Intermittent Hypoxic Conditioning Alleviates Post-Traumatic Stress Disorder-Induced Damage and Dysfunction of Rat Visceral Organs and Brain

 ,
,
Abstract
1. Introduction
2. Results
2.1. IHC Alleviated Behavioral Markers of PTSD
2.2. IHC Prevented Detrimental Effects of PTSD on the Heart
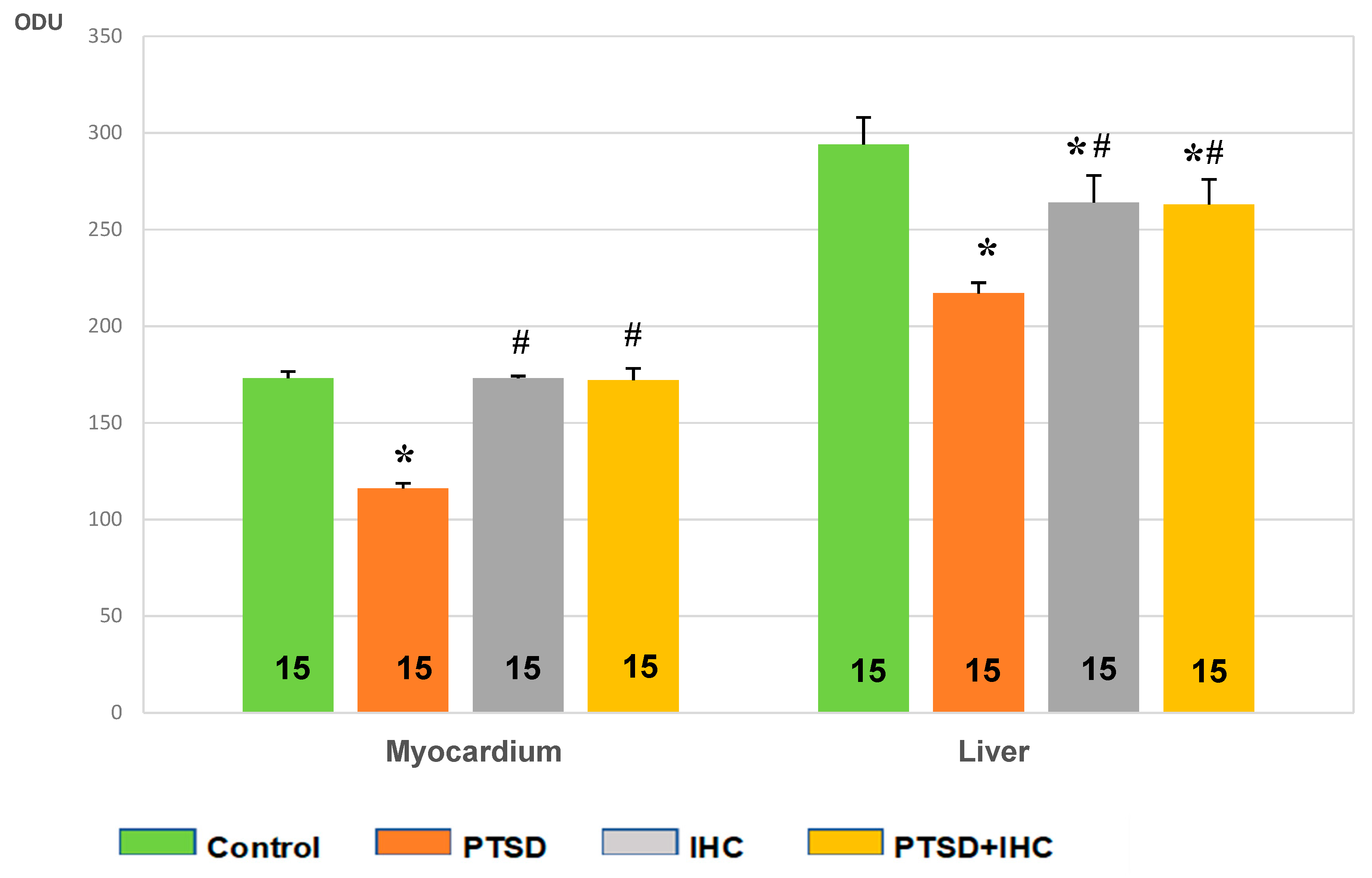
2.2.1. IHC Prevented PTSD-Mediated Decrease in Myocardial Glycogen
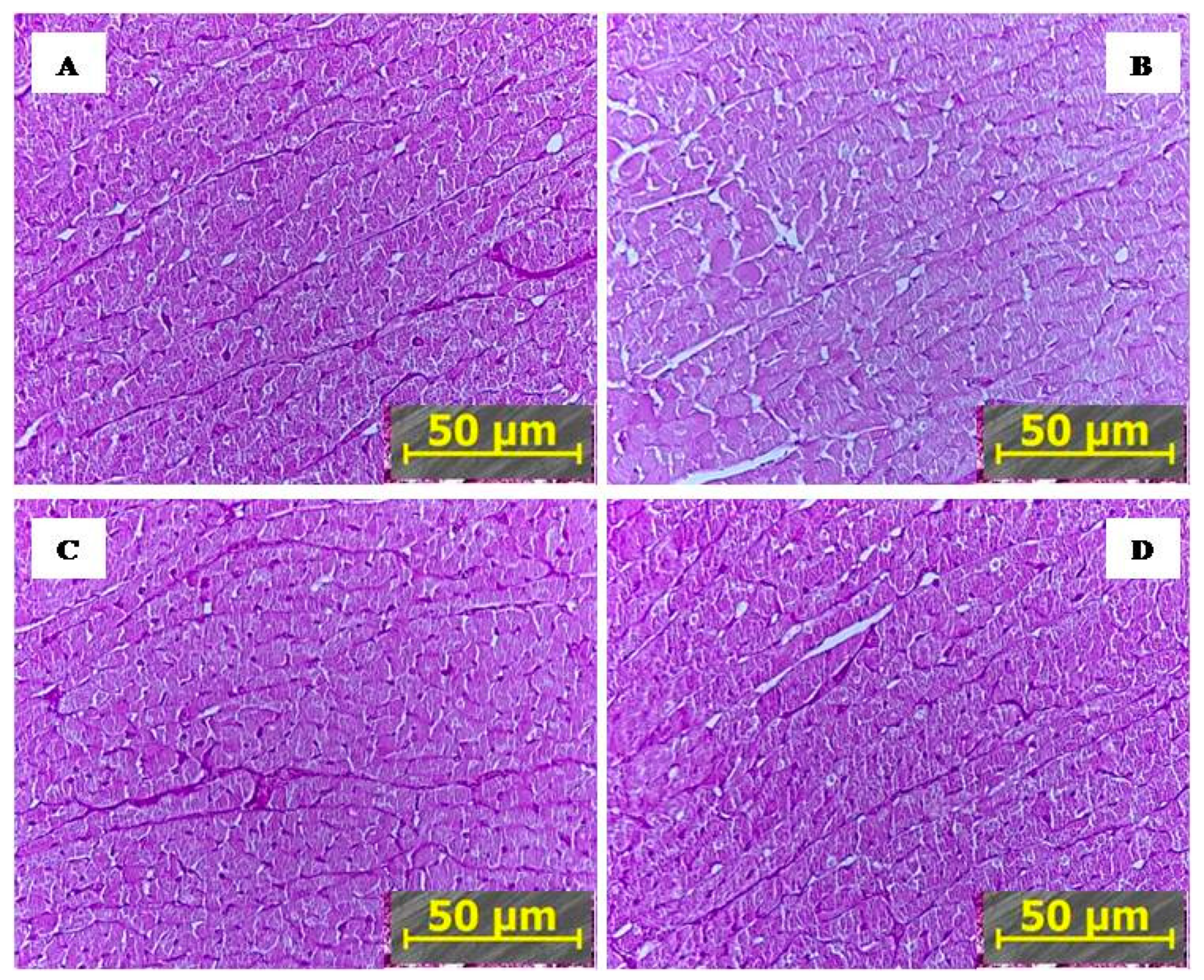
2.2.2. IHC Markedly Reduced PTS-Mediated Myocardial Damage
2.2.3. IHC Alleviated Oxidative Stress Induced by PTSD in the Heart
2.3. IHC Prevented Detrimental Effects of PTSD on the Liver
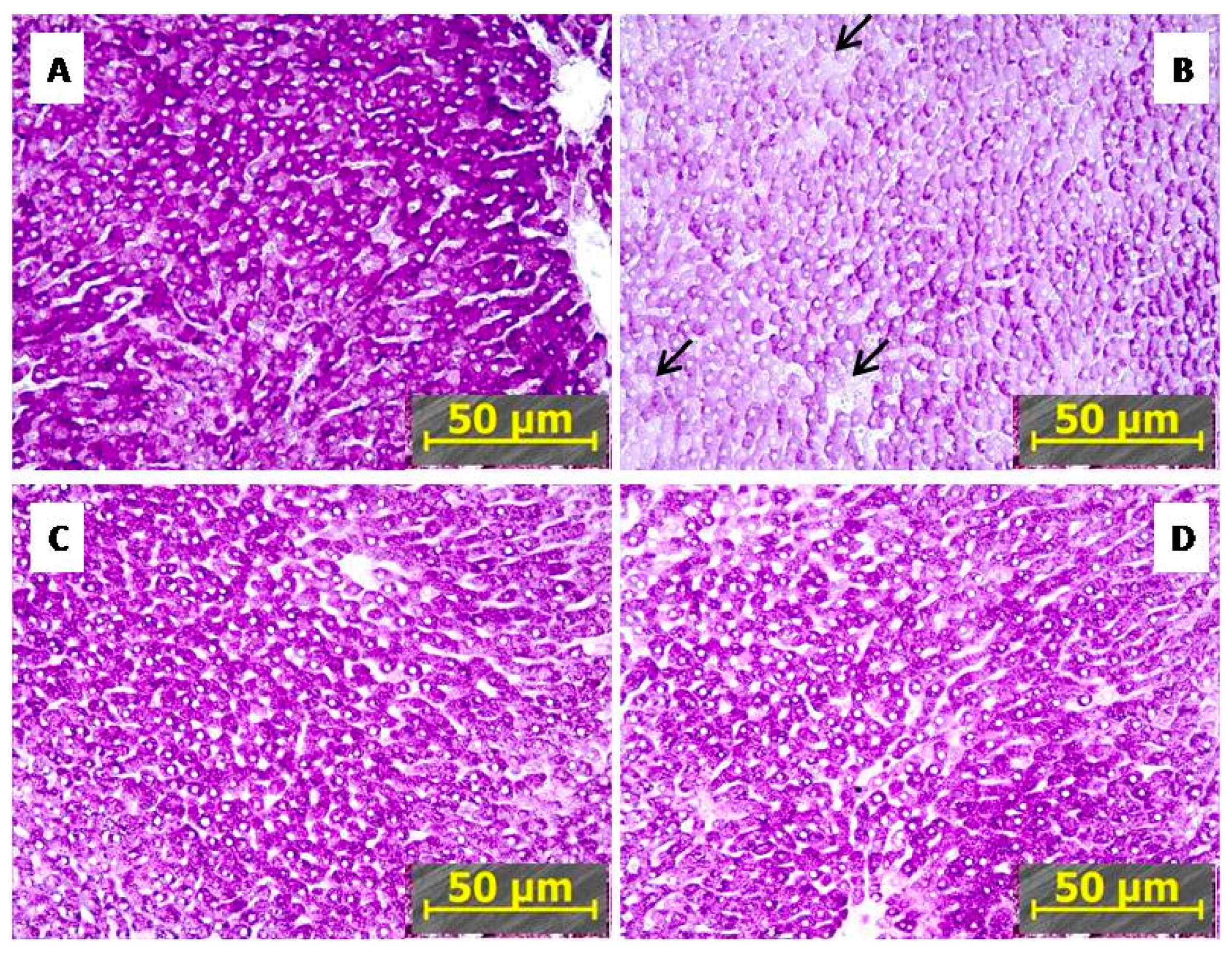
2.3.1. IHC Reduced PTSD-Mediated Depletion of Hepatic Glycogen
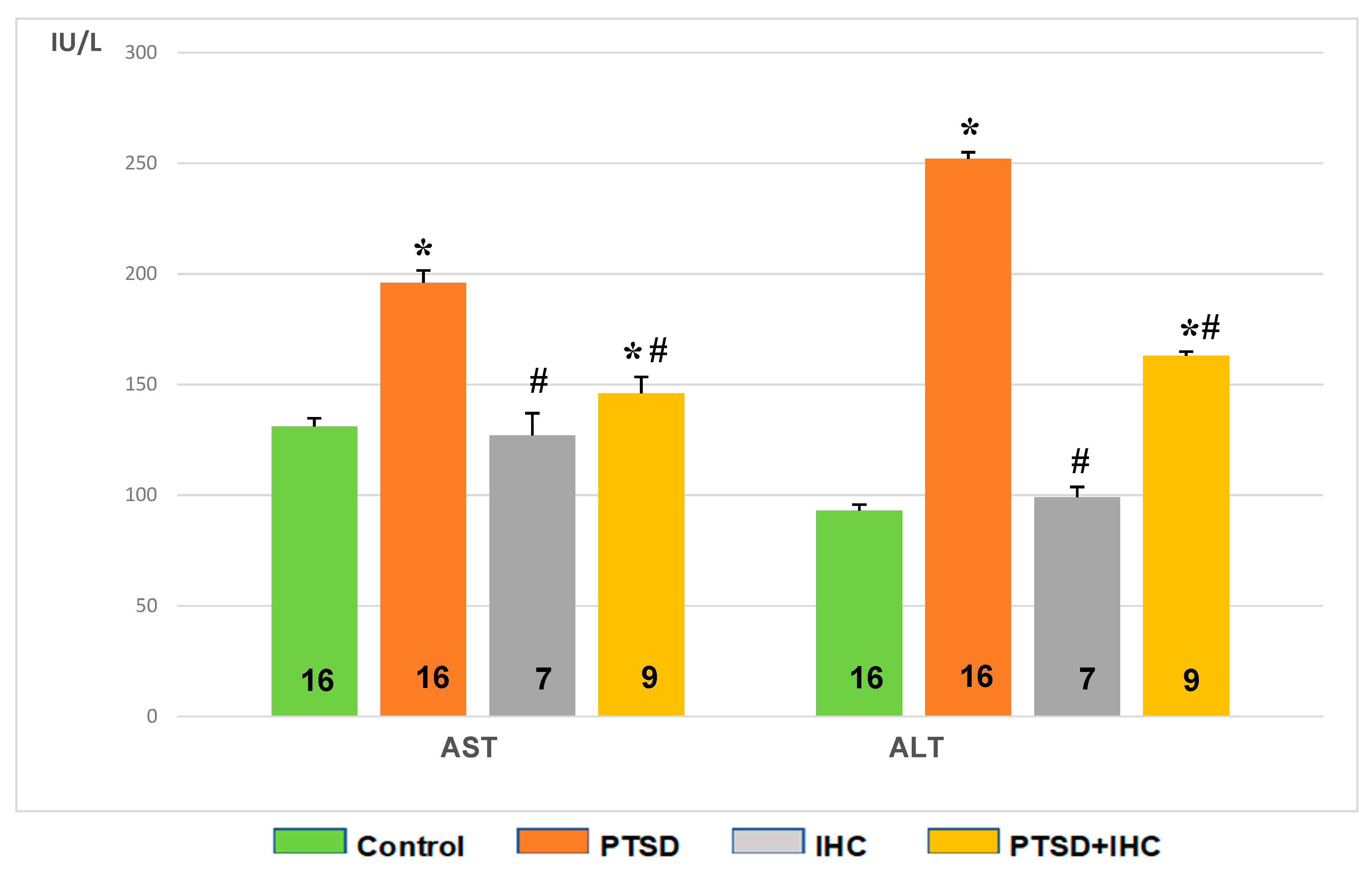
2.3.2. IHC Attenuated PTSD-Mediated Increases in Aspartate Aminotransferase (AST) and Alanine Aminotransferase (ALT) in Rat Liver and Blood
2.3.3. IHC Alleviated Oxidative Stress Induced by PTSD in the Liver
2.4. IHC Prevented Detrimental Effects of PTSD on the Brain
2.4.1. IHC Attenuated PTSD-Mediated Increase in NE Concentration and Decrease in MAO-A Activity
2.4.2. IHC Alleviated Oxidative Stress Induced by PTSD in the Brain
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. Modeling PTSD
4.3. Intermittent Hypoxia Conditioning (IHC)
4.4. Behavioral Testing
4.5. Blood and Tissue Collection and Storage
4.6. Measurement of Norepinephrine
4.7. Measurement of MAO-A Activity
4.8. Measurement of Alanine (AST) and Aspartate Aminotransferase (ALT) Activities
4.9. Histochemical Analysis
4.10. Evaluation of Oxidative Stress
4.10.1. Measurement of Lipid Peroxidation Products
4.10.2. Measurement of Protein Oxidation Products
4.11. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PTSD | Posttraumatic stress disorder |
| IHC | Intermittent hypoxia conditioning |
| AST | Aspartate aminotransferase |
| ALT | Alanine aminotransferase |
| NE | Norepinephrine |
| MAO | Monoaminoxidase |
References
- Stander, V.A.; Thomsen, C.J.; Highfill-McRoy, R.M. Etiology of depression comorbidity in combat-related PTSD: A review of the literature. Clin. Psychol. Rev. 2014, 34, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Malarbi, S.; Abu-Rayya, H.M.; Muscara, F.; Stargatt, R. Neuropsychological functioning of childhood trauma and post-traumatic stress disorder: A meta-analysis. Neurosci. Biobehav. Rev. 2017, 72, 68–86. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, D.; von Känel, R. Post-traumatic stress disorder and cardiovascular disease. Lancet Psychiatry 2017, 4, 320–329. [Google Scholar] [CrossRef]
- Pietrzak, R.H.; Goldstein, R.B.; Southwick, S.M.; Grant, B.F. Medical comorbidity of full and partial posttraumatic stress disorder in US adults: Results from Wave 2 of the National Epidemiologic Survey on Alcohol and Related Conditions. Psychosom. Med. 2011, 73, 697–707. [Google Scholar] [CrossRef]
- Breslau, N. The epidemiology of posttraumatic stress disorder: What is the extent of the problem? J. Clin. Psychiatry 2001, 62, 16–22. [Google Scholar]
- Horn, S.R.; Charney, D.S.; Feder, A. Understanding resilience: New approaches for preventing and treating PTSD. Exp. Neurol. 2016, 284, 119–132. [Google Scholar] [CrossRef]
- Osório, C.; Probert, T.; Jones, E.; Young, A.H.; Robbins, I. Adapting to stress: Understanding the neurobiology of resilience. Behav. Med. 2017, 43, 307–322. [Google Scholar] [CrossRef]
