes. FASEB J. 2002, 16, 1418–1420. [Google Scholar] [CrossRef]
- Pozzi, P.; Bendotti, C.; Simeoni, S.; Piccioni, F.; Guerini, V.; Marron, T.U.; Martini, L.; Poletti, A. Androgen 5-alpha-reductase type 2 is highly expressed and active in rat spinal cord motor neurones. J. Neuroendocr. 2003, 15, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Crippa, V.; Sau, D.; Rusmini, P.; Boncoraglio, A.; Onesto, E.; Bolzoni, E.; Galbiati, M.; Fontana, E.; Marino, M.; Carra, S.; et al. The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum. Mol. Genet. 2010, 19, 3440–3456. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rusmini, P.; Cristofani, R.; Tedesco, B.; Ferrari, V.; Messi, E.; Piccolella, M.; Casarotto, E.; Chierichetti, M.; Cicardi, M.E.; Galbiati, M.; et al. Enhanced Clearance of Neurotoxic Misfolded Proteins by the Natural Compound Berberine and Its Derivatives. Int. J. Mol. Sci. 2020, 21, 3443. https://doi.org/10.3390/ijms21103443
Rusmini P, Cristofani R, Tedesco B, Ferrari V, Messi E, Piccolella M, Casarotto E, Chierichetti M, Cicardi ME, Galbiati M, et al. Enhanced Clearance of Neurotoxic Misfolded Proteins by the Natural Compound Berberine and Its Derivatives. International Journal of Molecular Sciences. 2020; 21(10):3443. https://doi.org/10.3390/ijms21103443
Chicago/Turabian StyleRusmini, Paola, Riccardo Cristofani, Barbara Tedesco, Veronica Ferrari, Elio Messi, Margherita Piccolella, Elena Casarotto, Marta Chierichetti, Maria Elena Cicardi, Mariarita Galbiati, and et al. 2020. "Enhanced Clearance of Neurotoxic Misfolded Proteins by the Natural Compound Berberine and Its Derivatives" International Journal of Molecular Sciences 21, no. 10: 3443. https://doi.org/10.3390/ijms21103443
APA StyleRusmini, P., Cristofani, R., Tedesco, B., Ferrari, V., Messi, E., Piccolella, M., Casarotto, E., Chierichetti, M., Cicardi, M. E., Galbiati, M., Geroni, C., Lombardi, P., Crippa, V., & Poletti, A. (2020). Enhanced Clearance of Neurotoxic Misfolded Proteins by the Natural Compound Berberine and Its Derivatives. International Journal of Molecular Sciences, 21(10), 3443. https://doi.org/10.3390/ijms21103443