Disrupted Calcium Signaling in Animal Models of Human Spinocerebellar Ataxia (SCA)



Abstract
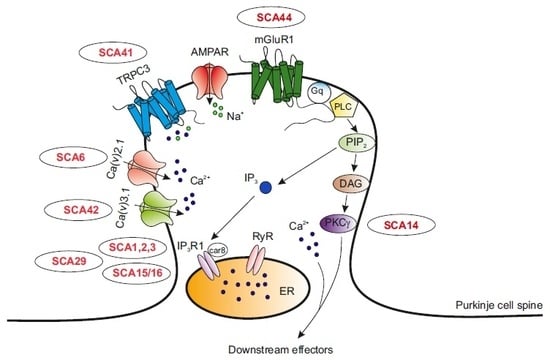
1. Introduction
2. Mutations in the CACNA1A Gene
3. Mutations in the CACNA1G Gene
4. Mutations in the GRID2 Gene
5. Mutations in the PRKCG Gene
6. Mutations in the TRPC3 Gene
7. Mutations in the Itpr1 Gene
8. Mutations in the ATXN2/ATXN3 Genes
9. Mutations in the ATXN1 Gene
10. Mutations in the GMR1 Gene
11. Prospects for Therapeutic Development
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- D’Angelo, E.; Mazzarello, P.; Prestori, F.; Mapelli, J.; Solinas, S.; Lombardo, P.; Cesana, E.; Gandolfi, D.; Congi, L. The cerebellar network: From structure to function and dynamics. Brain Res. Rev. 2011, 66, 5–15. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, E.; Mapelli, L.; Casellato, C.; Garrido, J.A.; Luque, N.; Monaco, J.; Prestori, F.; Pedrocchi, A.; Ros, E. Distributed Circuit Plasticity: New Clues for the Cerebellar Mechanisms of Learning. Cerebellum 2016, 15, 139–151. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, E. The cerebellum gets social. Science 2019, 363, 229. [Google Scholar] [CrossRef] [PubMed]
- Schmahmann, J.D.; Sherman, J.C. The cerebellar cognitive affective syndrome. Brain 1998, 121 Pt 4, 561–579. [Google Scholar] [CrossRef]
- Strick, P.L.; Dum, R.P.; Fiez, J.A. Cerebellum and nonmotor function. Annu. Rev. Neurosci. 2009, 32, 413–434. [Google Scholar] [CrossRef]
- Cendelin, J. From mice to men: Lessons from mutant ataxic mice. Cerebellum Ataxias 2014, 1, 4. [Google Scholar] [CrossRef]
- Manto, M.U. The wide spectrum of spinocerebellar ataxias (SCAs). Cerebellum 2005, 4, 2–6. [Google Scholar] [CrossRef]
- Jayadev, S.; Bird, T.D. Hereditary ataxias: Overview. Genet. Med. 2013, 15, 673–683. [Google Scholar] [CrossRef]
- Sullivan, R.; Yau, W.Y.; O’Connor, E.; Houlden, H. Spinocerebellar ataxia: An update. J. Neurol. 2019, 266, 533–544. [Google Scholar] [CrossRef]
- Paulson, H.L. The spinocerebellar ataxias. J. Neuroophthalmol. 2009, 29, 227–237. [Google Scholar] [CrossRef]
- Matilla-Duenas, A.; Sanchez, I.; Corral-Juan, M.; Davalos, A.; Alvarez, R.; Latorre, P. Cellular and molecular pathways triggering neurodegeneration in the spinocerebellar ataxias. Cerebellum 2010, 9, 148–166. [Google Scholar] [CrossRef] [PubMed]
- Shimobayashi, E.; Kapfhammer, J.P. Calcium Signaling, PKC Gamma, IP3R1 and CAR8 Link Spinocerebellar Ataxias and Purkinje Cell Dendritic Development. Curr. Neuropharmacol. 2018, 16, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Underwood, B.R.; Rubinsztein, D.C. Spinocerebellar ataxias caused by polyglutamine expansions: A review of therapeutic strategies. Cerebellum 2008, 7, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, M.; Banno, H.; Suzuki, K.; Takeuchi, Y.; Kawashima, M.; Tanaka, F.; Adachi, H.; Sobue, G. Molecular genetics and biomarkers of polyglutamine diseases. Curr. Mol. Med. 2008, 8, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Jankovic, J. Spinocerebellar ataxia 8: Variable phenotype and unique pathogenesis. Parkinsonism Relat. Disord. 2009, 15, 621–626. [Google Scholar] [CrossRef]
- Ikeda, Y.; Daughters, R.S.; Ranum, L.P. Bidirectional expression of the SCA8 expansion mutation: One mutation, two genes. Cerebellum 2008, 7, 150–158. [Google Scholar] [CrossRef]
- Klebe, S.; Durr, A.; Rentschler, A.; Hahn-Barma, V.; Abele, M.; Bouslam, N.; Schols, L.; Jedynak, P.; Forlani, S.; Denis, E.; et al. New mutations in protein kinase Cgamma associated with spinocerebellar ataxia type 14. Ann. Neurol. 2005, 58, 720–729. [Google Scholar] [CrossRef]
- Ohata, T.; Yoshida, K.; Sakai, H.; Hamanoue, H.; Mizuguchi, T.; Shimizu, Y.; Okano, T.; Takada, F.; Ishikawa, K.; Mizusawa, H.; et al. A −16C>T substitution in the 5′ UTR of the puratrophin-1 gene is prevalent in autosomal dominant cerebellar ataxia in Nagano. J. Hum. Genet. 2006, 51, 461–466. [Google Scholar] [CrossRef]
- Chen, D.H.; Raskind, W.H.; Bird, T.D. Spinocerebellar ataxia type 14. Handb. Clin. Neurol. 2012, 103, 555–559. [Google Scholar]
- Liu, J.; Tang, T.S.; Tu, H.; Nelson, O.; Herndon, E.; Huynh, D.P.; Pulst, S.M.; Bezprozvanny, I. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J. Neurosci. 2009, 29, 9148–9162. [Google Scholar] [CrossRef]
- Schorge, S.; van de Leemput, J.; Singleton, A.; Houlden, H.; Hardy, J. Human ataxias: A genetic dissection of inositol triphosphate receptor (ITPR1)-dependent signaling. Trends Neurosci. 2010, 33, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Kasumu, A.; Bezprozvanny, I. Deranged calcium signaling in Purkinje cells and pathogenesis in spinocerebellar ataxia 2 (SCA2) and other ataxias. Cerebellum 2012, 11, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Grusser-Cornehls, U.; Baurle, J. Mutant mice as a model for cerebellar ataxia. Prog. Neurobiol. 2001, 63, 489–540. [Google Scholar] [CrossRef]
- Shakkottai, V.G.; Chou, C.H.; Oddo, S.; Sailer, C.A.; Knaus, H.G.; Gutman, G.A.; Barish, M.E.; LaFerla, F.M.; Chandy, K.G. Enhanced neuronal excitability in the absence of neurodegeneration induces cerebellar ataxia. J. Clin. Investig. 2004, 113, 582–590. [Google Scholar] [CrossRef]
- Manto, M.; Marmolino, D. Animal models of human cerebellar ataxias: A cornerstone for the therapies of the twenty-first century. Cerebellum 2009, 8, 137–154. [Google Scholar] [CrossRef]
- Ingram, M.A.; Orr, H.T.; Clark, H.B. Genetically engineered mouse models of the trinucleotide-repeat spinocerebellar ataxias. Brain Res. Bull. 2012, 88, 33–42. [Google Scholar] [CrossRef]
- Ophoff, R.A.; Terwindt, G.M.; Vergouwe, M.N.; van Eijk, R.; Oefner, P.J.; Hoffman, S.M.; Lamerdin, J.E.; Mohrenweiser, H.W.; Bulman, D.E.; Ferrari, M.; et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 1996, 87, 543–552. [Google Scholar] [CrossRef]
- Zhuchenko, O.; Bailey, J.; Bonnen, P.; Ashizawa, T.; Stockton, D.W.; Amos, C.; Dobyns, W.B.; Subramony, S.H.; Zoghbi, H.Y.; Lee, C.C. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat. Genet. 1997, 15, 62–69. [Google Scholar] [CrossRef]
- Pietrobon, D. Biological science of headache channels. Handb. Clin. Neurol. 2010, 97, 73–83. [Google Scholar]
- Tsien, R.W.; Tsien, R.Y. Calcium channels, stores, and oscillations. Annu. Rev. Cell Biol. 1990, 6, 715–760. [Google Scholar] [CrossRef]
- Tsien, R.W.; Ellinor, P.T.; Horne, W.A. Molecular diversity of voltage-dependent Ca2+ channels. Trends Pharmacol. Sci. 1991, 12, 349–354. [Google Scholar] [CrossRef]
- Llinas, R.R.; Sugimori, M.; Cherksey, B. Voltage-dependent calcium conductances in mammalian neurons. The P channel. Ann. N. Y. Acad. Sci. 1989, 560, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Usowicz, M.M.; Sugimori, M.; Cherksey, B.; Llinas, R. P-type calcium channels in the somata and dendrites of adult cerebellar Purkinje cells. Neuron 1992, 9, 1185–1199. [Google Scholar] [CrossRef]
- Mintz, I.M.; Adams, M.E.; Bean, B.P. P-type calcium channels in rat central and peripheral neurons. Neuron 1992, 9, 85–95. [Google Scholar] [CrossRef]
- Westenbroek, R.E.; Sakurai, T.; Elliott, E.M.; Hell, J.W.; Starr, T.V.; Snutch, T.P.; Catterall, W.A. Immunochemical identification and subcellular distribution of the alpha 1A subunits of brain calcium channels. J. Neurosci. 1995, 15, 6403–6418. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Takagi, H.; Miyasho, T.; Inoue, M.; Kirino, Y.; Kudo, Y.; Miyakawa, H. Differential roles of two types of voltage-gated Ca2+ channels in the dendrites of rat cerebellar Purkinje neurons. Brain Res. 1998, 791, 43–55. [Google Scholar] [CrossRef]
- Miyazaki, T.; Hashimoto, K.; Shin, H.S.; Kano, M.; Watanabe, M. P/Q-Type Ca2+ Channel α1A Regulates Synaptic Competition on Developing Cerebellar Purkinje Cells. J. Neurosci. 2004, 24, 1734–1743. [Google Scholar] [CrossRef]
- Noebels, J.L.; Sidman, R.L. Inherited epilepsy: Spike-wave and focal motor seizures in the mutant mouse tottering. Science 1979, 204, 1334–1336. [Google Scholar] [CrossRef]
- Herrup, K.; Wilczynski, S.L. Cerebellar cell degeneration in the leaner mutant mouse. Neuroscience 1982, 7, 2185–2196. [Google Scholar] [CrossRef]
- Campbell, D.B.; North, J.B.; Hess, E.J. Tottering mouse motor dysfunction is abolished on the Purkinje cell degeneration (pcd) mutant background. Exp. Neurol. 1999, 160, 268–278. [Google Scholar] [CrossRef]