- Meerson, F.Z. Essential of Adaptive Medicine: Protective Effects of Adaptation; Hypoxia Medical LTD: Moscow, Russia, 1994. [Google Scholar]
- Pshennikova, M.G. Inherent efficiency of stress-limiting systems as a factor of the resistance to stress-induced disorders. Usp. Fiziol. Nauk. 2003, 34, 55–67. (In Russian) [Google Scholar]
- Malyshev, I.Y.; Manukhina, E.B. Stress, adaptation, and nitric oxide. Biochemistry (Mosc) 1998, 63, 840–853. [Google Scholar]
- Mallet, R.T.; Manukhina, E.B.; Ruelas, S.S.; Caffrey, J.L.; Downey, H.F. Cardioprotection by intermittent hypoxia conditioning: Evidence, mechanisms and therapeutic potential. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H216–H232. [Google Scholar] [CrossRef]
- Manukhina, E.B.; Downey, H.F.; Shi, X.; Mallet, R.T. Intermittent hypoxia training protects cerebrovascular function in Alzheimer’s disease. Exp. Biol. Med. 2016, 241, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Zhen, J.; Wang, W.; Zhou, J.; Qu, Z.; Fang, H.; Zhao, R.; Lu, Y.; Wang, H.; Zang, H. Chronic intermittent hypoxic preconditioning suppresses pilocarpine-induced seizures and associated hippocampal neurodegeneration. Brain Res. 2014, 1563, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikova, E.E.; Serebrovskaya, T.V. Parkinson’s disease and intermittent hypoxia training. In Intermittent Hypoxia: From Molecular Mechanisms to Clinical Applications; Xi, L., Serebrovskaya, T.V., Eds.; Nova Science Publ.: New York, NY, USA, 2009; pp. 549–560. [Google Scholar]
- Manukhina, E.B.; Goryacheva, A.V.; Smirin, B.V.; Malyshev, I.Y.; Budanova, O.P.; Downey, H.F. Prevention of cerebrovascular and neurodegenerative disorders by adaptation to hypoxia in Alzheimer’s disease. In Adaptation Biology and Medicine: Current Trends; Kawai, Y., Hargens, A.R., Singal, P.K., Eds.; Narosa Publishing House: New Delhi, India, 2017; Volume 8, pp. 241–260. [Google Scholar]
- Rybnikova, E.A.; Samoilov, M.O.; Mironova, V.I.; Tyul’kova, E.I.; Pivina, S.G.; Vataeva, L.A.; Ordyan, N.E.; Abritalin, E.Yu.; Kolchev, A.I. The possible use of hypoxic preconditioning for the prophylaxis of post-stress depressive episodes. Neurosci. Behav. Physiol. 2008, 38, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Rybnikova, E.A.; Baranova, K.A.; Glushchenko, T.S.; Vetrovoĭ, O.V.; Sidorova, M.V.; Portnichenko, A.V. Involvement of transcriptional factor induced by hypoxia in the neuronal mechanisms of adaptation to psychoemotional and hypoxic stress. Fiziol. Zh. 2013, 59, 88–97. (In Russian) [Google Scholar] [CrossRef]
- Manukhina, E.B.; Tseilikman, V.E.; Tseilikman, O.B.; Komelkova, M.V.; Kondashevskaya, M.V.; Goryacheva, A.V.; Lapshin, M.S.; Platkovskii, P.O.; Alliluev, A.V.; Downey, H.F. Intermittent hypoxia improves behavioral and adrenal gland dysfunction induced by post-traumatic stress disorder in rats. J. Appl. Physiol. 2018, 125, 931–937. [Google Scholar] [CrossRef]
- Hashmi, S.; Al-Salam, S. Acute myocardial infarction and myocardial ischemia-reperfusion injury: A comparison. Int. J. Clin. Exp. Pathol. 2015, 8, 8786–8796. [Google Scholar]
- O’Toole, B.I.; Catts, S.V. Trauma, PTSD, and physical health: An epidemiological study of Australian Vietnam veterans. J. Psychosom. Res. 2008, 64, 33–40. [Google Scholar] [CrossRef]
