The Effect of Proline cis-trans Isomerization on the Folding of the C-Terminal SH2 Domain from p85



Abstract
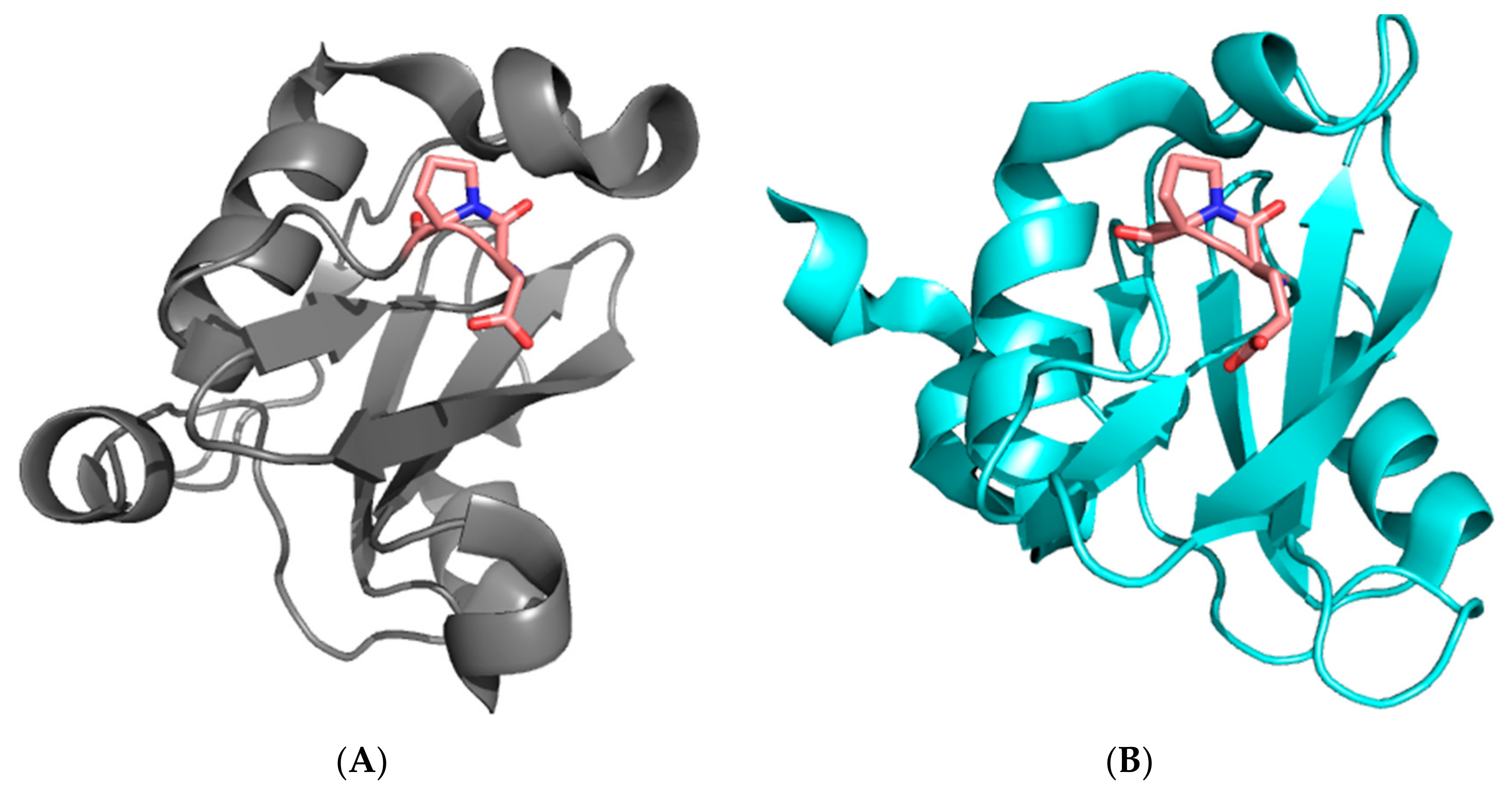
1. Introduction