- Hoebeek, F.E.; Stahl, J.S.; van Alphen, A.M.; Schonewille, M.; Luo, C.; Rutteman, M.; van den Maagdenberg, A.M.; Molenaar, P.C.; Goossens, H.H.; Frens, M.A.; et al. Increased noise level of purkinje cell activities minimizes impact of their modulation during sensorimotor control. Neuron 2005, 45, 953–965. [Google Scholar] [CrossRef] [PubMed]
- Stahl, J.S.; James, R.A.; Oommen, B.S.; Hoebeek, F.E.; De Zeeuw, C.I. Eye movements of the murine P/Q calcium channel mutant tottering, and the impact of aging. J. Neurophysiol. 2006, 95, 1588–1607. [Google Scholar] [CrossRef] [PubMed]
- Wakamori, M.; Yamazaki, K.; Matsunodaira, H.; Teramoto, T.; Tanaka, I.; Niidome, T.; Sawada, K.; Nishizawa, Y.; Sekiguchi, N.; Mori, E.; et al. Single tottering mutations responsible for the neuropathic phenotype of the P-type calcium channel. J. Biol. Chem. 1998, 273, 34857–34867. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, K.; Wakamori, M.; Rhyu, I.J.; Arii, T.; Oda, S.; Mori, Y.; Imoto, K. Bidirectional alterations in cerebellar synaptic transmission of tottering and rolling Ca2+ channel mutant mice. J. Neurosci. 2002, 22, 4388–4398. [Google Scholar] [CrossRef]
- Meier, H.; MacPike, A.D. Three syndromes produced by two mutant genes in the mouse. Clinical, pathological, and ultrastructural bases of tottering, leaner, and heterozygous mice. J. Hered. 1971, 62, 297–302. [Google Scholar] [CrossRef]
- Isaacs, K.R.; Abbott, L.C. Cerebellar volume decreases in the tottering mouse are specific to the molecular layer. Brain Res. Bull. 1995, 36, 309–314. [Google Scholar] [CrossRef]
- Rhyu, I.J.; Abbott, L.C.; Walker, D.B.; Sotelo, C. An ultrastructural study of granule cell/Purkinje cell synapses in tottering (tg/tg), leaner (tg(la)/tg(la)) and compound heterozygous tottering/leaner (tg/tg(la)) mice. Neuroscience 1999, 90, 717–728. [Google Scholar] [CrossRef]
- Oda, S. The observation of rolling mouse Nagoya (rol), a new neurological mutant, and its maintenance (author’s transl). Jikken Dobutsu 1973, 22, 281–288. [Google Scholar]
- Plomp, J.J.; van den Maagdenberg, A.M.; Kaja, S. The ataxic Cacna1a-mutant mouse rolling nagoya: An overview of neuromorphological and electrophysiological findings. Cerebellum 2009, 8, 222–230. [Google Scholar] [CrossRef][Green Version]
- Heckroth, J.A.; Abbott, L.C. Purkinje cell loss from alternating sagittal zones in the cerebellum of leaner mutant mice. Brain Res. 1994, 658, 93–104. [Google Scholar] [CrossRef]
- Fukumoto, N.; Kitamura, N.; Niimi, K.; Takahashi, E.; Itakura, C.; Shibuya, I. Ca2+ channel currents in dorsal root ganglion neurons of P/Q-type voltage-gated Ca2+ channel mutant mouse, rolling mouse Nagoya. Neurosci. Res. 2012, 73, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Wakamori, M.; Oda, S.; Fletcher, C.F.; Sekiguchi, N.; Mori, E.; Copeland, N.G.; Jenkins, N.A.; Matsushita, K.; Matsuyama, Z.; et al. Reduced voltage sensitivity of activation of P/Q-type Ca2+ channels is associated with the ataxic mouse mutation rolling Nagoya (tg(rol)). J. Neurosci. 2000, 20, 5654–5662. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, S.; Kuramoto, T.; Tanaka, K.; Kaneko, S.; Takeuchi, I.K.; Sasa, M.; Serikawa, T. The ataxic groggy rat has a missense mutation in the P/Q-type voltage-gated Ca2+ channel alpha1A subunit gene and exhibits absence seizures. Brain Res. 2007, 1133, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Clapcote, S.J.; Nieman, B.J.; Tallerico, T.; Huang, Y.; Vukobradovic, I.; Cordes, S.P.; Osborne, L.R.; Rossant, J.; Sled, J.G.; et al. Forward genetic screen of mouse reveals dominant missense mutation in the P/Q-type voltage-dependent calcium channel, CACNA1A. Genes Brain Behav. 2007, 6, 717–727. [Google Scholar] [CrossRef]
- Kojic, M.; Gaik, M.; Kiska, B.; Salerno-Kochan, A.; Hunt, S.; Tedoldi, A.; Mureev, S.; Jones, A.; Whittle, B.; Genovesi, L.A.; et al. Elongator mutation in mice induces neurodegeneration and ataxia-like behavior. Nat. Commun. 2018, 9, 3195. [Google Scholar] [CrossRef]
- Miki, T.; Zwingman, T.A.; Wakamori, M.; Lutz, C.M.; Cook, S.A.; Hosford, D.A.; Herrup, K.; Fletcher, C.F.; Mori, Y.; Frankel, W.N.; et al. Two novel alleles of tottering with distinct Ca(v)2.1 calcium channel neuropathologies. Neuroscience 2008, 155, 31–44. [Google Scholar] [CrossRef]
- Matsuyama, Z.; Wakamori, M.; Mori, Y.; Kawakami, H.; Nakamura, S.; Imoto, K. Direct alteration of the P/Q-type Ca2+ channel property by polyglutamine expansion in spinocerebellar ataxia 6. J. Neurosci. 1999, 19, Rc14. [Google Scholar] [CrossRef]
- Toru, S.; Murakoshi, T.; Ishikawa, K.; Saegusa, H.; Fujigasaki, H.; Uchihara, T.; Nagayama, S.; Osanai, M.; Mizusawa, H.; Tanabe, T. Spinocerebellar ataxia type 6 mutation alters P-type calcium channel function. J. Biol. Chem. 2000, 275, 10893–10898. [Google Scholar] [CrossRef]
- Saegusa, H.; Wakamori, M.; Matsuda, Y.; Wang, J.; Mori, Y.; Zong, S.; Tanabe, T. Properties of human Cav2.1 channel with a spinocerebellar ataxia type 6 mutation expressed in Purkinje cells. Mol. Cell. Neurosci. 2007, 34, 261–270. [Google Scholar] [CrossRef]
- Watase, K.; Barrett, C.F.; Miyazaki, T.; Ishiguro, T.; Ishikawa, K.; Hu, Y.; Unno, T.; Sun, Y.; Kasai, S.; Watanabe, M.; et al. Spinocerebellar ataxia type 6 knockin mice develop a progressive neuronal dysfunction with age-dependent accumulation of mutant CaV2.1 channels. Proc. Natl. Acad. Sci. USA 2008, 105, 11987–11992. [Google Scholar] [CrossRef]
- Watase, K. Spinocerebellar ataxia type 6: Lessons from faithfull knck-in mouse models. Neurol. Clin. Neurosci. 2015, 3, 14–17. [Google Scholar] [CrossRef]
- Jayabal, S.; Ljungberg, L.; Erwes, T.; Cormier, A.; Quilez, S.; El Jaouhari, S.; Watt, A.J. Rapid Onset of Motor Deficits in a Mouse Model of Spinocerebellar Ataxia Type 6 Precedes Late Cerebellar Degeneration1,2,3. eNeuro 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Jayabal, S.; Ljungberg, L.; Watt, A.J. Transient cerebellar alterations during development prior to obvious motor phenotype in a mouse model of spinocerebellar ataxia type 6. J. Physiol. 2017, 595, 949–966. [Google Scholar] [CrossRef] [PubMed]
- Coutelier, M.; Blesneac, I.; Monteil, A.; Monin, M.L.; Ando, K.; Mundwiller, E.; Brusco, A.; Le Ber, I.; Anheim, M.; Castrioto, A.; et al. A Recurrent Mutation in CACNA1G Alters Cav3.1 T-Type Calcium-Channel Conduction and Causes Autosomal-Dominant Cerebellar Ataxia. Am. J. Hum. Genet. 2015, 97, 726–737. [Google Scholar] [CrossRef]
- Morino, H.; Matsuda, Y.; Muguruma, K.; Miyamoto, R.; Ohsawa, R.; Ohtake, T.; Otobe, R.; Watanabe, M.; Maruyama, H.; Hashimoto, K.; et al. A mutation in the low voltage-gated calcium channel CACNA1G alters the physiological properties of the channel, causing spinocerebellar ataxia. Mol. Brain 2015, 8, 89. [Google Scholar] [CrossRef]
- Kimura, M.; Yabe, I.; Hama, Y.; Eguchi, K.; Ura, S.; Tsuzaka, K.; Tsuji, S.; Sasaki, H. SCA42 mutation analysis in a case series of Japanese patients with spinocerebellar ataxia. J. Hum. Genet. 2017, 62, 857–859. [Google Scholar] [CrossRef]
- Ngo, K.; Aker, M.; Petty, L.E.; Chen, J.; Cavalcanti, F.; Nelson, A.B.; Hassin-Baer, S.; Geschwind, M.D.; Perlman, S.; Italiano, D.; et al. Expanding the global prevalence of spinocerebellar ataxia type 42. Neurol. Genet. 2018, 4, e232. [Google Scholar] [CrossRef]
- Talley, E.M.; Cribbs, L.L.; Lee, J.H.; Daud, A.; Perez-Reyes, E.; Bayliss, D.A. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J. Neurosci. 1999, 19, 1895–1911. [Google Scholar] [CrossRef]
- Yunker, A.M. Modulation and pharmacology of low voltage-activated (“T-Type”) calcium channels. J. Bioenerg. Biomembr. 2003, 35, 577–598. [Google Scholar] [CrossRef]
- Craig, P.J.; Beattie, R.E.; Folly, E.A.; Banerjee, M.D.; Reeves, M.B.; Priestley, J.V.; Carney, S.L.; Sher, E.; Perez-Reyes, E.; Volsen, S.G. Distribution of the voltage-dependent calcium channel alpha1G subunit mRNA and protein throughout the mature rat brain. Eur. J. Neurosci. 1999, 11, 2949–2964. [Google Scholar] [CrossRef]
- Molineux, M.L.; McRory, J.E.; McKay, B.E.; Hamid, J.; Mehaffey, W.H.; Rehak, R.; Snutch, T.P.; Zamponi, G.W.; Turner, R.W. Specific T-type calcium channel isoforms are associated with distinct burst phenotypes in deep cerebellar nuclear neurons. Proc. Natl. Acad. Sci. USA 2006, 103, 5555–5560. [Google Scholar] [CrossRef] [PubMed]