- McLeay, S.C.; Harvey, W.M.; Romaniuk, M.N.; Crawford, D.H.; Colquhoun, D.M.; Young, R.M.; Dwyer, M.; Gibson, J.M.; O’Sullivan, R.A.; Cooksley, G.; et al. Physical comorbidities of post-traumatic stress disorder in Australian Vietnam War veterans. Med. J. Aust. 2017, 206, 251–257. [Google Scholar] [CrossRef]
- Boscarino, J.A. Posttraumatic stress disorder and physical illness: Results from clinical and epidemiologic studies. Ann. N. Y. Acad. Sci. 2004, 1032, 141–153. [Google Scholar] [CrossRef]
- Wachen, J.S.; Shipherd, J.C.; Michael, S.; Dawne, V.; King, L.A.; King, D.W. Posttraumatic stress symptomatology as a mediator of the relationship between warzone exposure and physical health symptoms in men and women. J. Trauma Stress 2013, 26, 319–328. [Google Scholar] [CrossRef]
- Ryder, A.L.; Azcarate, P.M.; Cohen, B.E. PTSD and physical health. Curr. Psychiatry Rep. 2018, 20, 116. [Google Scholar] [CrossRef] [PubMed]
- Cohen, H.; Kozlovsky, N.; Alona, C.; Matar, M.A.; Joseph, Z. Animal model for PTSD: From clinical concept to translational research. Neuropharmacology 2012, 62, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Kubzansky, L.D.; Koenen, K.C.; Spiro, A., III; Vokonas, P.S.; Sparrow, D. Prospective study of posttraumatic stress disorder symptoms and coronary heart disease in the Normative Aging Study. Arch. Gen. Psychiatry 2007, 64, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, D.; Kronish, I.M.; Shaffer, J.A.; Falzon, L.; Burg, M.M. Posttraumatic stress disorder and risk for coronary heart disease: A meta-analytic review. Am. Heart J. 2013, 166, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.H.; Neylan, T.C.; Schiller, N.B.; Li, Y.; Cohen, B.E. Objective evidence of myocardial ischemia in patients with posttraumatic stress disorder. Biol. Psychiatry 2013, 74, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Paulus, E.J.; Argo, T.R.; Egge, J.A. The impact of posttraumatic stress disorder on blood pressure and heart rate in a veteran population. J. Trauma Stress 2013, 26, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.H.; Pan, T.L.; Li, C.T.; Lin, W.C.; Chen, Y.S.; Lee, Y.C.; Tsai, S.J.; Hsu, J.W.; Huang, K.L.; Tsai, C.F.; et al. Risk of stroke among patients with post-traumatic stress disorder: Nationwide longitudinal study. Br. J. Psychiatry 2015, 206, 302–307. [Google Scholar] [CrossRef]
- Vaccarino, V.; Goldberg, J.; Rooks, C.; Shah, A.J.; Veledar, E.; Faber, T.L.; Votaw, J.R.; Forsberg, C.W.; Bremner, J.D. Post-traumatic stress disorder and incidence of coronary heart disease. A twin study. J. Am. Coll. Cardiology 2013, 62, 970–978. [Google Scholar] [CrossRef]
- Dimsdale, J.E.; Ruberman, W.; Carleton, R.A.; DeQuattro, V.; Eaker, E.; Eliot, R.S.; Furberg, C.D.; Irvin, C.W., Jr.; Lown, B.; Shapiro, A.P. Sudden cardiac death. Stress and cardiac arrhythmias. Circulation 1987, 76, I198–I201. [Google Scholar]
- Schwartz, P.J.; La Rovere, M.T.; Vanoli, E. Autonomic nervous system and sudden cardiac death. Experimental basis and clinical observations for post–myocardial infarction risk stratification. Circulation 1992, 85, 177–191. [Google Scholar]
- Reznik, A.G. Morphology of acute myocardial infarction at prenecrotic stage. Kardiologiia 2010, 50, 4–8. (In Russian) [Google Scholar] [PubMed]
