Evolving a Peptide: Library Platforms and Diversification Strategies


Abstract
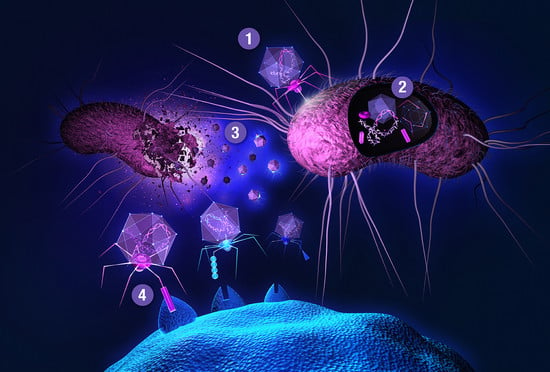
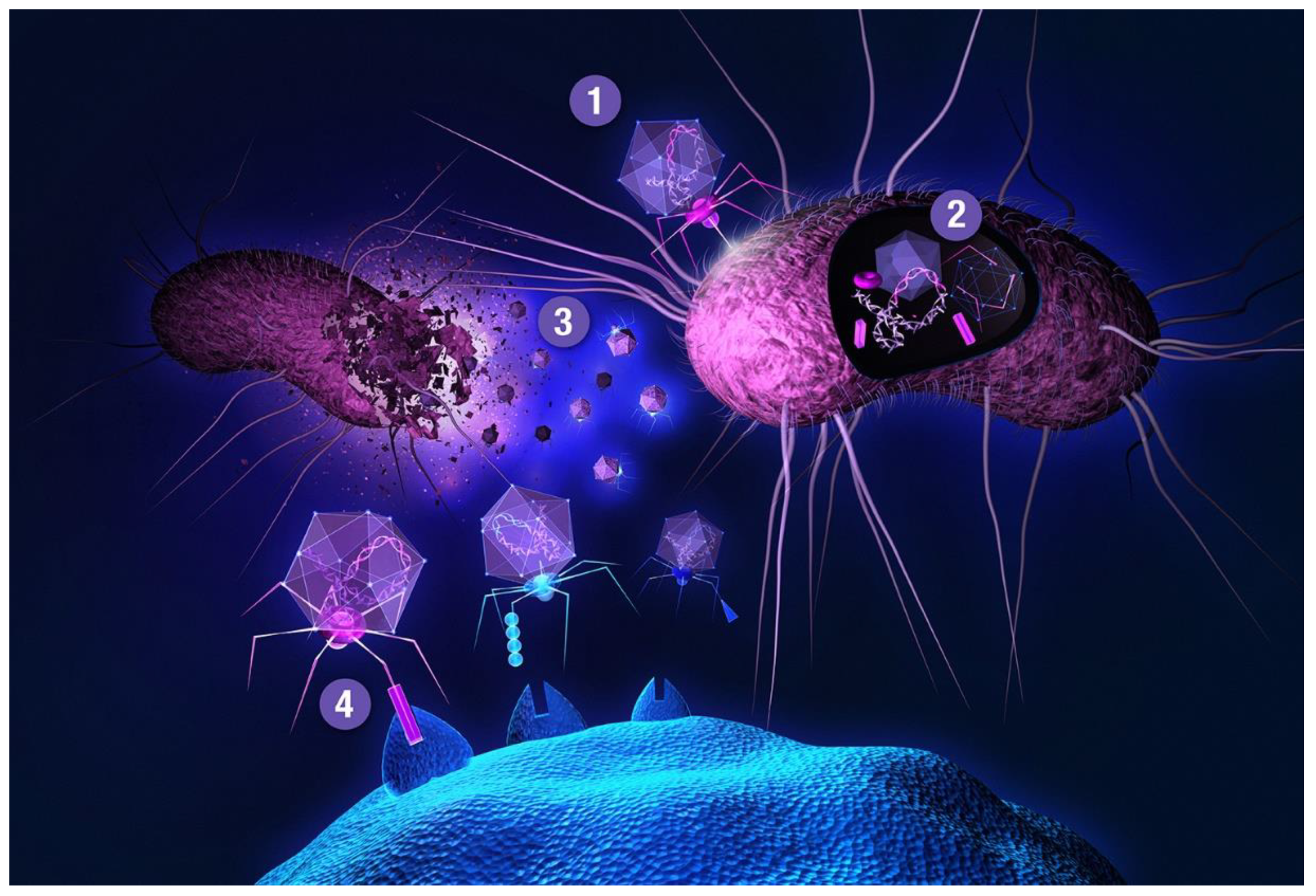
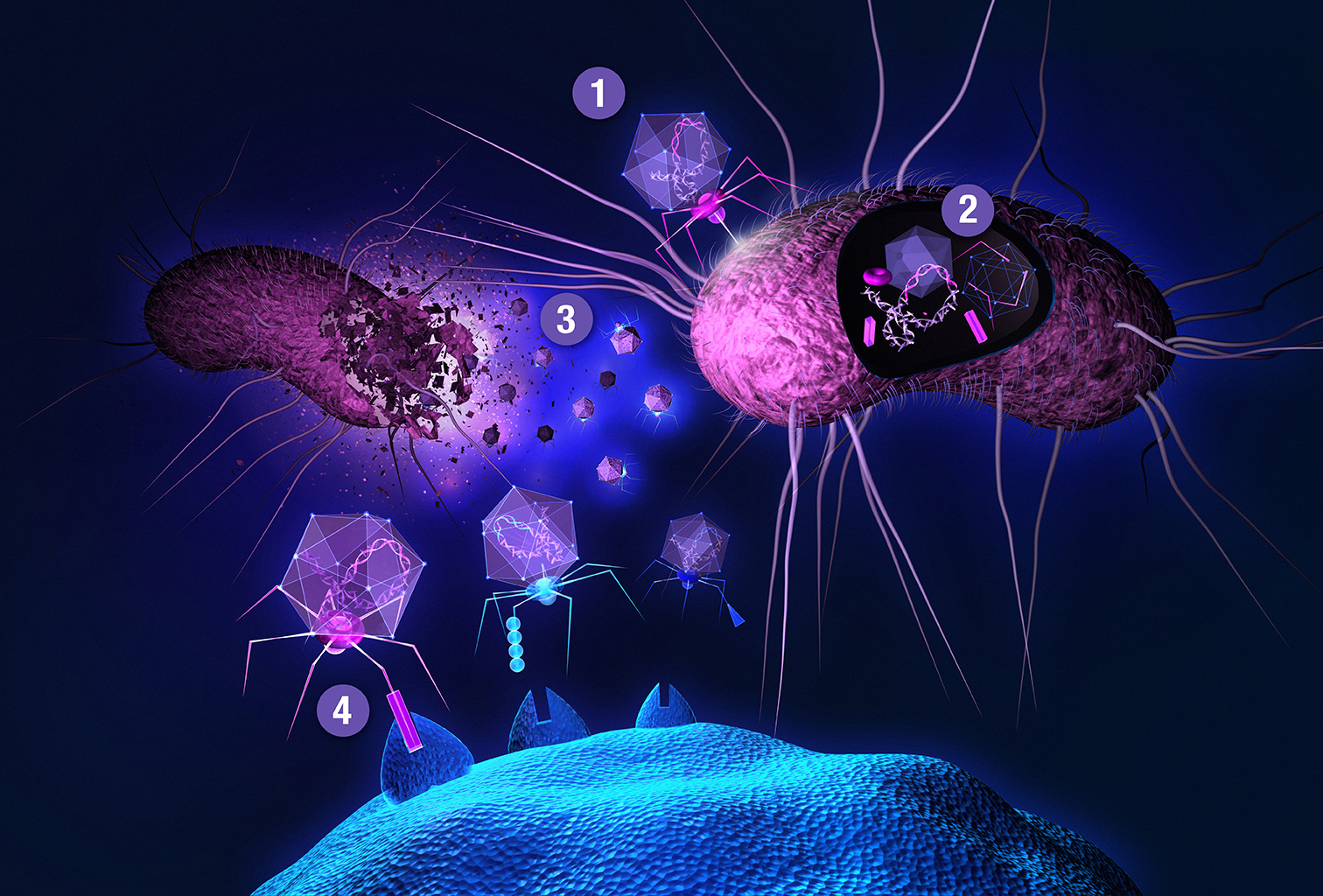{kind=link}
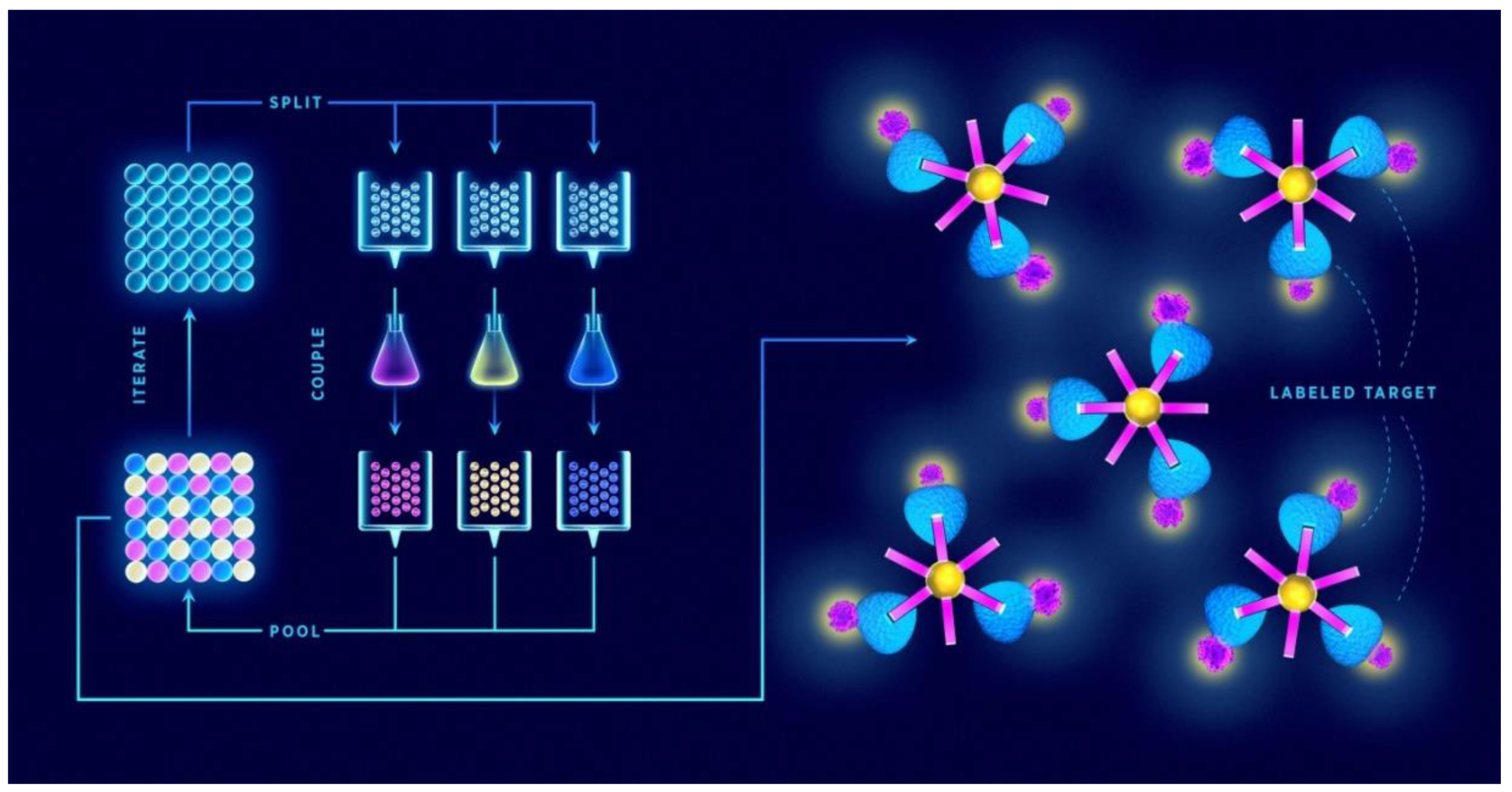{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Combinatorial Peptide Libraries
2.1. Chemical Peptide Libraries
2.2. Biological Peptide Libraries
2.2.1. Cellular Approach
2.2.2. Acellular Approach
3. Incorporating Unnatural Amino Acids and Constraints into (Library) Peptides
4. Peptide Library Design and Construction
4.1. Random Mutagenesis
4.2. Focused Mutagenesis
4.2.1. Enzyme-Based Approaches
4.2.2. Chemical-Based Mutagenesis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated virus |
| AcMNPV | Autographa californica multiple nuclear polyhedrosis virus |
| APEx | Anchored periplasmic expression |
| ARS | Aminoacyl-tRNA synthetase |
| cPTM | Chemical post-translational modifications |
| Cvap | Cv RNA-associating protein |
| DEL | DNA-encoded library |
| dNTP | deoxyribonucleoside triphosphate |
| epPCR | Error-prone PCR |
| FACS | Fluorescence activated cell sorting |
| FIT | Flexible in vitro translation |
| Fmoc | Fluorenylmethyloxycarbonyl |
| IVA | In vivo assembly |
| IVC | In vitro compartmentalization |
| MACS | Magnetic-activated cell sorting |
| MHC | Major histocompatibility complex |
| MORPHING | Mutagenic organized recombination process by homologous in vivo grouping |
| MP | Mutagenesis plasmid |
| MS | Mass spectrometry |
| NGS | Next-generation sequencing |
| OBOC | One-bead-one-compound |
| PERISS | intra periplasm secretion and selection system |
| PPI | Protein-protein interaction |
| PURE | Protein synthesis using recombinant elements |
| RaPID | Random non-standard peptide integrated discovery |
| RCA | Rolling circle amplification |
| SAR | Structure-activity relationship |
| SeSaM | Sequence saturation mutagenesis |
| SICLOPPS | Split-intein circular ligation of peptides and proteins |
| SLAY | Surface localized antimicrobial display |
| SLIP | Site-directed ligase-independent mutagenesis |
| SPPS | Solid-phase peptide synthesis |
| StEP | staggered extension process |
| VLP | Virus-like particle |
References
- Guzmán, F.; Barberis, S.; Illanes, A. Peptide synthesis: Chemical or enzymatic. Electron. J. Biotechnol. 2007, 10, 279–314. [Google Scholar] [CrossRef]
- Sun, E.; Belanger, C.R.; Haney, E.F.; Hancock, R.E.W. Host defense (antimicrobial) peptides. In Peptide Applications in Biomedicine, Biotechnology and Bioengineering; Woodhead Publishing: Cambridge, UK, 2017; pp. 253–285. [Google Scholar]
- Gribble, F.M.; Reimann, F. Function and mechanisms of enteroendocrine cells and gut hormones in metabolism. Nat. Rev. Endocrinol. 2019, 15, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Nusbaum, M.P. Neurotransmission: Peptide transmitters turn 36. J. Exp. Biol. 2017, 220, 2492–2494. [Google Scholar] [CrossRef] [PubMed]
- Clark, G.C.; Casewell, N.R.; Elliott, C.T.; Harvey, A.L.; Jamieson, A.G.; Strong, P.N.; Turner, A.D. Friends or foes? Emerging impacts of biological toxins. Trends Biochem. Sci. 2019, 44, 365–379. [Google Scholar] [CrossRef]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef]
- Slominsky, P.A.; Shadrina, M.I. Peptide pharmaceuticals: Opportunities, prospects, and limitations. Mol. Genet. Microbiol. Virol. 2018, 33, 8–14. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Buchholz, K.; Collins, J. Concepts in Biotechnology: History, Science and Business; John Wiley & Sons: Hoboken, NJ, USA, 2013; Volume 49, ISBN 352769109X. [Google Scholar]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The future of peptide-based drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef]
- Erak, M.; Bellmann-Sickert, K.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide chemistry toolbox—Transforming natural peptides into peptide therapeutics. Bioorg. Med. Chem. 2018, 26, 2759–2765. [Google Scholar] [CrossRef] [PubMed]
- Millward, S.W.; Fiacco, S.; Austin, R.J.; Roberts, R.W. Design of cyclic peptides that bind protein surfaces with antibody-like affinity. ACS Chem. Biol. 2007, 2, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Mimmi, S.; Maisano, D.; Quinto, I.; Iaccino, E. Phage display: An overview in context to drug discovery. Trends Pharmacol. Sci. 2019, 40, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Damayanti, N.P.; Buno, K.; Voytik Harbin, S.L.; Irudayaraj, J.M.K. Epigenetic process monitoring in live cultures with peptide biosensors. ACS Sens. 2019, 4, 562–565. [Google Scholar] [CrossRef] [PubMed]
- Care, A.; Bergquist, P.L.; Sunna, A. Solid-binding peptides: Smart tools for nanobiotechnology. Trends Biotechnol. 2015, 33, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Schoch, G.A.; Attias, R.; Belghazi, M.; Dansette, P.M.; Werck-Reichhart, D. Engineering of a water-soluble plant cytochrome P450, CYP73A1, and NMR-based orientation of natural and alternate substrates in the active site. Plant Physiol. 2003, 133, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Corin, K.; Baaske, P.; Wienken, C.J.; Jerabek-Willemsen, M.; Duhr, S.; Braun, D.; Zhang, S. Peptide surfactants for cell-free production of functional G protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2011, 108, 9049–9054. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.I.; Du, S.; Tortajada, A.; Paulo, J.; Zhang, S. Peptergents: Peptide detergents that improve stability and functionality of a membrane protein, glycerol-3-phosphate dehydrogenase. Biochemistry 2005, 44, 16912–16919. [Google Scholar] [CrossRef]
- Matsumoto, K.; Vaughn, M.; Bruce, B.D.; Koutsopoulos, S.; Zhang, S. Designer peptide surfactants stabilize functional Photosystem-I membrane complex in aqueous solution for extended time. J. Phys. Chem. B 2009, 113, 75–83. [Google Scholar] [CrossRef]
- Bukowska, M.A.; Grütter, M.G. New concepts and aids to facilitate crystallization. Curr. Opin. Struct. Biol. 2013, 23, 409–416. [Google Scholar] [CrossRef]
- Wei, Z.; Maeda, Y.; Matsui, H. Discovery of catalytic peptides for inorganic nanocrystal synthesis by a combinatorial phage display approach. Angew. Chem. Int. Ed. 2011, 50, 10585–10588. [Google Scholar] [CrossRef][Green Version]
- Kruljec, N.; Bratkovič, T. Alternative affinity ligands for immunoglobulins. Bioconjug. Chem. 2017, 28, 2009–2030. [Google Scholar] [CrossRef]
- Kish, W.S.; Roach, M.K.; Sachi, H.; Naik, A.D.; Menegatti, S.; Carbonell, R.G. Purification of human erythropoietin by affinity chromatography using cyclic peptide ligands. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1085, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Reverdatto, S.; Burz, D.; Shekhtman, A. Peptide aptamers: Development and applications. Curr. Top. Med. Chem. 2015, 15, 1082–1101. [Google Scholar] [CrossRef] [PubMed]
- Kotz, J. Phenotypic screening, take two. Sci. Exch. 2012, 5, 380. [Google Scholar] [CrossRef]
- Croston, G.E. The utility of target-based discovery. Expert Opin. Drug Discov. 2017, 12, 427–429. [Google Scholar] [CrossRef]
- Mersich, C.; Jungbauer, A. Generation of bioactive peptides by biological libraries. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 861, 160–170. [Google Scholar] [CrossRef]
- Yoshida, H.; Baik, S.H.; Harayama, S. An effective peptide screening system using recombinant fluorescent bacterial surface display. Biotechnol. Lett. 2002, 24, 1715–1722. [Google Scholar] [CrossRef]
- Stratis-Cullum, D.N. Method for discovery of peptide reagents using a commercial magnetic separation platform and bacterial cell surface display technology. J. Anal. Bioanal. Tech. 2015, 6, 1. [Google Scholar] [CrossRef]