- Weiss, N.; Zamponi, G.W. T-type calcium channels: From molecule to therapeutic opportunities. Int. J. Biochem. Cell. Biol. 2019, 108, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Swensen, A.M.; Bean, B.P. Ionic Mechanisms of Burst Firing in Dissociated Purkinje Neurons. J. Neurosci. 2003, 23, 9650–9663. [Google Scholar] [CrossRef] [PubMed]
- Womack, M.D.; Khodakhah, K. Dendritic control of spontaneous bursting in cerebellar Purkinje cells. J. Neurosci. 2004, 24, 3511–3521. [Google Scholar] [CrossRef]
- Hildebrand, M.E.; Isope, P.; Miyazaki, T.; Nakaya, T.; Garcia, E.; Feltz, A.; Schneider, T.; Hescheler, J.; Kano, M.; Sakimura, K.; et al. Functional coupling between mGluR1 and Cav3.1 T-type calcium channels contributes to parallel fiber-induced fast calcium signaling within Purkinje cell dendritic spines. J. Neurosci. 2009, 29, 9668–9682. [Google Scholar] [CrossRef]
- Ly, R.; Bouvier, G.; Schonewille, M.; Arabo, A.; Rondi-Reig, L.; Lena, C.; Casado, M.; De Zeeuw, C.I.; Feltz, A. T-type channel blockade impairs long-term potentiation at the parallel fiber-Purkinje cell synapse and cerebellar learning. Proc. Natl. Acad. Sci. USA 2013, 110, 20302–20307. [Google Scholar] [CrossRef]
- Hashiguchi, S.; Doi, H.; Kunii, M.; Nakamura, Y.; Shimuta, M.; Suzuki, E.; Koyano, S.; Okubo, M.; Kishida, H.; Shiina, M.; et al. Ataxic phenotype with altered CaV3.1 channel property in a mouse model for spinocerebellar ataxia 42. Neurobiol. Dis. 2019, 130, 104516. [Google Scholar] [CrossRef]
- Matsumoto-Makidono, Y.; Nakayama, H.; Yamasaki, M.; Miyazaki, T.; Kobayashi, K.; Watanabe, M.; Kano, M.; Sakimura, K.; Hashimoto, K. Ionic Basis for Membrane Potential Resonance in Neurons of the Inferior Olive. Cell Rep. 2016, 16, 994–1004. [Google Scholar] [CrossRef]
- Choi, S.; Yu, E.; Kim, D.; Urbano, F.J.; Makarenko, V.; Shin, H.S.; Llinas, R.R. Subthreshold membrane potential oscillations in inferior olive neurons are dynamically regulated by P/Q- and T-type calcium channels: A study in mutant mice. J. Physiol. 2010, 588 Pt 16, 3031–3043. [Google Scholar] [CrossRef]
- Kim, D.; Song, I.; Keum, S.; Lee, T.; Jeong, M.J.; Kim, S.S.; McEnery, M.W.; Shin, H.S. Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking alpha(1G) T-type Ca(2+) channels. Neuron 2001, 31, 35–45. [Google Scholar] [CrossRef]
- Park, Y.G.; Park, H.Y.; Lee, C.J.; Choi, S.; Jo, S.; Choi, H.; Kim, Y.H.; Shin, H.S.; Llinas, R.R.; Kim, D. Ca(V)3.1 is a tremor rhythm pacemaker in the inferior olive. Proc. Natl. Acad. Sci. USA 2010, 107, 10731–10736. [Google Scholar] [CrossRef] [PubMed]
- Coutelier, M.; Burglen, L.; Mundwiller, E.; Abada-Bendib, M.; Rodriguez, D.; Chantot-Bastaraud, S.; Rougeot, C.; Cournelle, M.A.; Milh, M.; Toutain, A.; et al. GRID2 mutations span from congenital to mild adult-onset cerebellar ataxia. Neurology 2015, 84, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R. “Lurcher”, a new gene in linkage group XI of the house mouse. J. Genet. 1960, 57, 35–42. [Google Scholar] [CrossRef]
- Takayama, C.; Nakagawa, S.; Watanabe, M.; Mishina, M.; Inoue, Y. Developmental changes in expression and distribution of the glutamate receptor channel delta 2 subunit according to the Purkinje cell maturation. Dev. Brain Res. 1996, 92, 147–155. [Google Scholar] [CrossRef]
- Landsend, A.S.; Amiry-Moghaddam, M.; Matsubara, A.; Bergersen, L.; Usami, S.; Wenthold, R.J.; Ottersen, O.P. Differential localization of delta glutamate receptors in the rat cerebellum: Coexpression with AMPA receptors in parallel fiber-spine synapses and absence from climbing fiber-spine synapses. J. Neurosci. 1997, 17, 834–842. [Google Scholar] [CrossRef]
- Yamazaki, M.; Mori, H.; Araki, K.; Mori, K.J.; Mishina, M. Cloning, expression and modulation of a mouse NMDA receptor subunit. FEBS Lett. 1992, 300, 39–45. [Google Scholar] [CrossRef]
- Hirai, H.; Miyazaki, T.; Kakegawa, W.; Matsuda, S.; Mishina, M.; Watanabe, M.; Yuzaki, M. Rescue of abnormal phenotypes of the delta2 glutamate receptor-null mice by mutant delta2 transgenes. EMBO Rep. 2005, 6, 90–95. [Google Scholar] [CrossRef]
- Kakegawa, W.; Miyazaki, T.; Kohda, K.; Matsuda, K.; Emi, K.; Motohashi, J.; Watanabe, M.; Yuzaki, M. The N-terminal domain of GluD2 (GluRdelta2) recruits presynaptic terminals and regulates synaptogenesis in the cerebellum in vivo. J. Neurosci. 2009, 29, 5738–5748. [Google Scholar] [CrossRef]
- Kashiwabuchi, N.; Ikeda, K.; Araki, K.; Hirano, T.; Shibuki, K.; Takayama, C.; Inoue, Y.; Kutsuwada, T.; Yagi, T.; Kang, Y.; et al. Impairment of motor coordination, Purkinje cell synapse formation, and cerebellar long-term depression in GluR delta 2 mutant mice. Cell 1995, 81, 245–252. [Google Scholar] [CrossRef]
- Uemura, T.; Mishina, M. The amino-terminal domain of glutamate receptor delta2 triggers presynaptic differentiation. Biochem. Biophys. Res. Commun. 2008, 377, 1315–1319. [Google Scholar] [CrossRef]
- Kurihara, H.; Hashimoto, K.; Kano, M.; Takayama, C.; Sakimura, K.; Mishina, M.; Inoue, Y.; Watanabe, M. Impaired parallel fiber→Purkinje cell synapse stabilization during cerebellar development of mutant mice lacking the glutamate receptor delta2 subunit. J. Neurosci. 1997, 17, 9613–9623. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, R.; Miyazaki, T.; Kano, M.; Hashikawa, T.; Tatsumi, H.; Sakimura, K.; Mishina, M.; Inoue, Y.; Watanabe, M. Distal extension of climbing fiber territory and multiple innervation caused by aberrant wiring to adjacent spiny branchlets in cerebellar Purkinje cells lacking glutamate receptor delta 2. J. Neurosci. 2002, 22, 8487–8503. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, Y.; Yoshioka, Y.; Suzuki, K.; Miyazaki, T.; Koura, M.; Saigoh, K.; Kajimura, N.; Monobe, Y.; Kusunoki, S.; Matsuda, J.; et al. A new mouse allele of glutamate receptor delta 2 with cerebellar atrophy and progressive ataxia. PLoS ONE 2014, 9, e107867. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.S.; Heintz, N. Massive loss of mid- and hindbrain neurons during embryonic development of homozygous lurcher mice. J. Neurosci. 1997, 17, 2400–2407. [Google Scholar] [CrossRef] [PubMed]
- Lalonde, R.; Botez, M.I.; Joyal, C.C.; Caumartin, M. Motor abnormalities in lurcher mutant mice. Physiol. Behav. 1992, 51, 523–525. [Google Scholar] [CrossRef]
- Vogel, M.W.; Caston, J.; Yuzaki, M.; Mariani, J. The Lurcher mouse: Fresh insights from an old mutant. Brain Res. 2007, 1140, 4–18. [Google Scholar] [CrossRef]
- Zuo, J.; De Jager, P.L.; Takahashi, K.A.; Jiang, W.; Linden, D.J.; Heintz, N. Neurodegeneration in Lurcher mice caused by mutation in delta2 glutamate receptor gene. Nature 1997, 388, 769–773. [Google Scholar] [CrossRef]
- Wollmuth, L.P.; Kuner, T.; Jatzke, C.; Seeburg, P.H.; Heintz, N.; Zuo, J. The Lurcher mutation identifies delta 2 as an AMPA/kainate receptor-like channel that is potentiated by Ca(2+). J. Neurosci. 2000, 20, 5973–5980. [Google Scholar] [CrossRef]
- Nishiyama, J.; Matsuda, K.; Kakegawa, W.; Yamada, N.; Motohashi, J.; Mizushima, N.; Yuzaki, M. Reevaluation of neurodegeneration in lurcher mice: Constitutive ion fluxes cause cell death with, not by, autophagy. J. Neurosci. 2010, 30, 2177–2187. [Google Scholar] [CrossRef]
- Nishiyama, J.; Yuzaki, M. Excitotoxicity and autophagy: Lurcher may not be a model of “autophagic cell death”. Autophagy 2010, 6, 568–570. [Google Scholar] [CrossRef][Green Version]
- Chen, D.H.; Brkanac, Z.; Verlinde, C.L.; Tan, X.J.; Bylenok, L.; Nochlin, D.; Matsushita, M.; Lipe, H.; Wolff, J.; Fernandez, M.; et al. Missense mutations in the regulatory domain of PKC gamma: A new mechanism for dominant nonepisodic cerebellar ataxia. Am. J. Hum. Genet. 2003, 72, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Guerra, G.; Lucariello, A.; Perna, A.; Botta, L.; De Luca, A.; Moccia, F. The Role of Endothelial Ca(2+) Signaling in Neurovascular Coupling: A View from the Lumen. Int. J. Mol. Sci. 2018, 19, 938. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Shirai, Y. Protein kinase C gamma (PKC gamma): Function of neuron specific isotype. J. Biochem. 2002, 132, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H. Protein Kinase C in the Cerebellum: Its Significance and Remaining Conundrums. Cerebellum 2018, 17, 23–27. [Google Scholar] [CrossRef]
- Moriya, M.; Tanaka, S. Prominent expression of protein kinase C (gamma) mRNA in the dendrite-rich neuropil of mice cerebellum at the critical period for synaptogenesis. Neuroreport 1994, 5, 929–932. [Google Scholar] [CrossRef]