- Kondashevskaya, M.V.; Tseylikman, V.E.; Komelkova, M.V.; Lapshin, M.S.; Sarapultsev, A.P.; Lazuko, S.S.; Kuzhel, O.P.; Manukhina, E.B.; Downey, H.F.; Chereshneva, M.V.; et al. Physical fatigue and morphofunctional state of the myocardium in experimental chronic stress. Dokl. Biol. Sci. 2019, 485, 30–32. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Xu, F.; Tao, T.; Song, D.; Li, D.; Li, Y.; Guo, Y.; Liu, X. Molecular mechanisms of stress-induced myocardial injury in a rat model simulating posttraumatic stress disorder. Psychosom. Med. 2016, 78, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Zhang, L.; Li, Y.Q.; Teng, X.; Tian, S.Y.; Wang, X.R.; Zhang, Y. Chronic intermittent hypobaric hypoxia improves cardiac function through inhibition of endoplasmic reticulum stress. Sci. Rep. 2017, 7, 7922. [Google Scholar] [CrossRef]
- Wang, Z.; Si, L.Y. Hypoxia-inducible factor-1α and vascular endothelial growth factor in the cardioprotective effects of intermittent hypoxia in rats. Ups. J. Med. Sci. 2013, 118, 65–74. [Google Scholar] [CrossRef]
- Lazo, M.; Rubin, J.; Clark, J.M.; Coresh, J.; Schneider, A.L.; Ndumele, C.; Hoogeveen, R.C.; Ballantyne, C.M.; Selvin, E. The association of liver enzymes with biomarkers of subclinical myocardial damage and structural heart disease. J. Hepatol. 2015, 62, 841–847. [Google Scholar] [CrossRef]
- Reinstadler, S.J.; Reindl, M.; Feistritzer, H.J.; Klug, G.; Mayr, A.; Kofler, M.; Tu, A.M.; Huybrechts, L.; Mair, J.; Franz, W.M.; et al. Prognostic significance of transaminases after acute ST-elevation myocardial infarction: Insights from a cardiac magnetic resonance study. Wien Klin. Wochenschr. 2015, 127, 843–850. [Google Scholar] [CrossRef]
- Moon, J.; Kang, W.; Oh, P.C.; Seo, S.Y.; Lee, K.; Han, S.H.; Ahn, T.; Shin, E. Serum transaminase determined in the emergency room predicts outcomes in patients with acute ST-segment elevation myocardial infarction who undergo primary percutaneous coronary intervention. Int. J. Cardiol. 2014, 177, 442–447. [Google Scholar] [CrossRef]
- Lazzeri, C.; Valente, S.; Boddi, M.; Mecarocci, V.; Chiostri, M.; Gensini, G.F. Clinical and prognostic significance of increased liver enzymes in ST-elevation myocardial infarction. Int. J. Cardiol. 2014, 177, 543–544. [Google Scholar] [CrossRef]
- Pratt, D.S.; Kaplan, M.M. Evaluation of abnormal liver-enzyme results in asymptomatic patients. N. Engl. J. Med. 2000, 342, 1266–1271. [Google Scholar] [CrossRef]
- Von Känel, R.; Abbas, C.C.; Begré, S.; Gander, M.L.; Saner, H.; Schmid, J.P. Association between posttraumatic stress disorder following myocardial infarction and liver enzyme levels: A prospective study. Dig. Dis. Sci. 2010, 55, 2614–2623. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, O.; Viladrich, M.; Ramı´rez, I.; Soley, M. Liver injury after an aggressive encounter in male mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1908–R1916. [Google Scholar] [CrossRef] [PubMed]
- Salas, M.; Tuchweber, B.; Kourounakis, P. Liver ultrastructure during acute stress. Pathol. Res. Pract. 1980, 167, 217–233. [Google Scholar] [CrossRef]
- Chida, Y.; Sudo, N.; Kubo, C. Does stress exacerbate liver diseases? J. Gastroenterol. Hepatol. 2006, 21, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Z.; Chen, J.K.; Zhang, Y.; Wang, X.; Qu, S.; Jiang, C.L. Chronic stress induces steatohepatitis while decreases visceral fat mass in mice. BMC Gastroenterol. 2014, 14, 106. [Google Scholar] [CrossRef]