- Erharuyi, O.; Simanski, S.; McEnaney, P.J.; Kodadek, T. Screening one bead one compound libraries against serum using a flow cytometer: Determination of the minimum antibody concentration required for ligand discovery. Bioorg. Med. Chem. Lett. 2018, 28, 2773–2778. [Google Scholar] [CrossRef]
- Matochko, W.L.; Derda, R. Next-generation sequencing of phage-displayed peptide libraries. Methods Mol. Biol. 2015, 1248, 249–266. [Google Scholar]
- Jalali-Yazdi, F.; Huong lai, L.; Takahashi, T.T.; Roberts, R.W. High-throughput measurement of binding kinetics by mRNA display and next-generation sequencing. Angew. Chem. Int. Ed. 2016, 55, 4007–4010. [Google Scholar] [CrossRef]
- Lagoutte, P.; Lugari, A.; Elie, C.; Potisopon, S.; Donnat, S.; Mignon, C.; Mariano, N.; Troesch, A.; Werle, B.; Stadthagen, G. Combination of ribosome display and next generation sequencing as a powerful method for identification of affibody binders against β-lactamase CTX-M15. New Biotechnol. 2019, 50, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Körbelin, J.; Sieber, T.; Michelfelder, S.; Lunding, L.; Spies, E.; Hunger, A.; Alawi, M.; Rapti, K.; Indenbirken, D.; Müller, O.J.; et al. Pulmonary targeting of adeno-associated viral vectors by next-generation sequencing-guided screening of random capsid displayed peptide libraries. Mol. Ther. 2016, 24, 1050–1061. [Google Scholar] [CrossRef] [PubMed]
- Mendes, K.R.; Malone, M.L.; Ndungu, J.M.; Suponitsky-Kroyter, I.; Cavett, V.J.; McEnaney, P.J.; MacConnell, A.B.; Doran, T.D.M.; Ronacher, K.; Stanley, K.; et al. High-throughput identification of DNA-encoded IgG ligands that distinguish active and latent mycobacterium tuberculosis infections. ACS Chem. Biol. 2017, 12, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Crook, Z.R.; Sevilla, G.P.; Friend, D.; Brusniak, M.Y.; Bandaranayake, A.D.; Clarke, M.; Gewe, M.; Mhyre, A.J.; Baker, D.; Strong, R.K.; et al. Mammalian display screening of diverse cystine-dense peptides for difficult to drug targets. Nat. Commun. 2017, 8, 2244. [Google Scholar] [CrossRef] [PubMed]
- Pantazes, R.J.; Reifert, J.; Bozekowski, J.; Ibsen, K.N.; Murray, J.A.; Daugherty, P.S. Identification of disease-specific motifs in the antibody specificity repertoire via next-generation sequencing. Sci. Rep. 2016, 6, 30312. [Google Scholar] [CrossRef]
- Suga, H. Max-Bergmann award lecture:A RaPID way to discover bioactive nonstandard peptides assisted by the flexizyme and FIT systems. J. Pept. Sci. 2018, 24, e3055. [Google Scholar] [CrossRef]
- Wang, Y.C.; Distefano, M.D. Synthesis and screening of peptide libraries with free C-termini. Curr. Top. Pept. Protein Res. 2014, 15, 1–23. [Google Scholar]
- Carpino, L.A.; Beyermann, M.; Wenschuh, H.; Bienert, M. Peptide synthesis via amino acid halides. Acc. Chem. Res. 1996, 29, 268–274. [Google Scholar] [CrossRef]
- Behrendt, R.; White, P.; Offer, J. Advances in Fmoc solid-phase peptide synthesis. J. Pept. Sci. 2016, 22, 4–27. [Google Scholar] [CrossRef]
- Houghten, R.A. General method for the rapid solid-phase synthesis of large numbers of peptides: Specificity of antigen-antibody interaction at the level of individual amino acids. Proc. Natl. Acad. Sci. USA 1985, 82, 5131–5135. [Google Scholar] [CrossRef]
- Furka, Á.; Sebestyén, F.; Asgedom, M.; Dibó, G. General method for rapid synthesis of multicomponent peptide mixtures. Int. J. Pept. Protein Res. 1991, 37, 487–493. [Google Scholar] [CrossRef]
- Lam, K.S.; Salmon, S.E.; Hersh, E.M.; Hruby, V.J.; Kazmierskit, W.M.; Knappt, R.J. A new type of synthetic peptide library for identifying ligand-binding activity. Nature 1991, 354, 82–84. [Google Scholar] [CrossRef]
- Ostresh, J.M.; Winkle, J.H.; Hamashin, V.T.; Houghten, R.A. Peptide libraries: Determination of relative reaction rates of protected amino acids in competitive couplings. Biopolymers 1994, 34, 1681–1689. [Google Scholar] [CrossRef]
- Wilson-Lingardo, L.; Davis, P.W.; Ecker, D.J.; Hébert, N.; Acevedo, O.; Sprankle, K.; Brennan, T.; Schwarcz, L.; Freier, S.M.; Wyatt, J.R. Deconvolution of combinatorial libraries for drug discovery: Experimental comparison of pooling strategies. J. Med. Chem. 1996, 39, 2720–2726. [Google Scholar] [CrossRef]
- Pinilla, C.; Appel, J.R.; Blanc, P.; Houghten, R.A.; Blanc, P.; Houghten, R.A. Rapid identification of high affinity peptide ligands using positional scanning synthetic peptide combinatorial libraries. Biotechniques 1993, 13, 901–905. [Google Scholar]
- Edman, P. Method for determination of the amino acid sequence in peptides. Acta Chem. Scand 1950, 4, 283–293. [Google Scholar] [CrossRef]
- Wang, P.; Arabaci, G.; Pei, D. Rapid sequencing of library-derived peptides by partial edman degradation and mass spectrometry. J. Comb. Chem. 2001, 3, 251–254. [Google Scholar] [CrossRef]
- Vinogradov, A.A.; Gates, Z.P.; Zhang, C.; Quartararo, A.J.; Halloran, K.H.; Pentelute, B.L. Library design-facilitated high-throughput sequencing of synthetic peptide libraries. ACS Comb. Sci. 2017, 19, 694–701. [Google Scholar] [CrossRef]
- Deutzmann, R. Structural characterization of proteins and peptides. Methods Mol. Med. 2004, 94, 269–297. [Google Scholar]
- Grant, G.A.; Crankshaw, M.W.; Gorka, J. Edman sequencing as tool for characterization of synthetic peptides. Methods Enzymol. 1997, 289, 395–419. [Google Scholar]
- Goodnow, R.A.; Dumelin, C.E.; Keefe, A.D. DNA-encoded chemistry: Enabling the deeper sampling of chemical space. Nat. Rev. Drug Discov. 2017, 16, 131–147. [Google Scholar] [CrossRef]
- Ottl, J.; Leder, L.; Schaefer, J.V.; Dumelin, C.E. Encoded library technologies as integrated lead finding platforms for drug discovery. Molecules 2019, 24, 1629. [Google Scholar] [CrossRef]
- Zhu, Z.; Shaginian, A.; Grady, L.S.C.; Davie, C.P.; Lind, K.; Pal, S.; Thansandote, P.; Simpson, G.L. DNA-encoded macrocyclic peptide library. Methods Mol. Biol. 2019, 2001, 273–284. [Google Scholar]
- Daguer, J.P.; Ciobanu, M.; Alvarez, S.; Barluenga, S.; Winssinger, N. DNA-templated combinatorial assembly of small molecule fragments amenable to selection/amplification cycles. Chem. Sci. 2011, 2, 625–632. [Google Scholar] [CrossRef]
- Svensen, N.; Díaz-Mochón, J.J.; Bradley, M. Encoded peptide libraries and the discovery of new cell binding ligands. Chem. Commun. 2011, 47, 7638–7640. [Google Scholar] [CrossRef]
- Stress, C.J.; Sauter, B.; Schneider, L.A.; Sharpe, T.; Gillingham, D. A DNA-encoded chemical library incorporating elements of natural macrocycles. Angew. Chem. Int. Ed. 2019, 58, 9570–9574. [Google Scholar] [CrossRef]
- Faver, J.C.; Riehle, K.; Lancia, D.R.; Milbank, J.B.J.; Kollmann, C.S.; Simmons, N.; Yu, Z.; Matzuk, M.M. Quantitative comparison of enrichment from DNA-encoded chemical library selections. ACS Comb. Sci. 2019, 21, 75–82. [Google Scholar] [CrossRef]
- Zimmermann, G.; Li, Y.; Rieder, U.; Mattarella, M.; Neri, D.; Scheuermann, J. Hit-validation methodologies for ligands isolated from DNA-encoded chemical libraries. ChemBioChem 2017, 18, 853–857. [Google Scholar] [CrossRef]
- Galán, A.; Comor, L.; Horvatić, A.; Kuleš, J.; Guillemin, N.; Mrljak, V.; Bhide, M. Library-based display technologies: Where do we stand? Mol. Biosyst. 2016, 12, 2342–2358. [Google Scholar] [CrossRef]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Molek, P.; Strukelj, B.; Bratkovic, T. Peptide phage display as a tool for drug discovery: Targeting membrane receptors. Molecules 2011, 16, 857–887. [Google Scholar] [CrossRef] [PubMed]
- Bratkovič, T. Progress in phage display: Evolution of the technique and its applications. Cell. Mol. Life Sci. 2010, 67, 749–767. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P.; Petrenko, V.A. Phage display. Chem. Rev. 1997, 97, 391–410. [Google Scholar] [CrossRef] [PubMed]