- Kano, M.; Hashimoto, K.; Chen, C.; Abeliovich, A.; Aiba, A.; Kurihara, H.; Watanabe, M.; Inoue, Y.; Tonegawa, S. Impaired synapse elimination during cerebellar development in PKC gamma mutant mice. Cell 1995, 83, 1223–1231. [Google Scholar] [CrossRef]
- Barmack, N.H.; Qian, Z.; Yoshimura, J. Regional and cellular distribution of protein kinase C in rat cerebellar Purkinje cells. J. Comp. Neurol. 2000, 427, 235–254. [Google Scholar] [CrossRef]
- Chen, C.; Kano, M.; Abeliovich, A.; Chen, L.; Bao, S.; Kim, J.J.; Hashimoto, K.; Thompson, R.F.; Tonegawa, S. Impaired motor coordination correlates with persistent multiple climbing fiber innervation in PKC gamma mutant mice. Cell 1995, 83, 1233–1242. [Google Scholar] [CrossRef]
- De Zeeuw, C.I.; Simpson, J.I.; Hoogenraad, C.C.; Galjart, N.; Koekkoek, S.K.; Ruigrok, T.J. Microcircuitry and function of the inferior olive. Trends Neurosci. 1998, 21, 391–400. [Google Scholar] [CrossRef]
- Van de Warrenburg, B.P.; Verbeek, D.S.; Piersma, S.J.; Hennekam, F.A.; Pearson, P.L.; Knoers, N.V.; Kremer, H.P.; Sinke, R.J. Identification of a novel SCA14 mutation in a Dutch autosomal dominant cerebellar ataxia family. Neurology 2003, 61, 1760–1765. [Google Scholar] [CrossRef]
- Brkanac, Z.; Bylenok, L.; Fernandez, M.; Matsushita, M.; Lipe, H.; Wolff, J.; Nochlin, D.; Raskind, W.H.; Bird, T.D. A new dominant spinocerebellar ataxia linked to chromosome 19q13.4-qter . Arch. Neurol. 2002, 59, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, D.S.; Knight, M.A.; Harmison, G.G.; Fischbeck, K.H.; Howell, B.W. Protein kinase C gamma mutations in spinocerebellar ataxia 14 increase kinase activity and alter membrane targeting. Brain 2005, 128 Pt 2, 436–442. [Google Scholar] [CrossRef]
- Adachi, N.; Kobayashi, T.; Takahashi, H.; Kawasaki, T.; Shirai, Y.; Ueyama, T.; Matsuda, T.; Seki, T.; Sakai, N.; Saito, N. Enzymological analysis of mutant protein kinase Cgamma causing spinocerebellar ataxia type 14 and dysfunction in Ca2+ homeostasis. J. Biol. Chem. 2008, 283, 19854–19863. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Lucariello, A.; Guerra, G. TRPC3-mediated Ca(2+) signals as a promising strategy to boost therapeutic angiogenesis in failing hearts: The role of autologous endothelial colony forming cells. J. Cell. Physiol. 2018, 233, 3901–3917. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, D.S.; Goedhart, J.; Bruinsma, L.; Sinke, R.J.; Reits, E.A. PKC gamma mutations in spinocerebellar ataxia type 14 affect C1 domain accessibility and kinase activity leading to aberrant MAPK signaling. J. Cell. Sci. 2008, 121 Pt 14, 2339–2349. [Google Scholar] [CrossRef]
- Toulouse, A.; Nolan, Y.M. A role for mitogen-activated protein kinase phosphatase 1 (MKP1) in neural cell development and survival. Neural Regen. Res. 2015, 10, 1748–1749. [Google Scholar] [CrossRef]
- Zhang, Y.; Snider, A.; Willard, L.; Takemoto, D.J.; Lin, D. Loss of Purkinje cells in the PKCgamma H101Y transgenic mouse. Biochem. Biophys. Res. Commun. 2009, 378, 524–528. [Google Scholar] [CrossRef]
- Ji, J.; Hassler, M.L.; Shimobayashi, E.; Paka, N.; Streit, R.; Kapfhammer, J.P. Increased protein kinase C gamma activity induces Purkinje cell pathology in a mouse model of spinocerebellar ataxia 14. Neurobiol. Dis. 2014, 70, 1–11. [Google Scholar] [CrossRef]
- Becker, E.B.E.; Oliver, P.L.; Glitsch, M.D.; Banks, G.T.; Achilli, F.; Hardy, A.; Nolan, P.M.; Fisher, E.M.C.; Davies, K.E. A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in moonwalker mice. Proc. Natl. Acad. Sci. USA 2009, 106, 6706–6711. [Google Scholar] [CrossRef]
- Dulneva, A.; Lee, S.; Oliver, P.L.; Di Gleria, K.; Kessler, B.M.; Davies, K.E.; Becker, E.B. The mutant Moonwalker TRPC3 channel links calcium signaling to lipid metabolism in the developing cerebellum. Hum. Mol. Genet. 2015, 24, 4114–4125. [Google Scholar] [CrossRef]
- Becker, E.B. The Moonwalker mouse: New insights into TRPC3 function, cerebellar development, and ataxia. Cerebellum 2014, 13, 628–636. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gees, M.; Colsoul, B.; Nilius, B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a003962. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.; Konnerth, A. TRPC3-dependent synaptic transmission in central mammalian neurons. J. Mol. Med. 2015, 93, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J. TRPC3 channel underlies cerebellar long-term depression. Cerebellum 2013, 12, 334–337. [Google Scholar] [CrossRef]
- Becker, E.B.E. From mice to men: TRPC3 in cerebellar ataxia. Cerebellum 2017, 16, 877–879. [Google Scholar] [CrossRef]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Lodola, F.; Bottino, C.; Guerra, G.; Borghesi, A.; Stronati, M.; Rosti, V.; Tanzi, F.; et al. Canonical transient receptor potential 3 channel triggers vascular endothelial growth factor-induced intracellular Ca2+ oscillations in endothelial progenitor cells isolated from umbilical cord blood. Stem Cells Dev. 2013, 22, 2561–2580. [Google Scholar] [CrossRef]
- Moccia, F.; Negri, S.; Shekha, M.; Faris, P.; Guerra, G. Endothelial Ca2+ Signaling, Angiogenesis and Vasculogenesis: Just What It Takes to Make a Blood Vessel. Int. J. Mol. Sci. 2019, 20, 3962. [Google Scholar] [CrossRef]
- Sekerková, G.; Kim, J.A.; Nigro, M.J.; Becker, E.B.E.; Hartmann, J.; Birnbaumer, L.; Mugnaini, E.; Martina, M. Early Onset of Ataxia in Moonwalker Mice Is Accompanied by Complete Ablation of Type II Unipolar Brush Cells and Purkinje Cell Dysfunction. J. Neurosci. 2013, 33, 19689–19694. [Google Scholar] [CrossRef]
- Hartmann, J.; Dragicevic, E.; Adelsberger, H.; Henning, H.A.; Sumser, M.; Abramowitz, J.; Blum, R.; Dietrich, A.; Freichel, M.; Flockerzi, V.; et al. TRPC3 Channels Are Required for Synaptic Transmission and Motor Coordination. Neuron 2008, 59, 392–398. [Google Scholar] [CrossRef]
- Huang, W.C.; Young, J.S.; Glitsch, M.D. Changes in TRPC channel expression during postnatal development of cerebellar neurons. Cell Calcium 2007, 42, 1–10. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Pellavio, G.; Botta, L.; Orgiu, M.; Forcaia, G.; Sancini, G.; Laforenza, U.; Moccia, F. Group 1 metabotropic glutamate receptors trigger glutamate-induced intracellular Ca2+ signals and nitric oxide release in human brain microvascular endothelial cells. Cell. Mol. Life Sci. 2019, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Zuccolo, E.; Soda, T.; Tanzi, F.; Guerra, G.; Mapelli, L.; Lodola, F.; D’Angelo, E. Stim and Orai proteins in neuronal Ca2+ signaling and excitability. Front. Cell. Neurosci. 2015, 9, 153. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.T.; Ong, H.L.; Liu, X.; Ambudkar, I.S. Contribution and Regulation of TRPC Channels in Store-Operated Ca2+ Entry. Curr. Top. Membr. 2013, 71, 149–179. [Google Scholar] [PubMed]
- Lockwich, T.; Makusky, A.; Kowalak, J.; Markey, S.; Ambudkar, I. Proteomic analysis of TRPC channels. In TRP Channels; Zhu, M., Ed.; CRC Press: Boca Raton, FL, USA, 2011. [Google Scholar]
- Spassova, M.A.; Soboloff, J.; He, L.P.; Xu, W.; Dziadek, M.A.; Gill, D.L. STIM1 has a plasma membrane role in the activation of store-operated Ca2+ channels. Proc. Natl. Acad. Sci. USA 2006, 103, 4040–4045. [Google Scholar] [CrossRef] [PubMed]
- Soboloff, J.; Spassova, T.; Hewavitharana, L.; HE, L.; Luncsford, W.; Xu, K.; Venkatachalam, D.; van Rossum, R.; Patterson, D.; Gill, L. TRPC channels: Integrators of multiple cellular signals. In Transient Receptor Potential (TRP) Channels; Flockerzi, V., Nilius, B., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; Volume 179, pp. 575–591. [Google Scholar]
- Zhou, H.; Lin, Z.; Voges, K.; Ju, C.; Gao, Z.; Bosman, L.W.; Ruigrok, T.J.; Hoebeek, F.E.; De Zeeuw, C.I.; Schonewille, M. Cerebellar modules operate at different frequencies. eLife 2014, 3, e02536. [Google Scholar] [CrossRef] [PubMed]
- Konur, S.; Ghosh, A. Calcium signaling and the control of dendritic development. Neuron 2005, 46, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Schrenk, K.; Kapfhammer, J.P.; Metzger, F. Altered dendritic development of cerebellar Purkinje cells in slice cultures from protein kinase Cgamma-deficient mice. Neuroscience 2002, 110, 675–689. [Google Scholar] [CrossRef]