- Kondashevskaya, M.V. Experimental evaluation of the effects of low-dose heparin on the behavior and morphofunctional status of the liver in Wistar rats with posttraumatic stress disorders. Bull. Exp. Biol. Med. 2018, 164, 488–492. (In Russian) [Google Scholar] [CrossRef]
- Kanungo, S.; Wells, K.; Tribett, T.; El-Gharbawy, A. Glycogen metabolism and glycogen storage disorders. Ann. Transl. Med. 2018, 6, 474. [Google Scholar] [CrossRef]
- Altuna, M.E.; Lelli, S.M.; San Martín de Viale, L.C.; Damasco, M.C. Effect of stress on hepatic 11beta-hydroxysteroid dehydrogenase activity and its influence on carbohydrate metabolism. Can. J. Physiol. Pharmacol. 2006, 84, 977–984. [Google Scholar] [CrossRef]
- Paterson, J.M.; Morton, N.M.; Fievet, C.; Kenyon, C.J.; Holmes, M.C.; Staels, B.; Seckl, J.R.; Mullins, J.J. Metabolic syndrome without obesity: Hepatic overexpression of 11b-hydroxysteroid dehydrogenase type 1 in transgenic mice. Proc. Natl. Acad Sci. USA 2004, 101, 7088–7093. [Google Scholar] [CrossRef]
- Takahashi, A.; Ohira, T.; Okazaki, K.; Yasumura, S.; Sakai, A.; Maeda, M.; Yabe, H.; Hosoya, M.; Ohtsuru, A.; Kawasaki, Y.; et al. Effects of lifestyle on hepatobiliary enzyme abnormalities following the Fukushima Daiichi nuclear power plant accident: The Fukushima health management survey. Medicine (Baltimore) 2018, 97, e12890. [Google Scholar] [CrossRef]
- Takahashi, A.; Ohira, T.; Hosoya, M.; Yasumura, S.; Nagai, M.; Ohira, H.; Hashimoto, S.; Satoh, H.; Sakai, A.; Ohtsuru, A.; et al. Effect of evacuation on liver function after the Fukushima Daiichi Nuclear Power Plant accident: The Fukushima Health Management Survey. J. Epidemiol. 2017, 27, 180–185. [Google Scholar] [CrossRef]
- Boscarino, J.A.; Galea, S.; Ahern, J.; Resnick, H.; Vlahov, D. Psychiatric medication use among Manhattan residents following the World Trade Center disaster. J. Trauma Stress 2003, 16, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Peters, R.J.Jr.; Meshack, A.; Amos, C.; Scott-Gurnell, K.; Savage, C.; Ford, K. The association of drug use and post-traumatic stress reactions due to Hurricane Ike among Fifth Ward Houstonian youth. J. Ethn. Subst. Abuse 2010, 9, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Uchinami, M.; Muraoka, R.; Horiuchi, T.; Tabo, T.; Kimura, N.; Naito, Y. Effect of intermittent hepatic pedicle clamping on free radical generation in the rat liver. Surgery 1998, 124, 49–56. [Google Scholar] [CrossRef]
- Crenesse, D.; Laurens, M.; Gugenheim, J.; Heurteaux, C.; Cursio, R.; Rossi, B.; Schmid-Alliana, A. Intermittent ischemia reduces warm hypoxia-reoxygenation-induced JNK(1)/SAPK(1) activation and apoptosis in rat hepatocytes. Hepatology 2001, 34, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.S.; Sindram, D.; Rerry, D.K.; Clavien, P.A. Ischemic preconditioning protects the mouse liver by inhibition of apoptosis through a caspase dependent pathway. Hepatology 1999, 30, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Gasbarrini, A.; Colantoni, A.; DiCampeli, C.; De Notariis, S.; Masetti, M.; Iovine, E.; Mazziotti, A.; Massari, I.; Gasbarrini, G.; Pola, P.; et al. Intermittent anoxia reduces oxygen free radical formation during reoxygenation in rat hepatocytes. Free Rad. Biol. Med. 1997, 23, 1067–1072. [Google Scholar] [CrossRef]