- González-Techera, A.; Umpiérrez-Failache, M.; Cardozo, S.; Obal, G.; Pritsch, O.; Last, J.A.; Gee, S.J.; Hammock, B.D.; González-Sapienza, G. High-throughput method for ranking the affinity of peptide ligands selected from phage display libraries. Bioconjug. Chem. 2008, 19, 993–1000. [Google Scholar] [CrossRef][Green Version]
- Kruljec, N.; Molek, P.; Hodnik, V.; Anderluh, G.; Bratkovič, T. Development and characterization of peptide ligands of immunoglobulin G Fc region. Bioconjug. Chem. 2018, 29, 2763–2775. [Google Scholar] [CrossRef]
- Gray, B.P.; Brown, K.C. Combinatorial peptide libraries: Mining for cell-binding peptides. Chem. Rev. 2014, 114, 1020–1081. [Google Scholar] [CrossRef]
- Chackerian, B.; Caldeira, J.D.C.; Peabody, J.; Peabody, D.S. Peptide epitope identification by affinity selection on bacteriophage MS2 virus-like particles. J. Mol. Biol. 2011, 409, 225–237. [Google Scholar] [CrossRef]
- He, B.; Tjhung, K.F.; Bennett, N.J.; Chou, Y.; Rau, A.; Huang, J.; Derda, R. Compositional bias in naïve and chemically-modified phage-displayed libraries uncovered by paired-end deep sequencing. Sci. Rep. 2018, 8, 1214. [Google Scholar] [CrossRef]
- Peters, E.A.; Schatz, P.J.; Johnson, S.S.; Dower, W.J. Membrane insertion defects caused by positive charges in the early mature region of protein pIII of filamentous phage fd can be corrected by prlA suppressors. J. Bacteriol. 1994, 176, 4296–4305. [Google Scholar] [CrossRef]
- Krumpe, L.R.H.; Atkinson, A.J.; Smythers, G.W.; Kandel, A.; Schumacher, K.M.; McMahon, J.B.; Makowski, L.; Mori, T. T7 lytic phage-displayed peptide libraries exhibit less sequence bias than M13 filamentous phage-displayed peptide libraries. Proteomics 2006, 6, 4210–4222. [Google Scholar] [CrossRef]
- Paschke, M.; Höhne, W. A twin-arginine translocation (Tat)-mediated phage display system. Gene 2005, 350, 79–88. [Google Scholar] [CrossRef]
- Speck, J.; Arndt, K.M.; Mller, K.M. Efficient phage display of intracellularly folded proteins mediated by the TAT pathway. Protein Eng. Des. Sel. 2011, 24, 473–484. [Google Scholar] [CrossRef]
- Feng, Z.; Xu, B. Inspiration from the mirror: D-amino acid containing peptides in biomedical approaches. Biomol. Concepts 2016, 7, 179–187. [Google Scholar] [CrossRef]
- Deyle, K.; Kong, X.D.; Heinis, C. Phage selection of cyclic peptides for application in research and drug development. Acc. Chem. Res. 2017, 50, 1866–1874. [Google Scholar] [CrossRef]
- Wada, A.; Terashima, T.; Kageyama, S.; Yoshida, T.; Narita, M.; Kawauchi, A.; Kojima, H. Efficient prostate cancer therapy with tissue-specific homing peptides identified by advanced phage display technology. Mol. Ther. Oncolytics 2019, 12, 138–146. [Google Scholar] [CrossRef]
- Vekris, A.; Pilalis, E.; Chatziioannou, A.; Petry, K.G. A computational pipeline for the extraction of actionable biological information from NGS-phage display experiments. Front. Physiol. 2019, 10, 1160. [Google Scholar] [CrossRef]
- Gillespie, J.W.; Yang, L.; De Plano, L.M.; Stackhouse, M.A.; Petrenko, V.A. Evolution of a landscape phage library in a mouse xenograft model of human breast cancer. Viruses 2019, 11, 988. [Google Scholar] [CrossRef]
- Bessette, P.H.; Rice, J.J.; Daugherty, P.S. Rapid isolation of high-affinity protein binding peptides using bacterial display. Protein Eng. Des. Sel. 2004, 17, 731–739. [Google Scholar] [CrossRef]
- Bessette, P.H.; Hu, X.; Soh, H.T.; Daugherty, P.S. Microfluidic library screening for mapping antibody epitopes. Anal. Chem. 2007, 79, 2174–2178. [Google Scholar] [CrossRef]
- Wong, R.S.Y.; Wirtz, R.A.; Hancock, R.E.W. Pseudomonas aeruginosa outer membrane protein OprF as an expression vector for foreign epitopes: The effects of positioning and length on the antigenicity of the epitope. Gene 1995, 158, 55–60. [Google Scholar] [CrossRef]
- Saffar, B.; Yakhchali, B.; Arbabi, M. Development of a bacterial surface display of hexahistidine peptide using CS3 pili for bioaccumulation of heavy metals. Curr. Microbiol. 2007, 55, 273–277. [Google Scholar] [CrossRef]
- Westerlund-Wikström, B. Peptide display on bacterial flagella: Principles and applications. Int. J. Med. Microbiol. 2000, 290, 223–230. [Google Scholar] [CrossRef]
- Dong, J.; Liu, C.; Zhang, J.; Xin, Z.T.; Yang, G.; Gao, B.; Mao, C.Q.; Le Liu, N.; Wang, F.; Shao, N.S.; et al. Selection of novel nickel-binding peptides from flagella displayed secondary peptide library. Chem. Biol. Drug Des. 2006, 68, 107–112. [Google Scholar] [CrossRef]
- Kimura, T. Screening techniques using the periplasmic expression of peptide libraries and target molecules. J. Bioanal. Biomed. 2017, 9, 263–268. [Google Scholar] [CrossRef]
- Harvey, B.R.; Georgiou, G.; Hayhurst, A.; Jeong, K.J.; Iverson, B.L.; Rogers, G.K. Anchored periplasmic expression, a versatile technology for the isolation of high-affinity antibodies from Escherichia coli-expressed libraries. Proc. Natl. Acad. Sci. USA 2004, 101, 9193–9198. [Google Scholar] [CrossRef]
- Guo, M.; Xu, L.M.; Zhou, B.; Yin, J.C.; Ye, X.L.; Ren, G.P.; Li, D.S. Anchored periplasmic expression (APEx)-based bacterial display for rapid and high-throughput screening of B cell epitopes. Biotechnol. Lett. 2014, 36, 609–616. [Google Scholar] [CrossRef]
- Cull, M.G.; Miller, J.F.; Schatz, P.J. Screening for receptor ligands using large libraries of peptides linked to the C terminus of the lac repressor. Proc. Natl. Acad. Sci. USA 1992, 89, 1865–1869. [Google Scholar] [CrossRef]
- Zhu, W.; Williams, R.S.; Kodadek, T. A CDC6 protein-binding peptide selected using a bacterial two-hybrid-like system is a cell cycle inhibitor. J. Biol. Chem. 2000, 275, 32098–32105. [Google Scholar] [CrossRef]
- Karimova, G.; Gauliard, E.; Davi, M.; Ouellette, S.P.; Ladant, D. Protein–protein interaction: Bacterial two-hybrid. Methods Mol. Biol. 2017, 1615, 159–176. [Google Scholar]
- Mehla, J.; Caufield, J.H.; Sakhawalkar, N.; Uetz, P. A comparison of two-hybrid approaches for detecting protein–protein interactions. Methods Enzymol. 2017, 586, 333–358. [Google Scholar]
- Tucker, A.T.; Leonard, S.P.; DuBois, C.D.; Knauf, G.A.; Cunningham, A.L.; Wilke, C.O.; Trent, M.S.; Davies, B.W. Discovery of next-generation antimicrobials through bacterial self-screening of surface-displayed peptide libraries. Cell 2018, 172, 618–628. [Google Scholar] [CrossRef] [PubMed]
- Dell, A.; Galadari, A.; Sastre, F.; Hitchen, P. Similarities and differences in the glycosylation mechanisms in prokaryotes and eukaryotes. Int. J. Microbiol. 2010, 2010, 148178. [Google Scholar] [CrossRef] [PubMed]
- Mehla, J.; Caufield, J.H.; Uetz, P. The yeast two-hybrid system: A tool for mapping protein-protein interactions. Cold Spring Harb. Protoc. 2015, 2015, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Barreto, K.; Aparicio, A.; Bharathikumar, V.M.; DeCoteau, J.F.; Geyer, C.R. Yeast two-hybrid screening of cyclic peptide libraries using a combination of random and PI-deconvolution pooling strategies. Protein Eng. Des. Sel. 2012, 25, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Aronheim, A. The Ras Recruitment System (RRS) for the identification and characterization of protein–protein interactions. Methods Mol. Biol. 2018, 1794, 61–73. [Google Scholar] [PubMed]
- Dirnberger, D.; Messerschmid, M.; Baumeister, R. An optimized split-ubiquitin cDNA-library screening system to identify novel interactors of the human Frizzled 1 receptor. Nucleic Acids Res. 2008, 36, e37. [Google Scholar] [CrossRef]
- Licitra, E.J.; Liu, J.O. A three-hybrid system for detecting small ligand-protein receptor interactions. Proc. Natl. Acad. Sci. USA 1996, 93, 12817–12821. [Google Scholar] [CrossRef]