- Lopez-Bendito, G.; Shigemoto, R.; Lujan, R.; Juiz, J.M. Developmental changes in the localisation of the mGluR1alpha subtype of metabotropic glutamate receptors in Purkinje cells. Neuroscience 2001, 105, 413–429. [Google Scholar] [CrossRef]
- Venkatachalam, K.; Zheng, F.; Gill, D.L. Regulation of canonical transient receptor potential (TRPC) channel function by diacylglycerol and protein kinase C. J. Biol. Chem. 2003, 278, 29031–29040. [Google Scholar] [CrossRef]
- Trebak, M.; Hempel, N.; Wedel, B.J.; Smyth, J.T.; Bird, G.S.; Putney, J.W., Jr. Negative regulation of TRPC3 channels by protein kinase C-mediated phosphorylation of serine 712. Mol. Pharmacol. 2005, 67, 558–563. [Google Scholar] [CrossRef]
- Glitsch, M.D. Activation of native TRPC3 cation channels by phospholipase D. FASEB J. 2010, 24, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Schmid, S.M.; Hollmann, M. To gate or not to gate: Are the delta subunits in the glutamate receptor family functional ion channels? Mol. Neurobiol. 2008, 37, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Ady, V.; Perroy, J.; Tricoire, L.; Piochon, C.; Dadak, S.; Chen, X.; Dusart, I.; Fagni, L.; Lambolez, B.; Levenes, C. Type 1 metabotropic glutamate receptors (mGlu1) trigger the gating of GluD2 delta glutamate receptors. EMBO Rep. 2014, 15, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.S.; Knierman, M.D.; Siuda, E.R.; Isaac, J.T.R.; Nisenbaum, E.S.; Bredt, D.S. Glutamate Receptor δ2 Associates with Metabotropic Glutamate Receptor 1 (mGluR1), Protein Kinase Cγ, and Canonical Transient Receptor Potential 3 and Regulates mGluR1-Mediated Synaptic Transmission in Cerebellar Purkinje Neurons. J. Neurosci. 2012, 32, 15296–15308. [Google Scholar] [CrossRef] [PubMed]
- Ingram, M.; Wozniak, E.A.L.; Duvick, L.; Yang, R.; Bergmann, P.; Carson, R.; O’Callaghan, B.; Zoghbi, H.Y.; Henzler, C.; Orr, H.T. Cerebellar Transcriptome Profiles of ATXN1 Transgenic Mice Reveal SCA1 Disease Progression and Protection Pathways. Neuron 2016, 89, 1194–1207. [Google Scholar] [CrossRef]
- Lin, X.; Antalffy, B.; Kang, D.; Orr, H.T.; Zoghbi, H.Y. Polyglutamine expansion down-regulates specific neuronal genes before pathologic changes in SCA1. Nat. Neurosci. 2000, 3, 157–163. [Google Scholar] [CrossRef]
- Pflieger, L.T.; Dansithong, W.; Paul, S.; Scoles, D.R.; Figueroa, K.P.; Meera, P.; Otis, T.S.; Facelli, J.C.; Pulst, S.M. Gene co-expression network analysis for identifying modules and functionally enriched pathways in SCA2. Hum. Mol. Genet. 2017, 26, 3069–3080. [Google Scholar] [CrossRef]
- Fogel, B.; Hanson, S.; Becker, E. Mutation of the Murine Ataxia Gene TRPC3 Causes Cerebellar Ataxia in Humans (P1.009). Neurology 2016, 86, P1.009. [Google Scholar]
- Fogel, B.L.; Hanson, S.M.; Becker, E.B.E. Do Mutations in the Murine Ataxia Gene TRPC3 Cause Cerebellar Ataxia in Humans? Mov. Disord. 2015, 30, 284–286. [Google Scholar] [CrossRef]
- Becker, E.B.; Fogel, B.L.; Rajakulendran, S.; Dulneva, A.; Hanna, M.G.; Perlman, S.L.; Geschwind, D.H.; Davies, K.E. Candidate screening of the TRPC3 gene in cerebellar ataxia. Cerebellum 2011, 10, 296–299. [Google Scholar] [CrossRef]
- Parys, J.B.; De Smedt, H. Inositol 1,4,5-trisphosphate and its receptors. Adv. Exp. Med. Biol. 2012, 740, 255–279. [Google Scholar]
- Foskett, J.K.; White, C.; Cheung, K.H.; Mak, D.O. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007, 87, 593–658. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.W.; Tovey, S.C. IP(3) receptors: Toward understanding their activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a004010. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, H.; Vervliet, T.; Missiaen, L.; Parys, J.B.; De Smedt, H.; Bultynck, G. Inositol 1,4,5-trisphosphate receptor-isoform diversity in cell death and survival. Biochim. Biophys. Acta 2014, 1843, 2164–2183. [Google Scholar] [CrossRef] [PubMed]
- Vermassen, E.; Parys, J.B.; Mauger, J.P. Subcellular distribution of the inositol 1,4,5-trisphosphate receptors: Functional relevance and molecular determinants. Biol. Cell 2004, 96, 3–17. [Google Scholar] [CrossRef]
- Sharp, A.H.; Nucifora, F.C., Jr.; Blondel, O.; Sheppard, C.A.; Zhang, C.; Snyder, S.H.; Russell, J.T.; Ryugo, D.K.; Ross, C.A. Differential cellular expression of isoforms of inositol 1,4,5-triphosphate receptors in neurons and glia in brain. J. Comp. Neurol. 1999, 406, 207–220. [Google Scholar] [CrossRef]
- Nakanishi, S.; Maeda, N.; Mikoshiba, K. Immunohistochemical localization of an inositol 1,4,5-trisphosphate receptor, P400, in neural tissue: Studies in developing and adult mouse brain. J. Neurosci. 1991, 11, 2075–2086. [Google Scholar] [CrossRef]
- Yamada, N.; Makino, Y.; Clark, R.A.; Pearson, D.W.; Mattei, M.G.; Guenet, J.L.; Ohama, E.; Fujino, I.; Miyawaki, A.; Furuichi, T.; et al. Human inositol 1,4,5-trisphosphate type-1 receptor, InsP3R1: Structure, function, regulation of expression and chromosomal localization. Biochem. J. 1994, 302 Pt 3, 781–790. [Google Scholar] [CrossRef]
- Bezprozvanny, I. Role of inositol 1,4,5-trisphosphate receptors in pathogenesis of Huntington’s disease and spinocerebellar ataxias. Neurochem. Res. 2011, 36, 1186–1197. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium signalling and Alzheimer’s disease. Neurochem. Res. 2011, 36, 1149–1156. [Google Scholar] [CrossRef]
- Synofzik, M.; Beetz, C.; Bauer, C.; Bonin, M.; Sanchez-Ferrero, E.; Schmitz-Hubsch, T.; Wullner, U.; Nagele, T.; Riess, O.; Schols, L.; et al. Spinocerebellar ataxia type 15: Diagnostic assessment, frequency, and phenotypic features. J. Med. Genet. 2011, 48, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Van de Leemput, J.; Chandran, J.; Knight, M.A.; Holtzclaw, L.A.; Scholz, S.; Cookson, M.R.; Houlden, H.; Gwinn-Hardy, K.; Fung, H.C.; Lin, X.; et al. Deletion at ITPR1 underlies ataxia in mice and spinocerebellar ataxia 15 in humans. PLoS Genet. 2007, 3, e108. [Google Scholar] [CrossRef] [PubMed]
- Storey, E.; Gardner, R.J.; Knight, M.A.; Kennerson, M.L.; Tuck, R.R.; Forrest, S.M.; Nicholson, G.A. A new autosomal dominant pure cerebellar ataxia. Neurology 2001, 57, 1913–1915. [Google Scholar] [CrossRef] [PubMed]
- Marelli, C.; van de Leemput, J.; Johnson, J.O.; Tison, F.; Thauvin-Robinet, C.; Picard, F.; Tranchant, C.; Hernandez, D.G.; Huttin, B.; Boulliat, J.; et al. SCA15 due to large ITPR1 deletions in a cohort of 333 Caucasian families with dominant ataxia. Arch. Neurol. 2011, 68, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Di Gregorio, E.; Orsi, L.; Godani, M.; Vaula, G.; Jensen, S.; Salmon, E.; Ferrari, G.; Squadrone, S.; Abete, M.C.; Cagnoli, C.; et al. Two Italian families with ITPR1 gene deletion presenting a broader phenotype of SCA15. Cerebellum 2010, 9, 115–123. [Google Scholar] [CrossRef]
- Hara, K.; Shiga, A.; Nozaki, H.; Mitsui, J.; Takahashi, Y.; Ishiguro, H.; Yomono, H.; Kurisaki, H.; Goto, J.; Ikeuchi, T.; et al. Total deletion and a missense mutation of ITPR1 in Japanese SCA15 families. Neurology 2008, 71, 547–551. [Google Scholar] [CrossRef]
- Iwaki, A.; Kawano, Y.; Miura, S.; Shibata, H.; Matsuse, D.; Li, W.; Furuya, H.; Ohyagi, Y.; Taniwaki, T.; Kira, J.; et al. Heterozygous deletion of ITPR1, but not SUMF1, in spinocerebellar ataxia type 16. J. Med. Genet. 2008, 45, 32–35. [Google Scholar] [CrossRef]
- Gardner, R.J. “SCA16” is really SCA15. J. Med. Genet. 2008, 45, 192. [Google Scholar] [CrossRef]
- Zambonin, J.L.; Bellomo, A.; Ben-Pazi, H.; Everman, D.B.; Frazer, L.M.; Geraghty, M.T.; Harper, A.D.; Jones, J.R.; Kamien, B.; Kernohan, K.; et al. Spinocerebellar ataxia type 29 due to mutations in ITPR1: A case series and review of this emerging congenital ataxia. Orphanet J. Rare Dis. 2017, 12, 121. [Google Scholar] [CrossRef]
- Huang, L.; Chardon, J.W.; Carter, M.T.; Friend, K.L.; Dudding, T.E.; Schwartzentruber, J.; Zou, R.; Schofield, P.W.; Douglas, S.; Bulman, D.E.; et al. Missense mutations in ITPR1 cause autosomal dominant congenital nonprogressive spinocerebellar ataxia. Orphanet J. Rare Dis. 2012, 7, 67. [Google Scholar] [CrossRef]
- Hirota, J.; Ando, H.; Hamada, K.; Mikoshiba, K. Carbonic anhydrase-related protein is a novel binding protein for inositol 1,4,5-trisphosphate receptor type 1. Biochem. J. 2003, 372, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Kerkhofs, M.; Seitaj, B.; Ivanova, H.; Monaco, G.; Bultynck, G.; Parys, J.B. Pathophysiological consequences of isoform-specific IP3 receptor mutations. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1707–1717. [Google Scholar] [CrossRef] [PubMed]