- Plock, J.; Frese, S.; Keogh, A.; Bisch-Knaden, S.; Ayuni, E.; Corazza, N.; Weikert, C.; Jakob, S.; Erni, D.; Dufour, J.F.; et al. Activation of non-ischemic, hypoxia-inducible signalling pathways up-regulate cytoprotective genes in the murine liver. J. Hepatol. 2007, 47, 538–545. [Google Scholar] [CrossRef]
- Hzhehots’kyĭ, M.P.; Konyk, U.V.; Kozak, L.P.; Kovalyshyn, V.I. Effect of amaranth oil and intermittent hypoxic training on ultrastructural and metabolic changes in the liver induced by fluorine and low doses of radiation. Fiziol. Zh. 2006, 52, 90–98. [Google Scholar]
- Lebkova, N.P.; Chizhov, A.; Bobkov, I. The adaptational intracellular mechanisms regulating energy homeostasis during intermittent normobaric hypoxia. Rossiiskii fiziologicheskii zhurnal imeni IM Sechenova 1999, 85, 403–411. (In Russian) [Google Scholar]
- Datta, D.; Arnsten, A.F.T. Loss of prefrontal cortical higher cognition with uncontrollable stress: Molecular mechanisms, changes with age, and relevance to treatment. Brain Sci. 2019, 9, 113. [Google Scholar] [CrossRef] [PubMed]
- Arnsten, A.F.; Raskind, M.A.; Taylor, F.B.; Connor, D.F. The effects of stress exposure on prefrontal cortex: Translating basic research into successful treatments for post-traumatic stress disorder. Neurobiol. Stress 2015, 1, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Hains, A.B.; Vu, M.A.; Maciejewski, P.K.; Van Dyck, C.H.; Gottron, M.; Arnsten, A.F. Inhibition of protein kinase C signaling protects prefrontal cortex dendritic spines and cognition from the effects of chronic stress. Proc. Natl. Acad. Sci. USA 2009, 106, 17957–17962. [Google Scholar] [CrossRef] [PubMed]
- Liston, C.; Miller, M.M.; Goldwater, D.S.; Radley, J.J.; Rocher, A.B.; Hof, P.R.; Morrison, J.H.; McEwen, B.S. Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J. Neurosci. 2006, 26, 7870–7874. [Google Scholar] [CrossRef]
- Johansson, L.; Guo, X.; Duberstein, P.R.; Hällström, T.; Waern, M.; Ostling, S.; Skoog, I. Midlife personality and risk of Alzheimer disease and distress: A 38-year follow-up. Neurology 2014, 83, 1538–1544. [Google Scholar] [CrossRef]
- Flatt, J.D.; Gilsanz, P.; Quesenberry, C.P.J.; Albers, K.B.; Whitmer, R.A. Post-traumatic stress disorder and risk of dementia among members of a health care delivery system. Alzheimers Dement. 2018, 14, 28–34. [Google Scholar] [CrossRef]
- Tseilikman, V.; Dremencov, E.; Maslennikova, E.; Ishmatova, A.; Manukhina, E.; Downey, H.F.; Klebanov, I.; Tseilikman, O.; Komelkova, M.; Lapshin, M.S.; et al. Post-traumatic stress disorder chronification via monoaminoxidase and cortisol metabolism. Horm. Metab. Res. 2019, 51, 618–622. [Google Scholar]
- Grunewald, M.; Johnson, S.; Lu, D.; Wang, Z.; Lomberk, G.; Albert, P.R.; Stockmeier, C.A.; Meyer, J.H.; Urrutia, R.; Miczek, K.A.; et al. Mechanistic role for a novel glucocorticoid-KLF11 (TIEG2) protein pathway in stress-induced monoamine oxidase A expression. J. Biol. Chem. 2012, 287, 24195–24206. [Google Scholar] [CrossRef]
- Zhu, X.H.; Yan, H.C.; Zhang, J.; Qu, H.D.; Qiu, X.S.; Chen, L.; Li, S.J.; Cao, X.; Bean, J.C.; Chen, L.H.; et al. Intermittent hypoxia promotes hippocampal neurogenesis and produces antidepressant-like effects in adult rats. J. Neurosci. 2010, 30, 12653–12663. [Google Scholar] [CrossRef]