- Gera, N.; Hussain, M.; Rao, B.M. Protein selection using yeast surface display. Methods 2013, 60, 15–26. [Google Scholar] [CrossRef]
- Ueda, M. Principle of cell surface engineering of yeast. In Yeast Cell Surface Engineering; Springer: Singapore, 2019; pp. 3–14. [Google Scholar]
- Kondo, A.; Ueda, M. Yeast cell-surface display—Applications of molecular display. Appl. Microbiol. Biotechnol. 2004, 64, 28–40. [Google Scholar] [CrossRef]
- Cherf, G.M.; Cochran, J.R. Applications of yeast surface display for protein engineering. Methods Mol. Biol. 2015, 1319, 155–175. [Google Scholar]
- Mersich, C.; Billes, W.; Pabinger, I.; Jungbauer, A. Peptides derived from a secretory yeast library restore factor VIII activity in the presence of an inhibitory antibody. Biotechnol. Bioeng. 2007, 98, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.J.; Cochran, J.R. Engineering knottins as novel binding agents. Methods Enzymol. 2012, 503, 223–251. [Google Scholar] [PubMed]
- Hetrick, K.J.; Walker, M.C.; Van Der Donk, W.A. Development and application of yeast and phage display of diverse lanthipeptides. ACS Cent. Sci. 2018, 4, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Kritzer, J.A.; Hamamichi, S.; McCaffery, J.M.; Santagata, S.; Naumann, T.A.; Caldwell, K.A.; Caldwell, G.A.; Lindquist, S. Rapid selection of cyclic peptides that reduce α-synuclein toxicity in yeast and animal models. Nat. Chem. Biol. 2009, 5, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Andreu, C.; del Olmo, M. Yeast arming systems: Pros and cons of different protein anchors and other elements required for display. Appl. Microbiol. Biotechnol. 2018, 102, 2543–2561. [Google Scholar] [CrossRef] [PubMed]
- Angelini, A.; Chen, T.F.; De Picciotto, S.; Yang, N.J.; Tzeng, A.; Santos, M.S.; Van Deventer, J.A.; Traxlmayr, M.W.; Dane Wittrup, K. Protein engineering and selection using yeast surface display. Methods Mol. Biol. 2015, 1319, 3–36. [Google Scholar]
- Benatuil, L.; Perez, J.M.; Belk, J.; Hsieh, C.M. An improved yeast transformation method for the generation of very large human antibody libraries. Protein Eng. Des. Sel. 2010, 23, 155–159. [Google Scholar] [CrossRef]
- Orcutt, K.D.; Wittrup, K.D. Yeast display and selections. In Antibody Engineering; Springer: Berlin/Heidelberg, Germany, 2010; pp. 207–233. [Google Scholar]
- Zhang, N.; Liu, L.; Dan Dumitru, C.; Cummings, N.R.H.; Cukan, M.; Jiang, Y.; Li, Y.; Li, F.; Mitchell, T.; Mallem, M.R.; et al. Glycoengineered pichia produced anti-HER2 is comparable to trastuzumab in preclinical study. MAbs 2011, 3, 289–298. [Google Scholar] [CrossRef]
- Grabherr, R.; Ernst, W. Baculovirus for eukaryotic protein display. Curr. Gene Ther. 2010, 10, 195–200. [Google Scholar] [CrossRef]
- Ernst, W.; Schinko, T.; Spenger, A.; Oker-Blom, C.; Grabherr, R. Improving baculovirus transduction of mammalian cells by surface display of a RGD-motif. J. Biotechnol. 2006, 126, 237–240. [Google Scholar] [CrossRef]
- Song, L.; Liu, Y.; Chen, J. Baculoviral capsid display of His-tagged ZnO inorganic binding peptide. Cytotechnology 2010, 62, 133–141. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ernst, W.; Grabherr, R.; Wegner, D.; Borth, N.; Grassauer, A.; Katinger, H. Baculovirus surface display: Construction and screening of a eukaryotic epitope library. Nucleic Acids Res. 1998, 26, 1718–1723. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, R.; Ernst, W.; Oker-Blom, C.; Jones, I. Developments in the use of baculovirusesfor the surface display of complex eukaryotic proteins. Trends Biotechnol. 2001, 19, 231–236. [Google Scholar] [CrossRef]
- Wang, Y.; Rubtsov, A.; Heiser, R.; White, J.; Crawford, F.; Marrack, P.; Kappler, J.W. Using a baculovirus display library to identify MHC class I mimotopes. Proc. Natl. Acad. Sci. USA 2005, 102, 2476–2481. [Google Scholar] [CrossRef]
- Crawford, F.; Jordan, K.R.; Stadinski, B.; Wang, Y.; Huseby, E.; Marrack, P.; Slansky, J.E.; Kappler, J.W. Use of baculovirus MHC/peptide display libraries to characterize T-cell receptor ligands. Immunol. Rev. 2006, 210, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Kost, T.A.; Kemp, C.W. Fundamentals of baculovirus expression and applications. Adv. Exp. Med. Biol. 2016, 896, 187–197. [Google Scholar] [PubMed]
- Chan, L.C.L.; Reid, S. Development of serum-free media for lepidopteran insect cell lines. Methods Mol. Biol. 2016, 1350, 161–196. [Google Scholar]
- Geisler, C.; Jarvis, D. Insect cell glycosylation patterns in the context of biopharmaceuticals. In Post-Translational Modification of Protein Biopharmaceuticals; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; pp. 165–191. ISBN 9783527320745. [Google Scholar]
- Possee, R.D.; King, L.A. Baculovirus transfer vectors. Methods Mol. Biol. 2016, 1350, 51–71. [Google Scholar]
- Khare, P.D.; Rosales, A.G.; Bailey, K.R.; Russell, S.J.; Federspiel, M.J. Epitope selection from an uncensored peptide library displayed on avian leukosis virus. Virology 2003, 315, 313–321. [Google Scholar] [CrossRef]
- Bupp, K.; Sarangi, A.; Roth, M.J. Probing sequence variation in the receptor-targeting domain of feline leukemia virus envelope proteins with peptide display libraries. J. Virol. 2005, 79, 1463–1469. [Google Scholar] [CrossRef]
- Sarangi, A.; Bupp, K.; Roth, M.J. Identification of a retroviral receptor used by an Envelope protein derived by peptide library screening. Proc. Natl. Acad. Sci. USA 2007, 104, 11032–11037. [Google Scholar] [CrossRef] [PubMed]
- Michelfelder, S.; Kohlschütter, J.; Skorupa, A.; Pfennings, S.; Müller, O.; Kleinschmidt, J.A.; Trepel, M. Successful expansion but not complete restriction of tropism of adeno-associated virus by in vivo biopanning of random virus display peptide libraries. PLoS ONE 2009, 4, e5122. [Google Scholar] [CrossRef] [PubMed]
- Müller, O.J.; Kaul, F.; Weitzman, M.D.; Pasqualini, R.; Arap, W.; Kleinschmidt, J.A.; Trepel, M. Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors. Nat. Biotechnol. 2003, 21, 1040–1046. [Google Scholar] [CrossRef]
- Waterkamp, D.A.; Müller, O.J.; Ying, Y.; Trepel, M.; Kleinschmidt, J.A. Isolation of targeted AAV2 vectors from novel virus display libraries. J. Gene Med. 2006, 8, 1307–1319. [Google Scholar] [CrossRef]
- Michelfelder, S.; Lee, M.K.; deLima-Hahn, E.; Wilmes, T.; Kaul, F.; Müller, O.; Kleinschmidt, J.A.; Trepel, M. Vectors selected from adeno-associated viral display peptide libraries for leukemia cell-targeted cytotoxic gene therapy. Exp. Hematol. 2007, 35, 1766–1776. [Google Scholar] [CrossRef]
- Varadi, K.; Michelfelder, S.; Korff, T.; Hecker, M.; Trepel, M.; Katus, H.A.; Kleinschmidt, J.A.; Müller, O.J. Novel random peptide libraries displayed on AAV serotype 9 for selection of endothelial cell-directed gene transfer vectors. Gene Ther. 2012, 19, 800–809. [Google Scholar] [CrossRef]
- Körbelin, J.; Hunger, A.; Alawi, M.; Sieber, T.; Binder, M.; Trepel, M. Optimization of design and production strategies for novel adeno-associated viral display peptide libraries. Gene Ther. 2017, 24, 470–481. [Google Scholar] [CrossRef]
- Marsic, D.; Zolotukhin, S. Altering tropism of rAAV by directed evolution. Methods Mol. Biol. 2016, 1382, 151–173. [Google Scholar]
- Büning, H.; Srivastava, A. Capsid modifications for targeting and improving the efficacy of AAV vectors. Mol. Ther. Methods Clin. Dev. 2019, 12, 248–265. [Google Scholar] [CrossRef]
- Wolkowicz, R.; Jager, G.C.; Nolan, G.P. A random peptide library fused to CCR5 for selection of mimetopes expressed on the mammalian cell surface via retroviral vectors. J. Biol. Chem. 2005, 280, 15195–15201. [Google Scholar] [CrossRef]