- Street, V.A.; Bosma, M.M.; Demas, V.P.; Regan, M.R.; Lin, D.D.; Robinson, L.C.; Agnew, W.S.; Tempel, B.L. The type 1 inositol 1,4,5-trisphosphate receptor gene is altered in the opisthotonos mouse. J. Neurosci. 1997, 17, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Patterson, R.L.; Boehning, D.; Snyder, S.H. Inositol 1,4,5-trisphosphate receptors as signal integrators. Annu. Rev. Biochem. 2004, 73, 437–465. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Miyakawa, T.; Wang, Z.; Glouchankova, L.; Iino, M.; Bezprozvanny, I. Functional characterization of the type 1 inositol 1,4,5-trisphosphate receptor coupling domain SII(+/−) splice variants and the Opisthotonos mutant form. Biophys. J. 2002, 82, 1995–2004. [Google Scholar] [CrossRef]
- Matsumoto, M.; Nakagawa, T.; Inoue, T.; Nagata, E.; Tanaka, K.; Takano, H.; Minowa, O.; Kuno, J.; Sakakibara, S.; Yamada, M.; et al. Ataxia and epileptic seizures in mice lacking type 1 inositol 1,4,5-trisphosphate receptor. Nature 1996, 379, 168–171. [Google Scholar] [CrossRef]
- Matsumoto, M.; Nagata, E. Type 1 inositol 1,4,5-trisphosphate receptor knock-out mice: Their phenotypes and their meaning in neuroscience and clinical practice. J. Mol. Med. 1999, 77, 406–411. [Google Scholar] [CrossRef]
- Inoue, T.; Kato, K.; Kohda, K.; Mikoshiba, K. Type 1 inositol 1,4,5-trisphosphate receptor is required for induction of long-term depression in cerebellar Purkinje neurons. J. Neurosci. 1998, 18, 5366–5373. [Google Scholar] [CrossRef]
- Hisatsune, C.; Miyamoto, H.; Hirono, M.; Yamaguchi, N.; Sugawara, T.; Ogawa, N.; Ebisui, E.; Ohshima, T.; Yamada, M.; Hensch, T.K.; et al. IP3R1 deficiency in the cerebellum/brainstem causes basal ganglia-independent dystonia by triggering tonic Purkinje cell firings in mice. Front. Neural Circuits 2013, 7, 156. [Google Scholar] [CrossRef]
- Klar, J.; Ali, Z.; Farooq, M.; Khan, K.; Wikström, J.; Iqbal, M.; Zulfiqar, S.; Faryal, S.; Baig, S.M.; Dahl, N. A missense variant in ITPR1 provides evidence for autosomal recessive SCA29 with asymptomatic cerebellar hypoplasia in carriers. Eur. J. Hum. Genet. 2017, 25, 848–853. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Okamoto, T.; Taniwaki, M.; Aizawa, M.; Inoue, M.; Katayama, S.; Kawakami, H.; Nakamura, S.; Nishimura, M.; Akiguchi, I.; et al. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat. Genet. 1994, 8, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Imbert, G.; Saudou, F.; Yvert, G.; Devys, D.; Trottier, Y.; Garnier, J.M.; Weber, C.; Mandel, J.L.; Cancel, G.; Abbas, N.; et al. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat. Genet. 1996, 14, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Pulst, S.M.; Nechiporuk, A.; Nechiporuk, T.; Gispert, S.; Chen, X.N.; Lopes-Cendes, I.; Pearlman, S.; Starkman, S.; Orozco-Diaz, G.; Lunkes, A.; et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat. Genet. 1996, 14, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Sanpei, K.; Takano, H.; Igarashi, S.; Sato, T.; Oyake, M.; Sasaki, H.; Wakisaka, A.; Tashiro, K.; Ishida, Y.; Ikeuchi, T.; et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat. Genet. 1996, 14, 277–284. [Google Scholar] [CrossRef]
- Paulson, H.L.; Das, S.S.; Crino, P.B.; Perez, M.K.; Patel, S.C.; Gotsdiner, D.; Fischbeck, K.H.; Pittman, R.N. Machado-Joseph disease gene product is a cytoplasmic protein widely expressed in brain. Ann. Neurol. 1997, 41, 453–462. [Google Scholar] [CrossRef]
- Trottier, Y.; Cancel, G.; An-Gourfinkel, I.; Lutz, Y.; Weber, C.; Brice, A.; Hirsch, E.; Mandel, J.L. Heterogeneous intracellular localization and expression of ataxin-3. Neurobiol. Dis. 1998, 5, 335–347. [Google Scholar] [CrossRef]
- Huynh, D.P.; Figueroa, K.; Hoang, N.; Pulst, S.M. Nuclear localization or inclusion body formation of ataxin-2 are not necessary for SCA2 pathogenesis in mouse or human. Nat. Genet. 2000, 26, 44–50. [Google Scholar] [CrossRef]
- Schols, L.; Bauer, P.; Schmidt, T.; Schulte, T.; Riess, O. Autosomal dominant cerebellar ataxias: Clinical features, genetics, and pathogenesis. Lancet Neurol. 2004, 3, 291–304. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Rub, U.; Auburger, G. Spinocerebellar ataxia 2 (SCA2). Cerebellum 2008, 7, 115–124. [Google Scholar] [CrossRef]
- Koeppen, A.H. The Neuropathology of Spinocerebellar Ataxia Type 3/Machado-Joseph Disease. Adv. Exp. Med. Biol. 2018, 1049, 233–241. [Google Scholar]
- Coutinho, P.; Andrade, C. Autosomal dominant system degeneration in Portuguese families of the Azores Islands. A new genetic disorder involving cerebellar, pyramidal, extrapyramidal and spinal cord motor functions. Neurology 1978, 28, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Pulst, S.M.; Santos, N.; Wang, D.; Yang, H.; Huynh, D.; Velazquez, L.; Figueroa, K.P. Spinocerebellar ataxia type 2: PolyQ repeat variation in the CACNA1A calcium channel modifies age of onset. Brain 2005, 128 Pt 10, 2297–2303. [Google Scholar] [CrossRef][Green Version]
- Kiehl, T.R.; Nechiporuk, A.; Figueroa, K.P.; Keating, M.T.; Huynh, D.P.; Pulst, S.M. Generation and characterization of Sca2 (ataxin-2) knockout mice. Biochem. Biophys. Res. Commun. 2006, 339, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Egorova, P.A.; Zakharova, O.A.; Vlasova, O.L.; Bezprozvanny, I.B. In vivo analysis of cerebellar Purkinje cell activity in SCA2 transgenic mouse model. J. Neurophysiol. 2016, 115, 2840–2851. [Google Scholar] [CrossRef] [PubMed]
- Kasumu, A.W.; Liang, X.; Egorova, P.; Vorontsova, D.; Bezprozvanny, I. Chronic suppression of inositol 1,4,5-triphosphate receptor-mediated calcium signaling in cerebellar purkinje cells alleviates pathological phenotype in spinocerebellar ataxia 2 mice. J. Neurosci. 2012, 32, 12786–12796. [Google Scholar] [CrossRef] [PubMed]
- Edgerton, J.R.; Reinhart, P.H. Distinct contributions of small and large conductance Ca2+-activated K+ channels to rat Purkinje neuron function. J. Physiol. 2003, 548 Pt 1, 53–69. [Google Scholar] [CrossRef]
- Womack, M.D.; Khodakhah, K. Somatic and dendritic small-conductance calcium-activated potassium channels regulate the output of cerebellar Purkinje neurons. J. Neurosci. 2003, 23, 2600–2607. [Google Scholar] [CrossRef]
- Kasumu, A.W.; Hougaard, C.; Rode, F.; Jacobsen, T.A.; Sabatier, J.M.; Eriksen, B.L.; Strobaek, D.; Liang, X.; Egorova, P.; Vorontsova, D.; et al. Selective positive modulator of calcium-activated potassium channels exerts beneficial effects in a mouse model of spinocerebellar ataxia type 2. Chem. Biol. 2012, 19, 1340–1353. [Google Scholar] [CrossRef]
- Huynh, D.P.; Del Bigio, M.R.; Ho, D.H.; Pulst, S.M. Expression of ataxin-2 in brains from normal individuals and patients with Alzheimer’s disease and spinocerebellar ataxia 2. Ann. Neurol. 1999, 45, 232–241. [Google Scholar] [CrossRef]
- La Spada, A.R.; Taylor, J.P. Repeat expansion disease: Progress and puzzles in disease pathogenesis. Nat. Rev. Genet. 2010, 11, 247–258. [Google Scholar] [CrossRef]
- Bauer, P.O.; Nukina, N. The pathogenic mechanisms of polyglutamine diseases and current therapeutic strategies. J. Neurochem. 2009, 110, 1737–1765. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tang, T.S.; Tu, H.; Nelson, O.; Pook, M.; Hammer, R.; Nukina, N.; Bezprozvanny, I. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 3. J. Neurosci. 2008, 28, 12713–12724. [Google Scholar] [CrossRef] [PubMed]
- Haacke, A.; Hartl, F.U.; Breuer, P. Calpain inhibition is sufficient to suppress aggregation of polyglutamine-expanded ataxin-3. J. Biol. Chem. 2007, 282, 18851–18856. [Google Scholar] [CrossRef] [PubMed]
- Chou, A.H.; Yeh, T.H.; Ouyang, P.; Chen, Y.L.; Chen, S.Y.; Wang, H.L. Polyglutamine-expanded ataxin-3 causes cerebellar dysfunction of SCA3 transgenic mice by inducing transcriptional dysregulation. Neurobiol. Dis. 2008, 31, 89–101. [Google Scholar] [CrossRef]
- Zoghbi, H.Y.; Orr, H.T. Spinocerebellar ataxia type 1. Semin. Cell Biol. 1995, 6, 29–35. [Google Scholar] [CrossRef]
- Orr, H.T.; Chung, M.Y.; Banfi, S.; Kwiatkowski, T.J., Jr.; Servadio, A.; Beaudet, A.L.; McCall, A.E.; Duvick, L.A.; Ranum, L.P.; Zoghbi, H.Y. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat. Genet. 1993, 4, 221–226. [Google Scholar] [CrossRef]