- Goryacheva, A.V.; Kruglov, S.V.; Pshennikova, M.G.; Smirin, B.V.; Malyshev, I.Yu.; Barskov, I.V.; Viktorov, I.V.; Downey, H.F.; Manukhina, E.B. Adaptation to intermittent hypoxia restricts nitric oxide overproduction and prevents beta-amyloid toxicity in rat brain. Nitric Oxide 2010, 23, 289–299. [Google Scholar] [CrossRef]
- Kushwah, N.; Jain, V.; Deep, S.; Prasad, D.; Singh, S.B.; Khan, N. Neuroprotective role of intermittent hypobaric hypoxia in unpredictable chronic mild stress induced depression in rats. PLoS ONE 2016, 11, e0149309. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.E.; Simpkins, J.W.; Wilson, A.M.; Downey, H.F.; Mallet, R.T. Intermittent hypoxia conditioning prevents behavioral deficit and brain oxidative stress in ethanol-withdrawn rats. J. Appl. Physiol. 2008, 105, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.W.; Lin, A.P.; Wolf, E.J.; Miller, D.R. Oxidative stress, inflammation, and neuroprogression in chronic PTSD. Harv. Rev. Psychiatry 2018, 26, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Black, C.N.; Bot, M.; Scheffer, P.G.; Cuijpers, P.; Penninx, B.W. Is depression associated with increased oxidative stress? A systematic review and meta-analysis. Psychoneuroendocrinology 2015, 51, 164–175. [Google Scholar] [CrossRef]
- Kamat, P.K.; Kalani, A.; Rai, S.; Swarnkar, S.; Tota, S.; Nath, C.; Tyagi, N. Mechanism of oxidative stress and synapse dysfunction in the pathogenesis of Alzheimer’s disease: Understanding the therapeutics strategies. Mol. Neurobiol. 2016, 53, 648–661. [Google Scholar] [CrossRef]
- Cohen, H.; Zohar, J. Animal models of post-traumatic stress disorder: The use of cut off behavioral criteria. Ann. N.Y. Acad. Sci. 2004, 1032, 167–178. [Google Scholar] [CrossRef]
- Lazuko, S.S.; Kuzhel, O.P.; Belyaeva, L.E.; Manukhina, E.B.; Downey, H.F.; Tseilikman, O.B.; Komelkova, M.V.; Tseilikman, V.E. Posttraumatic stress disorder disturbs coronary tone and its regulatory mechanisms. Cell. Mol. Neurobiol. 2018, 38, 209–217. [Google Scholar] [CrossRef]
- Pshennikova, M.G.; Popkova, E.V.; Pokidyshev, D.A.; Khomenko, I.P.; Zelenina, O.M.; Kruglov, S.V.; Manukhina, E.B.; Shimkovich, M.V.; Malyshev, I.Yu. Resistance to stress and neurodegenerative damage in rats of different genetic strains: Protective effects of adaptation to hypoxia. In Adaptation Biology and Medicine. Health Potentials; Lukyanova, L., Takeda, N., Singal, P.K., Eds.; Narosa Publishing House: New Delhi, India, 2008; Volume 5, pp. 153–163. [Google Scholar]
- Cohen, H.; Matar, M.A.; Buskila, D.; Kaplan, Z.; Zohar, J. Early post-stressor intervention with high-dose corticosterone attenuates posttraumatic stress response in an animal model of posttraumatic stress disorder. Biol. Psychiatry. 2008, 64, 708–717. [Google Scholar] [CrossRef]
- Tipton, K.F.; Davey, G.; Motherway, M. Monoamine oxidase assays. Curr. Protoc. Toxicol. 2006, 30, 4–21. [Google Scholar] [CrossRef]
- Satav, J.G.; Katyare, S.S. Effect of streptozotocin-induced diabetes on oxidative energy metabolism in rat liver mitochondria-A comparative study of early and late effects. Indian J. Clin. Biochem. 2004, 19, 23–31. [Google Scholar] [CrossRef]