- Xu, X.; Leo, C.; Jang, Y.; Chan, E.; Padilla, D.; Huang, B.C.B.; Lin, T.; Gururaja, T.; Hitoshi, Y.; Lorens, J.B.; et al. Dominant effector genetics in mammalian cells. Nat. Genet. 2001, 27, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Peelle, B.; Lorens, J.; Li, W.; Bogenberger, J.; Payan, D.G.; Anderson, D.C. Intracellular protein scaffold-mediated display of random peptide libraries for phenotypic screens in mammalian cells. Chem. Biol. 2001, 8, 521–534. [Google Scholar] [CrossRef]
- Peelle, B.; Gururaja, T.L.; Payan, D.G.; Anderson, D.C. Characterization and use of green fluorescent proteins from Renilla mulleri and Ptilosarcus guernyi for the human cell display of functional peptides. J. Protein Chem. 2001, 20, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, T.M.; Ohashi, C.T.; Harder, A.G.; Yam, G.C.; Li, W.; Peelle, B.; Pali, E.S.; Bennett, M.K.; Molineaux, S.M.; Anderson, D.A.; et al. Retrovirally delivered random cyclic peptide libraries yield inhibitors of interleukin-4 signaling in human B cells. J. Biol. Chem. 2002, 277, 37512–37518. [Google Scholar] [CrossRef]
- Tavassoli, A. SICLOPPS cyclic peptide libraries in drug discovery. Curr. Opin. Chem. Biol. 2017, 38, 30–35. [Google Scholar] [CrossRef]
- Pavankumar, T. Inteins: Localized distribution, gene regulation, and protein engineering for biological applications. Microorganisms 2018, 6, 19. [Google Scholar] [CrossRef]
- Horswill, A.R.; Savinov, S.N.; Benkovic, S.J. A systematic method for identifying small-molecule modulators of protein-protein interactions. Proc. Natl. Acad. Sci. USA 2004, 101, 15591–15596. [Google Scholar] [CrossRef]
- Mistry, I.N.; Tavassoli, A. Reprogramming the transcriptional response to hypoxia with a chromosomally encoded cyclic peptide HIF-1 inhibitor. ACS Synth. Biol. 2017, 6, 518–527. [Google Scholar] [CrossRef]
- Castillo, F.; Tavassoli, A. Genetic selections with SICLOPPS Libraries: Toward the identification of novel protein-protein interaction inhibitors and chemical tools. Methods Mol. Biol. 2019, 2001, 317–328. [Google Scholar]
- Obexer, R.; Walport, L.J.; Suga, H. Exploring sequence space: Harnessing chemical and biological diversity towards new peptide leads. Curr. Opin. Chem. Biol. 2017, 38, 52–61. [Google Scholar] [CrossRef]
- Li, R.; Kang, G.; Hu, M.; Huang, H. Ribosome display: A potent display technology used for selecting and evolving specific binders with desired properties. Mol. Biotechnol. 2019, 61, 60–71. [Google Scholar] [CrossRef]
- Gersuk, G.M.; Corey, M.J.; Corey, E.; Stray, J.E.; Kawasaki, G.H.; Vessella, R.L. High-affinity peptide ligands to prostate-specific antigen identified by polysome selection. Biochem. Biophys. Res. Commun. 1997, 232, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, H.; Kanamori, T.; Osada, E.; Akbar, B.K.; Ueda, T. Peptide screening using pure ribosome display. Methods Mol. Biol. 2012, 805, 251–259. [Google Scholar] [PubMed]
- Mattheakis, L.C.; Bhatt, R.R.; Dower, W.J. An in vitro polysome display system for identifying ligands from very large peptide libraries. Proc. Natl. Acad. Sci. USA 1994, 91, 9022–9026. [Google Scholar] [CrossRef]
- Lamla, T.; Erdmann, V.A. Searching sequence space for high-affinity binding peptides using ribosome display. J. Mol. Biol. 2003, 329, 381–388. [Google Scholar] [CrossRef]
- Wada, A.; Sawata, S.Y.; Ito, Y. Ribosome display selection of a metal-binding motif from an artificial peptide library. Biotechnol. Bioeng. 2008, 101, 1102–1107. [Google Scholar] [CrossRef]
- Dreier, B.; Plückthun, A. Ribosome display: A technology for selecting and evolving proteins from large libraries. Methods Mol. Biol. 2011, 687, 283–306. [Google Scholar]
- Kanamori, T.; Fujino, Y.; Ueda, T. PURE ribosome display and its application in antibody technology. Biochim. Biophys. Acta Proteins Proteom. 2014, 1844, 1925–1932. [Google Scholar] [CrossRef]
- Wada, A. Ribosome display technology for selecting peptide and protein ligands. In Biomedical Applications of Functionalized Nanomaterials: Concepts, Development and Clinical Translation; Elsevier: Amsterdam, The Netherlands, 2018; pp. 89–104. ISBN 9780323508797. [Google Scholar]
- Barendt, P.A.; Ng, D.T.W.; McQuade, C.N.; Sarkar, C.A. Streamlined protocol for mRNA display. ACS Comb. Sci. 2013, 15, 77–81. [Google Scholar] [CrossRef]
- Wilson, D.S.; Keefe, A.D.; Szostak, J.W. The use of mRNA display to select high-affinity protein-binding peptides. Proc. Natl. Acad. Sci. USA 2001, 98, 3750–3755. [Google Scholar] [CrossRef]
- Horiya, S.; Bailey, J.K.; Krauss, I.J. Directed evolution of glycopeptides using mRNA display. Methods Enzymol. 2017, 597, 83–141. [Google Scholar]
- Valencia, C.A.; Zou, J.; Liu, R. In vitro selection of proteins with desired characteristics using mRNA-display. Methods 2013, 60, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Gold, L. mRNA display: Diversity matters during in vitro selection. Proc. Natl. Acad. Sci. USA 2001, 98, 4825–4826. [Google Scholar] [CrossRef] [PubMed]
- Lamboy, J.A.; Tam, P.Y.; Lee, L.S.; Jackson, P.J.; Avrantinis, S.K.; Lee, H.J.; Corn, R.M.; Weiss, G.A. Chemical and genetic wrappers for improved phage and RNA display. Chembiochem 2008, 9, 2846–2852. [Google Scholar] [CrossRef]
- Yamaguchi, J.; Naimuddin, M.; Biyani, M.; Sasaki, T.; Machida, M.; Kubo, T.; Funatsu, T.; Husimi, Y.; Nemoto, N. cDNA display: A novel screening method for functional disulfide-rich peptides by solid-phase synthesis and stabilization of mRNA-protein fusions. Nucleic Acids Res. 2009, 37, e108. [Google Scholar] [CrossRef]
- Nemoto, N.; Tsutsui, C.; Yamaguchi, J.; Ueno, S.; Machida, M.; Kobayashi, T.; Sakai, T. Antagonistic effect of disulfide-rich peptide aptamers selected by cDNA display on interleukin-6-dependent cell proliferation. Biochem. Biophys. Res. Commun. 2012, 421, 129–133. [Google Scholar] [CrossRef]
- Ueno, S.; Yoshida, S.; Mondal, A.; Nishina, K.; Koyama, M.; Sakata, I.; Miura, K.; Hayashi, Y.; Nemoto, N.; Nishigaki, K.; et al. In vitro selection of a peptide antagonist of growth hormone secretagogue receptor using cDNA display. Proc. Natl. Acad. Sci. USA 2012, 109, 11121–11126. [Google Scholar] [CrossRef]
- Mochizuki, Y.; Nishigaki, K.; Nemoto, N. Amino group binding peptide aptamers with double disulphide-bridged loops selected by in vitro selection using cDNA display. Chem. Commun. 2014, 50, 5608–5610. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Matsuno, M.; Tanaka, M.; Wada, A.; Kitamura, K.; Takei, O.; Sasaki, R.; Mizukami, T.; Hasegawa, M. Complementary DNA display selection of high-affinity peptides binding the vacuolating toxin (VacA) of Helicobacter pylori. J. Pept. Sci. 2015, 21, 710–716. [Google Scholar] [CrossRef]
- Doi, N.; Yamakawa, N.; Matsumoto, H.; Yamamoto, Y.; Nagano, T.; Matsumura, N.; Horisawa, K.; Yanagawa, H. DNA display selection of peptide ligands for a full-length human G protein-coupled receptor on CHO-K1 cells. PLoS ONE 2012, 7, e30084. [Google Scholar] [CrossRef]
- Odegrip, R.; Coomber, D.; Eldridge, B.; Hederer, R.; Kuhlman, P.A.; Ullman, C.; FitzGerald, K.; McGregor, D. CIS display: In vitro selection of peptides from libraries of protein-DNA complexes. Proc. Natl. Acad. Sci. USA 2004, 101, 2806–2810. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, B.; Cooley, R.N.; Odegrip, R.; McGregor, D.P.; FitzGerald, K.J.; Ullman, C.G. An in vitro selection strategy for conferring protease resistance to ligand binding peptides. Protein Eng. Des. Sel. 2009, 22, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Mathonet, P.; Jaulent, A.M.; Ullman, C.G. Selection of a high-affinity WW domain against the extracellular region of VEGF receptor isoform-2 from a combinatorial library using CIS display. Protein Eng. Des. Sel. 2013, 26, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Tawfik, D.S.; Griffiths, A.D. Man-made cell-like compartments for molecular evolution. Nat. Biotechnol. 1998, 16, 652–656. [Google Scholar] [CrossRef]