- Banfi, S.; Servadio, A.; Chung, M.Y.; Kwiatkowski, T.J., Jr.; McCall, A.E.; Duvick, L.A.; Shen, Y.; Roth, E.J.; Orr, H.T.; Zoghbi, H.Y. Identification and characterization of the gene causing type 1 spinocerebellar ataxia. Nat. Genet. 1994, 7, 513–520. [Google Scholar] [CrossRef]
- Gusella, J.F.; MacDonald, M.E. Molecular genetics: Unmasking polyglutamine triggers in neurodegenerative disease. Nat. Rev. Neurosci. 2000, 1, 109–115. [Google Scholar] [CrossRef]
- Matilla-Duenas, A.; Goold, R.; Giunti, P. Clinical, genetic, molecular, and pathophysiological insights into spinocerebellar ataxia type 1. Cerebellum 2008, 7, 106–114. [Google Scholar] [CrossRef]
- Servadio, A.; Koshy, B.; Armstrong, D.; Antalffy, B.; Orr, H.T.; Zoghbi, H.Y. Expression analysis of the ataxin-1 protein in tissues from normal and spinocerebellar ataxia type 1 individuals. Nat. Genet. 1995, 10, 94–98. [Google Scholar] [CrossRef]
- Durr, A. Autosomal dominant cerebellar ataxias: Polyglutamine expansions and beyond. Lancet Neurol. 2010, 9, 885–894. [Google Scholar] [CrossRef]
- Seidel, K.; Siswanto, S.; Brunt, E.R.; den Dunnen, W.; Korf, H.W.; Rub, U. Brain pathology of spinocerebellar ataxias. Acta Neuropathol. 2012, 124, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Ramirez, R.L.; Bjork, S.T.; Bauer, P.; Feustel, P.J. The reciprocal cerebellar circuitry in human hereditary ataxia. Cerebellum 2013, 12, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Robitaille, Y.; Schut, L.; Kish, S.J. Structural and immunocytochemical features of olivopontocerebellar atrophy caused by the spinocerebellar ataxia type 1 (SCA-1) mutation define a unique phenotype. Acta Neuropathol. 1995, 90, 572–581. [Google Scholar] [CrossRef]
- Skinner, P.J.; Koshy, B.T.; Cummings, C.J.; Klement, I.A.; Helin, K.; Servadio, A.; Zoghbi, H.Y.; Orr, H.T. Ataxin-1 with an expanded glutamine tract alters nuclear matrix-associated structures. Nature 1997, 389, 971–974. [Google Scholar] [CrossRef]
- Duyckaerts, C.; Durr, A.; Cancel, G.; Brice, A. Nuclear inclusions in spinocerebellar ataxia type 1. Acta Neuropathol. 1999, 97, 201–207. [Google Scholar] [CrossRef]
- Hendelman, W.J.; Aggerwal, A.S. The Purkinje neuron: I. A Golgi study of its development in the mouse and in culture. J. Comp. Neurol. 1980, 193, 1063–1079. [Google Scholar] [CrossRef]
- Banfi, S.; Servadio, A.; Chung, M.; Capozzoli, F.; Duvick, L.A.; Elde, R.; Zoghbi, H.Y.; Orr, H.T. Cloning and developmental expression analysis of the murine homolog of the spinocerebellar ataxia type 1 gene (Sca1). Hum. Mol. Genet. 1996, 5, 33–40. [Google Scholar] [CrossRef][Green Version]
- Matilla, A.; Roberson, E.D.; Banfi, S.; Morales, J.; Armstrong, D.L.; Burright, E.N.; Orr, H.T.; Sweatt, J.D.; Zoghbi, H.Y.; Matzuk, M.M. Mice lacking ataxin-1 display learning deficits and decreased hippocampal paired-pulse facilitation. J. Neurosci. 1998, 18, 5508–5516. [Google Scholar] [CrossRef]
- Burright, E.N.; Clark, H.B.; Servadio, A.; Matilla, T.; Feddersen, R.M.; Yunis, W.S.; Duvick, L.A.; Zoghbi, H.Y.; Orr, H.T. SCA1 transgenic mice: A model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell 1995, 82, 937–948. [Google Scholar] [CrossRef]
- Clark, H.B.; Burright, E.N.; Yunis, W.S.; Larson, S.; Wilcox, C.; Hartman, B.; Matilla, A.; Zoghbi, H.Y.; Orr, H.T. Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J. Neurosci. 1997, 17, 7385–7395. [Google Scholar] [CrossRef]
- Clark, H.B.; Orr, H.T. Spinocerebellar ataxia type 1—Modeling the pathogenesis of a polyglutamine neurodegenerative disorder in transgenic mice. J. Neuropathol. Exp. Neurol. 2000, 59, 265–270. [Google Scholar] [CrossRef][Green Version]
- Barnes, J.A.; Ebner, B.A.; Duvick, L.A.; Gao, W.; Chen, G.; Orr, H.T.; Ebner, T.J. Abnormalities in the Climbing Fiber-Purkinje Cell Circuitry Contribute to Neuronal Dysfunction in ATXN1[82Q] Mice. J. Neurosci. 2011, 31, 12778–12789. [Google Scholar] [CrossRef] [PubMed]
- Vig, P.J.; Subramony, S.H.; Burright, E.N.; Fratkin, J.D.; McDaniel, D.O.; Desaiah, D.; Qin, Z. Reduced immunoreactivity to calcium-binding proteins in Purkinje cells precedes onset of ataxia in spinocerebellar ataxia-1 transgenic mice. Neurology 1998, 50, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Vig, P.J.; Subramony, S.H.; McDaniel, D.O. Calcium homeostasis and spinocerebellar ataxia-1 (SCA-1). Brain Res. Bull. 2001, 56, 221–225. [Google Scholar] [CrossRef]
- Rossi, P.I.; Vaccari, C.M.; Terracciano, A.; Doria-Lamba, L.; Facchinetti, S.; Priolo, M.; Ayuso, C.; De Jorge, L.; Gimelli, S.; Santorelli, F.M.; et al. The metabotropic glutamate receptor 1, GRM1: Evaluation as a candidate gene for inherited forms of cerebellar ataxia. J. Neurol. 2010, 257, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Guergueltcheva, V.; Azmanov, D.N.; Angelicheva, D.; Smith, K.R.; Chamova, T.; Florez, L.; Bynevelt, M.; Nguyen, T.; Cherninkova, S.; Bojinova, V.; et al. Autosomal-recessive congenital cerebellar ataxia is caused by mutations in metabotropic glutamate receptor 1. Am. J. Hum. Genet. 2012, 91, 553–564. [Google Scholar] [CrossRef]
- Watson, L.M.; Bamber, E.; Schnekenberg, R.P.; Williams, J.; Bettencourt, C.; Lickiss, J.; Jayawant, S.; Fawcett, K.; Clokie, S.; Wallis, Y.; et al. Dominant Mutations in GRM1 Cause Spinocerebellar Ataxia Type 44. Am. J. Hum. Genet. 2017, 101, 451–458. [Google Scholar] [CrossRef]
- Zu, T.; Duvick, L.A.; Kaytor, M.D.; Berlinger, M.S.; Zoghbi, H.Y.; Clark, H.B.; Orr, H.T. Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice. J. Neurosci. 2004, 24, 8853–8861. [Google Scholar] [CrossRef]
- Shuvaev, A.N.; Hosoi, N.; Sato, Y.; Yanagihara, D.; Hirai, H. Progressive impairment of cerebellar mGluR signalling and its therapeutic potential for cerebellar ataxia in spinocerebellar ataxia type 1 model mice. J. Physiol. 2017, 595, 141–164. [Google Scholar] [CrossRef]
- Hoxha, E.; Balbo, I.; Miniaci, M.C.; Tempia, F. Purkinje Cell Signaling Deficits in Animal Models of Ataxia. Front. Synaptic Neurosci. 2018, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Konno, A.; Shuvaev, A.N.; Miyake, N.; Miyake, K.; Iizuka, A.; Matsuura, S.; Huda, F.; Nakamura, K.; Yanagi, S.; Shimada, T.; et al. Mutant ataxin-3 with an abnormally expanded polyglutamine chain disrupts dendritic development and metabotropic glutamate receptor signaling in mouse cerebellar Purkinje cells. Cerebellum 2014, 13, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.T.; Meera, P.; Otis, T.S.; Pulst, S.M. Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum. Mol. Genet. 2013, 22, 271–283. [Google Scholar] [CrossRef]
- Meera, P.; Pulst, S.; Otis, T. A positive feedback loop linking enhanced mGluR function and basal calcium in spinocerebellar ataxia type 2. Elife 2017, 6, e26377. [Google Scholar] [CrossRef]
- Bezprozvanny, I.; Klockgether, T. Therapeutic prospects for spinocerebellar ataxia type 2 and 3. Drugs Future 2009, 34. [Google Scholar] [CrossRef]
- Kimura, T.; Sugimori, M.; Llinas, R.R. Purkinje cell long-term depression is prevented by T-588, a neuroprotective compound that reduces cytosolic calcium release from intracellular stores. Proc. Natl. Acad. Sci. USA 2005, 102, 17160–17165. [Google Scholar] [CrossRef]
- Goncalves, N.; Simoes, A.T.; Prediger, R.D.; Hirai, H.; Cunha, R.A.; Pereira de Almeida, L. Caffeine alleviates progressive motor deficits in a transgenic mouse model of spinocerebellar ataxia. Ann. Neurol. 2017, 81, 407–418. [Google Scholar] [CrossRef]
- Ristori, G.; Romano, S.; Visconti, A.; Cannoni, S.; Spadaro, M.; Frontali, M.; Pontieri, F.E.; Vanacore, N.; Salvetti, M. Riluzole in cerebellar ataxia: A randomized, double-blind, placebo-controlled pilot trial. Neurology 2010, 74, 839–845. [Google Scholar] [CrossRef]
- Keiser, M.S.; Kordasiewicz, H.B.; McBride, J.L. Gene suppression strategies for dominantly inherited neurodegenerative diseases: Lessons from Huntington’s disease and spinocerebellar ataxia. Hum. Mol. Genet. 2016, 25, R53–R64. [Google Scholar] [CrossRef]
- Persengiev, S.; Kondova, I.; Otting, N.; Koeppen, A.H.; Bontrop, R.E. Genome-wide analysis of miRNA expression reveals a potential role for miR-144 in brain aging and spinocerebellar ataxia pathogenesis. Neurobiol. Aging 2011, 32, 2316.e17–2316.e27. [Google Scholar] [CrossRef]