- Reitman, S.; Frankel, S. A colorimetric method for the determination of serum glutamic oxalacetic and glutamic pyruvic transaminases. Am. J. Clin. Pathol. 1957, 28, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Meyerholz, D.K.; Rodgers, J.; Castilow, E.M.; Varga, S.M. Alcian Blue and Pyronine Y histochemical stains permit assessment of multiple parameters in pulmonary disease models. Vet. Pathol. 2009, 46, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Meyerholz, D.K.; Beck, A.P.; Goeken, A.; Leidinger, M.R.; Ofori‑Amanfo, G.K.; Brown, H.C.; Businga, T.R.; Stoltz, D.A.; Reznikov, L.R.; Flaherty, H.A. Glycogen depletion can increase the specificity of mucin detection in airway tissues. BMC Res. Notes 2018, 11, 763. [Google Scholar] [CrossRef] [PubMed]
- Volchegorskii, I.A.; Rassokhina, L.M.; Miroshnichenko, I.Y. Dynamics of lipid peroxidation—Antioxidant defense system during alloxan diabetes in rats. Bull. Exp. Biol. Med. 2013, 155, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Garland, D.; Oliver, C.N.; Amici, A.; Climent, I.; Lenz, A.G.; Ahn, B.W.; Shaltiel, S.; Stadtman, E.R. Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol. 1990, 186, 464–478. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control (n = 20) | PTSD (n = 20) | IHC (n = 20) | PTSD+IHC (n = 20) | |
|---|---|---|---|---|
| Number of entries into closed arms | 6.4 ± 2.7 | 6.9 ± 2.8 | 7.8 ± 2.6 | 7.0 ± 2.5 |
| Number of entries into open arms | 3.3 ± 1.7 | 3.1 ± 1.6 | 4.2 ± 2.1 | 3.3 ± 0.7 |
| Time spent in closed arms (s) | 525.4 ± 28.3 | 570.0 ± 19.6 *** | 488 ± 72.8 * | 537 ± 15.2 ### |
| Time spent in open arms (s) | 75.6 ± 28.3 | 30.0 ± 19.6 *** | 112 ± 72.8 * | 62.7 ± 15.7 ### |
| Anxiety index | 0.76 ± 0.073 | 0.83 ± 0.051 * | 0.73 ± 0.11 | 0.78 ± 0.043 # |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manukhina, E.B.; Tseilikman, V.E.; Karpenko, M.N.; Pestereva, N.S.; Tseilikman, O.B.; Komelkova, M.V.; Kondashevskaya, M.V.; Goryacheva, A.V.; Lapshin, M.S.; Platkovskii, P.O.; et al. Intermittent Hypoxic Conditioning Alleviates Post-Traumatic Stress Disorder-Induced Damage and Dysfunction of Rat Visceral Organs and Brain. Int. J. Mol. Sci. 2020, 21, 345. https://doi.org/10.3390/ijms21010345
Manukhina EB, Tseilikman VE, Karpenko MN, Pestereva NS, Tseilikman OB, Komelkova MV, Kondashevskaya MV, Goryacheva AV, Lapshin MS, Platkovskii PO, et al. Intermittent Hypoxic Conditioning Alleviates Post-Traumatic Stress Disorder-Induced Damage and Dysfunction of Rat Visceral Organs and Brain. International Journal of Molecular Sciences. 2020; 21(1):345. https://doi.org/10.3390/ijms21010345
Chicago/Turabian StyleManukhina, Eugenia B., Vadim E. Tseilikman, Marina N. Karpenko, Nina S. Pestereva, Olga B. Tseilikman, Maria V. Komelkova, Marina V. Kondashevskaya, Anna V. Goryacheva, Maxim S. Lapshin, Pavel O. Platkovskii, and et al. 2020. "Intermittent Hypoxic Conditioning Alleviates Post-Traumatic Stress Disorder-Induced Damage and Dysfunction of Rat Visceral Organs and Brain" International Journal of Molecular Sciences 21, no. 1: 345. https://doi.org/10.3390/ijms21010345
APA StyleManukhina, E. B., Tseilikman, V. E., Karpenko, M. N., Pestereva, N. S., Tseilikman, O. B., Komelkova, M. V., Kondashevskaya, M. V., Goryacheva, A. V., Lapshin, M. S., Platkovskii, P. O., Sarapultsev, A. P., Alliluev, A. V., & Downey, H. F. (2020). Intermittent Hypoxic Conditioning Alleviates Post-Traumatic Stress Disorder-Induced Damage and Dysfunction of Rat Visceral Organs and Brain. International Journal of Molecular Sciences, 21(1), 345. https://doi.org/10.3390/ijms21010345