- Bernath, K.; Hai, M.; Mastrobattista, E.; Griffiths, A.D.; Magdassi, S.; Tawfik, D.S. In vitro compartmentalization by double emulsions: Sorting and gene enrichment by fluorescence activated cell sorting. Anal. Biochem. 2004, 325, 151–157. [Google Scholar] [CrossRef]
- Doi, N.; Yanagawa, H. STABLE: Protein-DNA fusion system for screening of combinatorial protein libraries in vitro. FEBS Lett. 1999, 457, 227–230. [Google Scholar] [CrossRef]
- Kaltenbach, M.; Hollfelder, F. SNAP display: In vitro protein evolution in microdroplets. Methods Mol. Biol. 2012, 805, 101–111. [Google Scholar]
- Contreras-Llano, L.E.; Tan, C. High-throughput screening of biomolecules using cell-free gene expression systems. Synth. Biol. 2018, 3. [Google Scholar] [CrossRef]
- Körfer, G.; Pitzler, C.; Vojcic, L.; Martinez, R.; Schwaneberg, U. In vitro flow cytometry-based screening platform for cellulase engineering. Sci. Rep. 2016, 6, 26128. [Google Scholar] [CrossRef]
- Yonezawa, M.; Doi, N.; Kawahashi, Y.; Higashinakagawa, T.; Yanagawa, H. DNA display for in vitro selection of diverse peptide libraries. Nucleic Acids Res. 2003, 31, e118. [Google Scholar] [CrossRef]
- Sepp, A.; Tawfik, D.S.; Griffiths, A.D. Microbead display by in vitro compartmentalisation: Selection for binding using flow cytometry. FEBS Lett. 2002, 532, 455–458. [Google Scholar] [CrossRef]
- Rothe, A.; Surjadi, R.N.; Power, B.E. Novel proteins in emulsions using in vitro compartmentalization. Trends Biotechnol. 2006, 24, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Sunami, T.; Matsuura, T.; Yomo, T. Directed evolution of proteins through in vitro protein synthesis in liposomes. J. Nucleic Acids 2012, 2012, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fallah-Araghi, A.; Baret, J.C.; Ryckelynck, M.; Griffiths, A.D. A completely in vitro ultrahigh-throughput droplet-based microfluidic screening system for protein engineering and directed evolution. Lab Chip 2012, 12, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Seelig, B. MRNA display for the selection and evolution of enzymes from in vitro-translated protein libraries. Nat. Protoc. 2011, 6, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Tjhung, K.F.; Kitov, P.I.; Ng, S.; Kitova, E.N.; Deng, L.; Klassen, J.S.; Derda, R. Silent encoding of chemical post-translational modifications in phage-displayed libraries. J. Am. Chem. Soc. 2016, 138, 32–35. [Google Scholar] [CrossRef] [PubMed]
- Heinis, C.; Winter, G. Encoded libraries of chemically modified peptides. Curr. Opin. Chem. Biol. 2015, 26, 89–98. [Google Scholar] [CrossRef]
- Roxin, Á.; Zheng, G. Flexible or fixed: A comparative review of linear and cyclic cancer-targeting peptides. Future Med. Chem. 2012, 4, 1601–1618. [Google Scholar] [CrossRef]
- Soudy, R.; Gill, A.; Sprules, T.; Lavasanifar, A.; Kaur, K. Proteolytically stable cancer targeting peptides with high affinity for breast cancer cells. J. Med. Chem. 2011, 54, 7523–7534. [Google Scholar] [CrossRef]
- Valentine, J.; Tavassoli, A. Genetically encoded cyclic peptide libraries: From hit to lead and beyond. Methods Enzymol. 2018, 610, 117–134. [Google Scholar]
- Gang, D.; Kim, D.W.; Park, H.S. Cyclic peptides: Promising scaffolds for biopharmaceuticals. Genes 2018, 9, 557. [Google Scholar] [CrossRef] [PubMed]
- Freire, F.; Gellman, S.H. Macrocyclic design strategies for small, stable parallel β-sheet scaffolds. J. Am. Chem. Soc. 2009, 131, 7970–7972. [Google Scholar] [CrossRef] [PubMed]
- Huyer, G.; Kelly, J.; Moffat, J.; Zamboni, R.; Jia, Z.; Gresser, M.J.; Ramachandran, C. Affinity selection from peptide libraries to determine substrate specificity of protein tyrosine phosphatases. Anal. Biochem. 1998, 258, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Moradi, S.V.; Hussein, W.M.; Varamini, P.; Simerska, P.; Toth, I. Glycosylation, an effective synthetic strategy to improve the bioavailability of therapeutic peptides. Chem. Sci. 2016, 7, 2492–2500. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.; Jafari, M.R.; Matochko, W.L.; Derda, R. Quantitative synthesis of genetically encoded glycopeptide libraries displayed on M13 phage. ACS Chem. Biol. 2012, 7, 1482–1487. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Tsao, M.L.; Schultz, P.G. A phage display system with unnatural amino acids. J. Am. Chem. Soc. 2004, 126, 15962–15963. [Google Scholar] [CrossRef]
- Dwyer, M.A.; Lu, W.; Dwyer, J.J.; Kossiakoff, A.A. Biosynthetic phage display: A novel protein engineering tool combining chemical and genetic diversity. Chem. Biol. 2000, 7, 263–274. [Google Scholar] [CrossRef][Green Version]
- Kale, S.S.; Villequey, C.; Kong, X.D.; Zorzi, A.; Deyle, K.; Heinis, C. Cyclization of peptides with two chemical bridges affords large scaffold diversities. Nat. Chem. 2018, 10, 715–723. [Google Scholar] [CrossRef]
- Heinis, C.; Rutherford, T.; Freund, S.; Winter, G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat. Chem. Biol. 2009, 5, 502–507. [Google Scholar] [CrossRef]
- Derda, R.; Ng, S. Genetically encoded fragment-based discovery. Curr. Opin. Chem. Biol. 2019, 50, 128–137. [Google Scholar] [CrossRef]
- Young, T.S.; Schultz, P.G. Beyond the canonical 20 amino acids: Expanding the genetic lexicon. J. Biol. Chem. 2010, 285, 11039–11044. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Katoh, T.; Suga, H. Flexizymes for genetic code reprogramming. Nat. Protoc. 2011, 6, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.S.; Liu, D.R. Methods for the directed evolution of proteins. Nat. Rev. Genet. 2015, 16, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.P.; Huang, J.; Wang, L.F.; Li, J.; Wu, Z.R. A new approach to random mutagenesis in vitro. Biotechnol. Bioeng. 2004, 86, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Myers, R.M.; Lerman, L.S.; Maniatis, T. A general method for saturation mutagenesis of cloned DNA fragments. Science 1985, 229, 242–247. [Google Scholar] [CrossRef]
- Cox, E.C. Bacterial mutator genes and the control of spontaneous mutation. Annu. Rev. Genet. 1976, 10, 135–156. [Google Scholar] [CrossRef]
- Greener, A.; Callahan, M.; Jerpseth, B. An efficient random mutagenesis technique using an E. coli mutator strain. Appl. Biochem. Biotechnol. Part B Mol. Biotechnol. 1997, 7, 189–195. [Google Scholar]
- Scheuermann, R.; Tam, S.; Burgers, P.M.J. Identification of the ε-subunit of Escherichia coli DNA polymerase III holoenzyme as the dnaQ gene product: A fidelity subunit for DNA replication. Proc. Natl. Acad. Sci. USA 1983, 80, 7085–7089. [Google Scholar] [CrossRef]
- Badran, A.H.; Liu, D.R. Development of potent in vivo mutagenesis plasmids with broad mutational spectra. Nat. Commun. 2015, 6, 8425. [Google Scholar] [CrossRef]
- Ravikumar, A.; Arrieta, A.; Liu, C.C. An orthogonal DNA replication system in yeast. Nat. Chem. Biol. 2014, 10, 175–177. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, H.; Jin, L.; Czornyj, E.; Hodes, A.; Hui, W.H.; Nieh, A.W.; Miller, J.F.; Hong Zhou, Z. A new topology of the HK97-like fold revealed in Bordetella bacteriophage by cryoEM at 3.5 Å resolution. Elife 2013, 2, e01299. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.Z.; Overstreet, C.M.; Moody, I.S.; Weiss, G.A. Protein Engineering with Biosynthesized Libraries from Bordetella bronchiseptica Bacteriophage. PLoS ONE 2013, 8, e55617. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.W.; Chen, E.; Goeddel, D.V. A Method for random mutagenesis of a defined DNA segment using a modified polymerase chain reaction. Technique 1989, 1, 11–15. [Google Scholar]