- Xia, H.; Mao, Q.; Eliason, S.L.; Harper, S.Q.; Martins, I.H.; Orr, H.T.; Paulson, H.L.; Yang, L.; Kotin, R.M.; Davidson, B.L. RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat. Med. 2004, 10, 816–820. [Google Scholar] [CrossRef] [PubMed]
- Scoles, D.R.; Meera, P.; Schneider, M.D.; Paul, S.; Dansithong, W.; Figueroa, K.P.; Hung, G.; Rigo, F.; Bennett, C.F.; Otis, T.S.; et al. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature 2017, 544, 362–366. [Google Scholar] [CrossRef] [PubMed]
- McLoughlin, H.S.; Moore, L.R.; Chopra, R.; Komlo, R.; McKenzie, M.; Blumenstein, K.G.; Zhao, H.; Kordasiewicz, H.B.; Shakkottai, V.G.; Paulson, H.L. Oligonucleotide therapy mitigates disease in spinocerebellar ataxia type 3 mice. Ann. Neurol. 2018, 84, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Kaemmerer, W.F.; Low, W.C. Cerebellar allografts survive and transiently alleviate ataxia in a transgenic model of spinocerebellar ataxia type-1. Exp. Neurol. 1999, 158, 301–311. [Google Scholar] [CrossRef]
- Chintawar, S.; Hourez, R.; Ravella, A.; Gall, D.; Orduz, D.; Rai, M.; Bishop, D.P.; Geuna, S.; Schiffmann, S.N.; Pandolfo, M. Grafting neural precursor cells promotes functional recovery in an SCA1 mouse model. J. Neurosci. 2009, 29, 13126–13135. [Google Scholar] [CrossRef]
- Mendonca, L.S.; Nobrega, C.; Hirai, H.; Kaspar, B.K.; Pereira de Almeida, L. Transplantation of cerebellar neural stem cells improves motor coordination and neuropathology in Machado-Joseph disease mice. Brain 2015, 138 Pt 2, 320–335. [Google Scholar] [CrossRef]
- Chang, Y.K.; Chen, M.H.; Chiang, Y.H.; Chen, Y.F.; Ma, W.H.; Tseng, C.Y.; Soong, B.W.; Ho, J.H.; Lee, O.K. Mesenchymal stem cell transplantation ameliorates motor function deterioration of spinocerebellar ataxia by rescuing cerebellar Purkinje cells. J. Biomed. Sci. 2011, 18, 54. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Buijsen, R.A.M.; Gardiner, S.L.; Bouma, M.J.; van der Graaf, L.M.; Boogaard, M.W.; Pepers, B.A.; Eussen, B.; de Klein, A.; Freund, C.; van Roon-Mom, W.M.C. Generation of 3 spinocerebellar ataxia type 1 (SCA1) patient-derived induced pluripotent stem cell lines LUMCi002-A, B, and C and 2 unaffected sibling control induced pluripotent stem cell lines LUMCi003-A and B. Stem Cell Res. 2018, 29, 125–128. [Google Scholar] [CrossRef]
- Ishida, Y.; Kawakami, H.; Kitajima, H.; Nishiyama, A.; Sasai, Y.; Inoue, H.; Muguruma, K. Vulnerability of Purkinje Cells Generated from Spinocerebellar Ataxia Type 6 Patient-Derived iPSCs. Cell Rep. 2016, 17, 1482–1490. [Google Scholar] [CrossRef]
- Tian, Z.M.; Chen, T.; Zhong, N.; Li, Z.C.; Yin, F.; Liu, S. Clinical study of transplantation of neural stem cells in therapy of inherited cerebellar atrophy. J. Peking Univ. Health Sci. 2009, 41, 456–458. [Google Scholar]
- Lee, P.H.; Lee, J.E.; Kim, H.S.; Song, S.K.; Lee, H.S.; Nam, H.S.; Cheong, J.W.; Jeong, Y.; Park, H.J.; Kim, D.J.; et al. A randomized trial of mesenchymal stem cells in multiple system atrophy. Ann. Neurol. 2012, 72, 32–40. [Google Scholar] [CrossRef]
- Duenas, A.M.; Goold, R.; Giunti, P. Molecular pathogenesis of spinocerebellar ataxias. Brain 2006, 129, 1357–1370. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | SCA | OMIM Number | Distinguishing Clinical Manifestations (1) | Gene | Type of Mutation |
|---|---|---|---|---|---|
| 6p22.3 | SCA1 | 164400 | Pyramidal signs, peripheral neuropathy, and ophthalmoparesis | ATXN1 | (CAG)n |
| 12q24.12 | SCA2 | 183090 | Hyporeflexia, tremor and slow eye movements | ATXN2 | (CAG)n |
| 14q32.12 | SCA3 | 109150 | Motor neuron involvement and Parkinsonian features | ATXN3 | (CAG)n |
| 16q22.1 | SCA4 | 600223 | Sensory peripheral neuropathy | Unknown | Unknown |
| 11q13.2 | SCA5 | 600224 | Early onset and very slow disease progression. | SPTBN2 | Point mutations |
| 19p13.13 | SCA6 | 183086 | Late-onset, very slow disease progression. and nystagmus. | CACNA1A | (CAG)n |
| 3p14.1 | SCA7 | 164500 | Visual loss | ATXN7 | (CAG)n |
| 13q21 | SCA8 | 608768 | Cognitive dysfunction, pyramidal and sensory signs | ATXN8 | (CTG * CAG)n |
| 22q13.31 | SCA10 | 603516 | Occasional epilepsy | ATXN10 | (ATTCT)n |
| 15q15.2 | SCA11 | 604432 | Pyramidal signs. | TTBK2 | Point mutations |
| 5q32 | SCA12 | 604326 | Tremor, Parkinsonian features and dementia | PPP2R2B | (CAG)n |
| 19q13.33 | SCA13 | 605259 | Delayed motor and cognitive development | KCNC3 | Point mutations |
| 19q13.42 | SCA14 | 605361 | Dystonia and myoclonus. | PRKCG | Point mutations |
| 3p26.1 | SCA15/16 | 606658 | Tremor and cognitive impairment. | ITPR1 | Point mutations |
| 6q27 | SCA17 | 607136 | Dementia and Parkinsonian features | TBP | (CAG)n |
| 7q22–q32 | SCA18 | 607458 | Sensory and motor neuropathy | IFRD1 | Point mutations |
| 1p13.2 | SCA19/22 | 607346 | Cognitive impairment and myoclonus | KCND3 | Point mutations |
| 11q12 | SCA20 | 608687 | Cerebellar dysarthria | Unknown | Genomic duplication |
| 1p36.33 | SCA21 | 607454 | Mild cognitive impairment, and Parkinsonian features | TMEM240 | Unknown |
| 20p13 | SCA23 | 610245 | Pyramidal signs | PDYN | Point mutations |
| 2p21–p13 | SCA25 | 608703 | Peripheral neuropathy, | Unknown | Unknown |
| 19p13.3 | SCA26 | 609306 | Eye movement abnormalities. | EEF2 | Point mutations |
| 13q33.1 | SCA27 | 609307 | Tremor and dystonia | FGF14 | Point mutations |
| 18p11.21 | SCA28 | 610246 | Spastic ataxia | AFG3L2 | Point mutations |
| 3p26.1 | SCA29 | 117360 | Intellectual disability. | ITPR1 | Point mutations |
| 4q34.3–q35.1 | SCA30 | 613371 | Pure ataxia. | ODZ3 | Unknown |
| 16q21 | SCA31 | 117210 | Abnormal sensation | BEAN1 | (TGGAA)n |
| 6q14.1 | SCA34 | 133190 | Hyperkeratosis | ELOVL4 | Unknown |
| 20p13 | SCA35 | 613908 | Ocular dysmetria, tremor and hyperreflexia | TGM6 | Point mutations |
| 20p13 | SCA36 | 614153 | Motor neuron involvement | NOP56 | (GGCCTG)n |
| 1p32.2 | SCA37 | 615945 | Altered vertical eye movements. | DAB1 | (GGCCTG)n |
| 6p12.1 | SCA38 | 615957 | Nystagmus and dysarthria | ELOVL5 | Point mutations |
| 14q32.11–q32.12 | SCA40 | 616053 | Ocular dysmetria and tremor | CCDC88C | Point mutations |
| 4q27 | SCA41 | 616410 | Imbalance and loss of coordination | TRPC3 | Point mutations |
| 17q21.33 | SCA42 | 618087 | Gait instability, dysarthria and nystagmus | CACNA1G | Point mutations |
| 3q25.2 | SCA43 | 617018 | Peripheral neuropathy | MME | Point mutations |
| 6q24.3 | SCA44 | 617691 | Dysarthria, dysphagia and dysmetria | GRM1 | Point mutations |
| 5q33.1 | SCA45 | 617769 | Nystagmus, and dysarthria. | FAT2 | Point mutations |
| 19q13.2 | SCA46 | 617770 | Sensory ataxic neuropathy | PLD3 | Point mutations |
| 12p13.31 | DRLPA | 125370 | Involuntary movements, mental and emotional problems | ATN1 | (CAG)n |
| 4q22.1–q22.2 | GRID2-related spinocerebellar ataxia | 616204 | Motor, speech and cognitive delay and eye movement abnormalities | GRID2Rarely AD inheritance | Point mutations |
| SCA | Gene | Protein | Effect on Ca2+ Signaling |
|---|---|---|---|
| SCA1 | ATXN1 | Ataxin-1 | Decrease |
| SCA2 | ATXN2 | Ataxin-2 | Increase |
| SCA3 | ATXN3 | Ataxin-3 | Increase |
| SCA6 | CACNA1A | Ca2+voltage-gated channel subunit α1A | Decrease |
| SCA14 | PRKCG | PKCγ | Increase/Decrease |
| SCA15/16 | ITPR1 | IP3 receptor | Increase/Decrease |
| SCA29 | ITPR1 | IP3 receptor | Decrease |
| SCA41 | TRPC3 | TRPC3 channel | Increase |
| SCA42 | CACNA1G | Ca2+ voltage-gated channel subunit α1G | Decrease |
| SCA44 | GMR1 | mGlu receptor 1 | Increase |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prestori, F.; Moccia, F.; D’Angelo, E. Disrupted Calcium Signaling in Animal Models of Human Spinocerebellar Ataxia (SCA). Int. J. Mol. Sci. 2020, 21, 216. https://doi.org/10.3390/ijms21010216
Prestori F, Moccia F, D’Angelo E. Disrupted Calcium Signaling in Animal Models of Human Spinocerebellar Ataxia (SCA). International Journal of Molecular Sciences. 2020; 21(1):216. https://doi.org/10.3390/ijms21010216
Chicago/Turabian StylePrestori, Francesca, Francesco Moccia, and Egidio D’Angelo. 2020. "Disrupted Calcium Signaling in Animal Models of Human Spinocerebellar Ataxia (SCA)" International Journal of Molecular Sciences 21, no. 1: 216. https://doi.org/10.3390/ijms21010216
APA StylePrestori, F., Moccia, F., & D’Angelo, E. (2020). Disrupted Calcium Signaling in Animal Models of Human Spinocerebellar Ataxia (SCA). International Journal of Molecular Sciences, 21(1), 216. https://doi.org/10.3390/ijms21010216