- Lin-Goerke, J.L.; Robbins, D.J.; Burczak, J.D. PCRr-based random mutagenesis using manganese and reduced DNTP concentration. Biotechniques 1997, 23, 409–412. [Google Scholar] [CrossRef]
- Zaccolo, M.; Williams, D.M.; Brown, D.M.; Gherardi, E. An approach to random mutagenesis of DNA using mixtures of triphosphate derivatives of nucleoside analogues. J. Mol. Biol. 1996, 255, 589–603. [Google Scholar] [CrossRef]
- McCullum, E.O.; Williams, B.A.R.; Zhang, J.; Chaput, J.C. Random mutagenesis by error-prone PCR. Methods Mol. Biol. 2010, 634, 103–109. [Google Scholar]
- Mondon, P.; Grand, D.; Souyris, N.; Emond, S.; Bouayadi, K.; Kharrat, H. MutagenTM: A random mutagenesis method providing a complementary diversity generated by human error-prone DNA polymerases. Methods Mol. Biol. 2010, 634, 373–386. [Google Scholar]
- Vanhercke, T.; Ampe, C.; Tirry, L.; Denolf, P. Reducing mutational bias in random protein libraries. Anal. Biochem. 2005, 339, 9–14. [Google Scholar] [CrossRef]
- Ye, J.; Wen, F.; Xu, Y.; Zhao, N.; Long, L.; Sun, H.; Yang, J.; Cooley, J.; Todd Pharr, G.; Webby, R.; et al. Error-prone pcr-based mutagenesis strategy for rapidly generating high-yield influenza vaccine candidates. Virology 2015, 482, 234–243. [Google Scholar] [CrossRef]
- Tee, K.L.; Wong, T.S. Polishing the craft of genetic diversity creation in directed evolution. Biotechnol. Adv. 2013, 31, 1707–1721. [Google Scholar] [CrossRef]
- Wong, T.S. Sequence saturation mutagenesis (SeSaM): A novel method for directed evolution. Nucleic Acids Res. 2004, 32, e26. [Google Scholar] [CrossRef] [PubMed]
- Mundhada, H.; Marienhagen, J.; Scacioc, A.; Schenk, A.; Roccatano, D.; Schwaneberg, U. SeSaM-Tv-II generates a protein sequence space that is unobtainable by epPCR. ChemBioChem 2011, 12, 1595–1601. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Xu, Z.; Wang, G.S.; Ji, F.Y.; Mei, C.X.; Liu, J.; Wu, G.M. Directed evolution of an LBP/CD14 inhibitory peptide and its anti-endotoxin activity. PLoS ONE 2014, 9, e101406. [Google Scholar] [CrossRef] [PubMed]
- Selas Castiñeiras, T.; Williams, S.G.; Hitchcock, A.; Cole, J.A.; Smith, D.C.; Overton, T.W. Development of a generic β-lactamase screening system for improved signal peptides for periplasmic targeting of recombinant proteins in Escherichia coli. Sci. Rep. 2018, 8, 6986. [Google Scholar] [CrossRef] [PubMed]
- Zahnd, C.; Spinelli, S.; Luginbühl, B.; Amstutz, P.; Cambillau, C.; Plückthun, A. Directed in vitro evolution and crystallographic analysis of a peptide-binding single chain antibody fragment (scFv) with low picomolar affinity. J. Biol. Chem. 2004, 279, 18870–18877. [Google Scholar] [CrossRef] [PubMed]
- Fujii, R.; Kitaoka, M.; Hayashi, K. One-step random mutagenesis by error-prone rolling circle amplification. Nucleic Acids Res. 2004, 32, e145. [Google Scholar] [CrossRef]
- Kunkel, T.A. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl. Acad. Sci. USA 1985, 82, 488–492. [Google Scholar] [CrossRef]
- Huovinen, T.; Brockmann, E.C.; Akter, S.; Perez-Gamarra, S.; Ylä-Pelto, J.; Liu, Y.; Lamminmäki, U. Primer extension mutagenesis powered by selective rolling circle amplification. PLoS ONE 2012, 7, e31817. [Google Scholar] [CrossRef]
- Meyer, A.J.; Ellefson, J.W.; Ellington, A.D. Library generation by gene shuffling. In Current Protocols in Molecular Biology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; Volume 105, pp. 15.12.1–15.12.7. [Google Scholar]
- Lim, C.C.; Choong, Y.S.; Lim, T.S. Cognizance of molecular methods for the generation of mutagenic phage display antibody libraries for affnity maturation. Int. J. Mol. Sci. 2019, 20, 1861. [Google Scholar] [CrossRef]
- Stemmer, W.P.C. DNA shuffling by random fragmentation and reassembly: In vitro recombination for molecular evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 10747–10751. [Google Scholar] [CrossRef]
- Reid, A.J. DNA shuffling: Modifying the hand that nature dealt. Vitr. Cell. Dev. Biol. Plant 2000, 36, 331–337. [Google Scholar] [CrossRef]
- Zhao, H.; Giver, L.; Shao, Z.; Affholter, J.A.; Arnold, F.H. Molecular evolution by staggered extension process (StEP) in vitro recombination. Nat. Biotechnol. 1998, 16, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Fujishima, K.; Venter, C.; Wang, K.; Ferreira, R.; Rothschild, L.J. An overhang-based DNA block shuffling method for creating a customized random library. Sci. Rep. 2015, 5, 9740. [Google Scholar] [CrossRef]
- Gonzalez-Perez, D.; Molina-Espeja, P.; Garcia-Ruiz, E.; Alcalde, M. Mutagenic organized recombination process by homologous in vivo grouping (MORPHING) for directed enzyme evolution. PLoS ONE 2014, 9, e90919. [Google Scholar] [CrossRef] [PubMed]
- García-Nafría, J.; Watson, J.F.; Greger, I.H. IVA cloning: A single-tube universal cloning system exploiting bacterial in vivo assembly. Sci. Rep. 2016, 6, 27459. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.H.; Potter, S.C.; Tanomrat, A.C.; Ravikumar, K.M.; Toney, M.D. Site-directed mutant libraries for isolating minimal mutations yielding functional changes. Protein Eng. Des. Sel. 2017, 30, 347–357. [Google Scholar] [CrossRef][Green Version]
- Zoller, M.J.; Smith, M. Oligonucleotide-Directed Mutagenesis: A Simple Method Using two Oligonucleotide Primers and a Single-Stranded DNA Template. Methods Enzymol. 1987, 154, 329–350. [Google Scholar]
- Walker, K.W. Site-directed mutagenesis. Encycl. Cell Biol. 2015, 1, 122–127. [Google Scholar]
- Rapley, R.; Braman, J.; Papworth, C.; Greener, A. Site-directed mutagenesis using double-stranded plasmid DNA templates. In The Nucleic Acid Protocols Handbook; Humana Press: Totowa, NJ, USA, 2003; Volume 9, pp. 835–844. [Google Scholar]
- Huang, R.; Fang, P.; Kay, B.K. Improvements to the Kunkel mutagenesis protocol for constructing primary and secondary phage-display libraries. Methods 2012, 58, 10–17. [Google Scholar] [CrossRef]
- Scholle, M.D.; Kehoe, J.W.; Kay, B.K. Efficient construction of a large collection of phage-displayed combinatorial peptide libraries. Comb. Chem. High Throughput Screen. 2005, 8, 545–551. [Google Scholar] [CrossRef]
- Lindahl, T. DNA glycosylases, endonucleases for apurinic/apyrimidinic sites, and base excision-repair. Prog. Nucleic Acid Res. Mol. Biol. 1979, 22, 135–192. [Google Scholar] [PubMed]
- Ho, S.N.; Hunt, H.D.; Horton, R.M.; Pullen, J.K.; Pease, L.R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 1989, 77, 51–59. [Google Scholar] [CrossRef]
- Chiu, J.; March, P.E.; Lee, R.; Tillett, D. Site-directed, ligase-independent mutagenesis (SLIM): A single-tube methodology approaching 100% efficiency in 4 h. Nucleic Acids Res. 2004, 32, e174. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Maynard, N.D.; Gill, J. DNA library construction using Gibson Assembly. Nat. Methods 2015, 12, 1–2. [Google Scholar] [CrossRef]
- Galka, P.; Jamez, E.; Joachim, G.; Soumillion, P. QuickLib, a method for building fully synthetic plasmid libraries by seamless cloning of degenerate oligonucleotides. PLoS ONE 2017, 12, e0175146. [Google Scholar] [CrossRef]
- Mitsis, P.G.; Kwagh, J.G. Characterization of the interaction of lambda exonuclease with the ends of DNA. Nucleic Acids Res. 1999, 27, 3057–3063. [Google Scholar] [CrossRef]
- Lim, B.N.; Choong, Y.S.; Ismail, A.; Glökler, J.; Konthur, Z.; Lim, T.S. Directed evolution of nucleotide-based libraries using lambda exonuclease. Biotechniques 2012, 53, 357–364. [Google Scholar] [CrossRef]
- Weiss, G.A.; Watanabe, C.K.; Zhong, A.; Goddard, A.; Sidhu, S.S. Rapid mapping of protein functional epitopes by combinatorial alanine scanning. Proc. Natl. Acad. Sci. USA 2000, 97, 8950–8954. [Google Scholar] [CrossRef]
- Morrison, K.L.; Weiss, G.A. Combinatorial alanine-scanning. Curr. Opin. Chem. Biol. 2001, 5, 302–307. [Google Scholar] [CrossRef]
- Chatellier, J.; Mazza, A.; Brousseau, R.; Vernet, T. Codon-based combinatorial alanine scanning site-directed mutagenesis: Design, implementation, and polymerase chain reaction screening. Anal. Biochem. 1995, 229, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Pál, G.; Fong, S.-Y.; Kossiakoff, A.A.; Sidhu, S.S. Alternative views of functional protein binding epitopes obtained by combinatorial shotgun scanning mutagenesis. Protein Sci. 2005, 14, 2405–2413. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.A.; Vasser, M.; Powers, D.B. Cassette mutagenesis: An efficient method for generation of multiple mutations at defined sites. Gene 1985, 34, 315–323. [Google Scholar] [CrossRef]
- Kegler-ebo, D.M.; Docktor, C.M.; Dimaio, D. Codon cassette mutagenesis: A general method to insert or replace individual codons by using universal mutagenic cassettes. Nucleic Acids Res. 1994, 22, 1593–1599. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Arkin, M. In vitro mutagenesis. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 46–50. ISBN 9780080961569. [Google Scholar]
- Lai, R.; Bekessy, A.; Chen, C.C.; Walsh, T.; Barnard, R. Megaprimer mutagenesis using very long primers. Biotechniques 2003, 34, 52–56. [Google Scholar] [CrossRef]
- Cárcamo, E.; Roldán-Salgado, A.; Osuna, J.; Bello-Sanmartin, I.; Yánez, J.A.; Saab-Rincón, G.; Viadiu, H.; Gaytán, P. Spiked genes: A method to introduce random point nucleotide mutations evenly throughout an entire gene using a complete set of spiked oligonucleotides for the assembly. ACS Omega 2017, 2, 3183–3191. [Google Scholar] [CrossRef]
- Hermes, J.D.; Parekh, S.M.; Blacklow, S.C.; Koster, H.; Knowles, J.R. A reliable method for random mutagenesis: The generation of mutant libraries using spiked oligodeoxyribonucleotide primers. Gene 1989, 84, 143–151. [Google Scholar] [CrossRef]
- Firnberg, E.; Ostermeier, M. PFunkel: Efficient, expansive, user-defined mutagenesis. PLoS ONE 2012, 7, e52031. [Google Scholar] [CrossRef]
- Valetti, F.; Gilardi, G. Improvement of biocatalysts for industrial and environmental purposes by saturation mutagenesis. Biomolecules 2013, 3, 778–811. [Google Scholar] [CrossRef]
- Sun, D.; Ostermaier, M.K.; Heydenreich, F.M.; Mayer, D.; Jaussi, R.; Standfuss, J.; Veprintsev, D.B. AAscan, PCRdesign and MutantChecker: A suite of programs for primer design and sequence analysis for high-throughput scanning mutagenesis. PLoS ONE 2013, 8, e78878. [Google Scholar] [CrossRef]
- Derbyshire, K.M.; Salvo, J.J.; Grindley, N.D.F. A simple and efficient procedure for saturation mutagenesis using mixed oligodeoxynucleotides. Gene 1986, 46, 145–152. [Google Scholar] [CrossRef]
- Arunachalam, T.S.; Wichert, C.; Appel, B.; Müller, S. Mixed oligonucleotides for random mutagenesis: Best way of making them. Org. Biomol. Chem. 2012, 10, 4641. [Google Scholar] [CrossRef] [PubMed]
- Siloto, R.M.P.; Weselake, R.J. Site saturation mutagenesis: Methods and applications in protein engineering. Biocatal. Agric. Biotechnol. 2012, 1, 181–189. [Google Scholar] [CrossRef]
- Nov, Y. When second best is good enough: Another probabilistic look at saturation mutagenesis. Appl. Environ. Microbiol. 2012, 78, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Gao, H.; Zhu, X.; Wang, X.; Zhou, M.; Jiang, R. Construction of “small-intelligent” focused mutagenesis libraries using well-designed combinatorial degenerate primers. Biotechniques 2012, 52, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Kille, S.; Acevedo-Rocha, C.G.; Parra, L.P.; Zhang, Z.G.; Opperman, D.J.; Reetz, M.T.; Acevedo, J.P. Reducing codon redundancy and screening effort of combinatorial protein libraries created by saturation mutagenesis. ACS Synth. Biol. 2013, 2, 83–92. [Google Scholar] [CrossRef]
- Gaytán, P.; Roldán-Salgado, A. Elimination of redundant and stop codons during the chemical synthesis of degenerate oligonucleotides. Combinatorial testing on the chromophore region of the red fluorescent protein mkate. ACS Synth. Biol. 2013, 2, 453–462. [Google Scholar] [CrossRef]
- Neuner, P.; Cortese, R.; Monaci, P. Codon-based mutagenesis using dimer-phosphoramidites. Nucleic Acids Res. 1998, 26, 1223–1227. [Google Scholar] [CrossRef]
- Ono, A.; Matsuda, A.; Zhao, J.; Santi, D.V. The synthesis of blocked triplet-phosphoramidites and their use in mutagenesis. Nucleic Acids Res. 1995, 23, 4677–4682. [Google Scholar] [CrossRef]
- Gaytán, P.; Contreras-Zambrano, C.; Ortiz-Alvarado, M.; Morales-Pablos, A.; Yáñez, J. TrimerDimer: An oligonucleotide-based saturation mutagenesis approach that removes redundant and stop codons. Nucleic Acids Res. 2009, 37, e125. [Google Scholar] [CrossRef]
- Hughes, M.D.; Nagel, D.A.; Santos, A.F.; Sutherland, A.J.; Hine, A.V. Removing the redundancy from randomised gene libraries. J. Mol. Biol. 2003, 331, 973–979. [Google Scholar] [CrossRef]
- Ashraf, M.; Frigotto, L.; Smith, M.E.; Patel, S.; Hughes, M.D.; Poole, A.J.; Hebaishi, H.R.M.; Ullman, C.G.; Hine, A.V. ProxiMAX randomization: A new technology for non-degenerate saturation mutagenesis of contiguous codons. Biochem. Soc. Trans. 2013, 41, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Pines, G.; Pines, A.; Garst, A.D.; Zeitoun, R.I.; Lynch, S.A.; Gill, R.T. Codon compression algorithms for saturation mutagenesis. ACS Synth. Biol. 2015, 4, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Wang, X.; Ru, B.; Sun, H.; Huang, J.; Gao, H. MDC-Analyzer: A novel degenerate primer design tool for the construction of intelligent mutagenesis libraries with contiguous sites. Biotechniques 2014, 56, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Acevedo-Rocha, C.G.; Reetz, M.T. Boosting the efficiency of site-saturation mutagenesis for a difficult-to-randomize gene by a two-step PCR strategy. Appl. Microbiol. Biotechnol. 2018, 102, 6095–6103. [Google Scholar] [CrossRef] [PubMed]
- Acevedo-Rocha, C.G.; Reetz, M.T.; Nov, Y. Economical analysis of saturation mutagenesis experiments. Sci. Rep. 2015, 5, 10654. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, A.; Silva, D.A.; Rocklin, G.J.; Hicks, D.R.; Vergara, R.; Murapa, P.; Bernard, S.M.; Zhang, L.; Lam, K.H.; Yao, G.; et al. Massively parallel de novo protein design for targeted therapeutics. Nature 2017, 550, 74–79. [Google Scholar] [CrossRef]
- Alford, R.F.; Leaver-Fay, A.; Jeliazkov, J.R.; O’Meara, M.J.; DiMaio, F.P.; Park, H.; Shapovalov, M.V.; Renfrew, P.D.; Mulligan, V.K.; Kappel, K.; et al. The Rosetta all-atom energy function for macromolecular modeling and design. J. Chem. Theory Comput. 2017, 13, 3031–3048. [Google Scholar] [CrossRef]
- Yang, K.K.; Wu, Z.; Arnold, F.H. Machine-learning-guided directed evolution for protein engineering. Nat. Methods 2019, 16, 687–694. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bozovičar, K.; Bratkovič, T. Evolving a Peptide: Library Platforms and Diversification Strategies. Int. J. Mol. Sci. 2020, 21, 215. https://doi.org/10.3390/ijms21010215
Bozovičar K, Bratkovič T. Evolving a Peptide: Library Platforms and Diversification Strategies. International Journal of Molecular Sciences. 2020; 21(1):215. https://doi.org/10.3390/ijms21010215
Chicago/Turabian StyleBozovičar, Krištof, and Tomaž Bratkovič. 2020. "Evolving a Peptide: Library Platforms and Diversification Strategies" International Journal of Molecular Sciences 21, no. 1: 215. https://doi.org/10.3390/ijms21010215
APA StyleBozovičar, K., & Bratkovič, T. (2020). Evolving a Peptide: Library Platforms and Diversification Strategies. International Journal of Molecular Sciences, 21(1), 215. https://doi.org/10.3390/ijms21010215