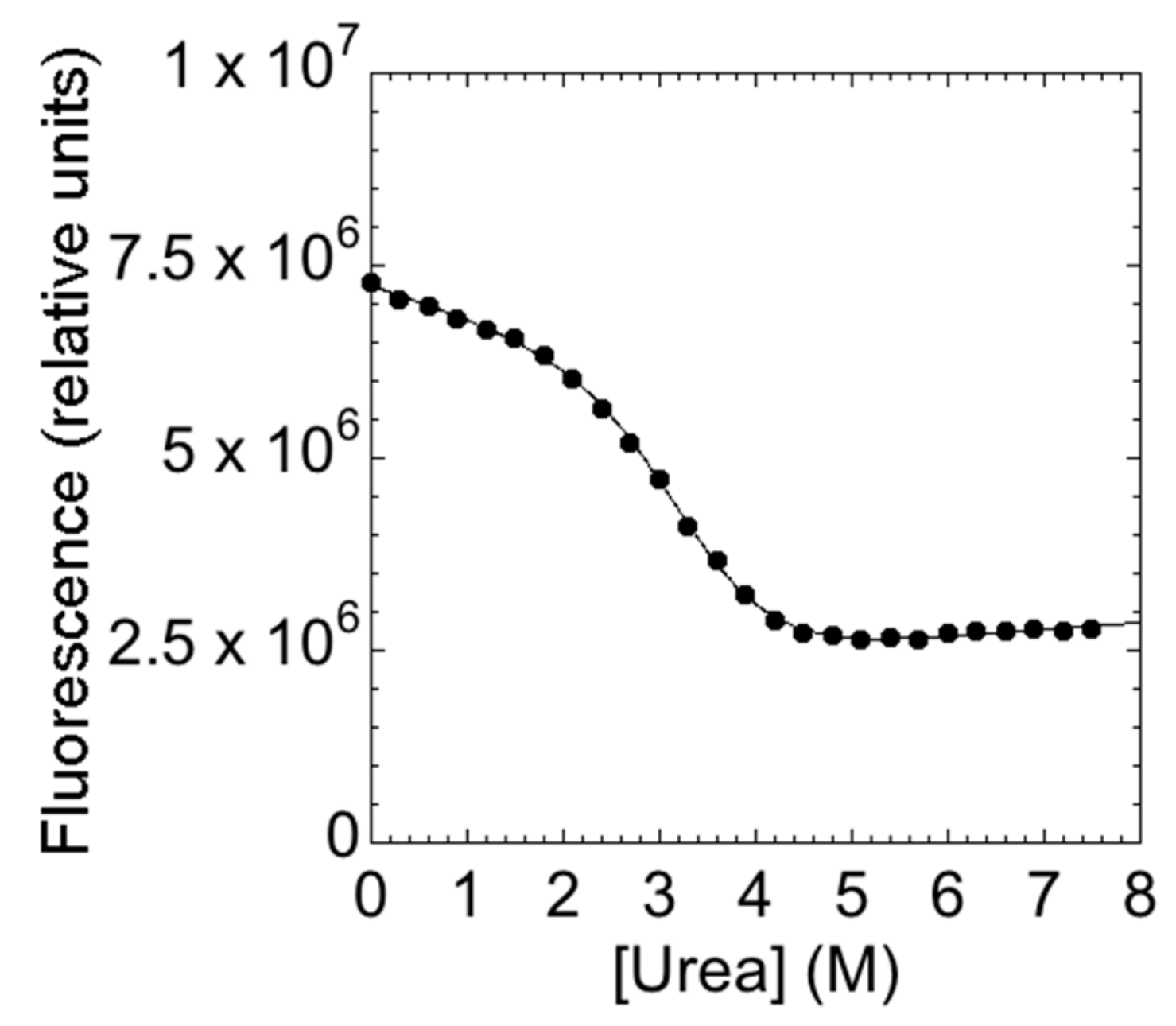
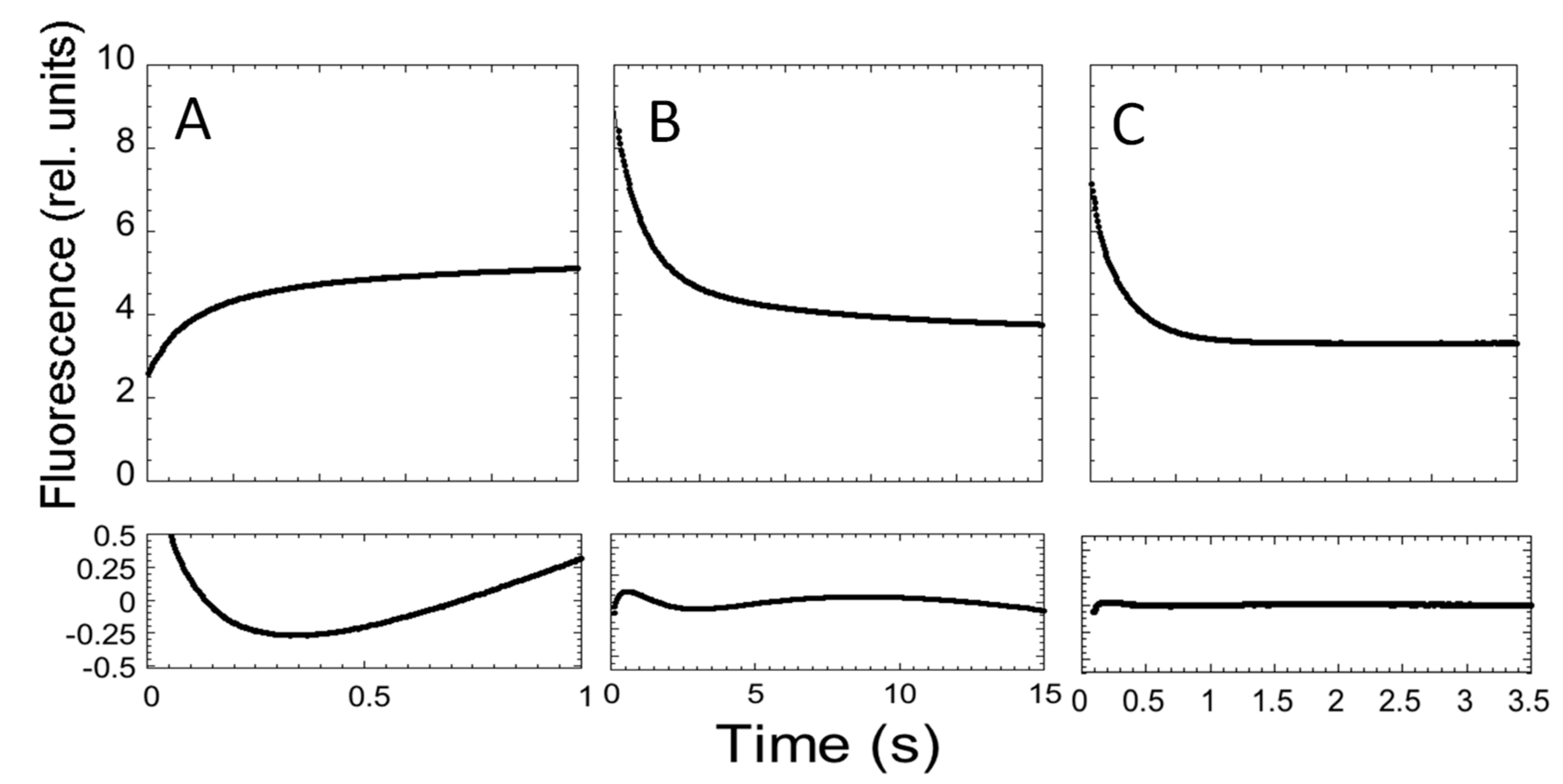
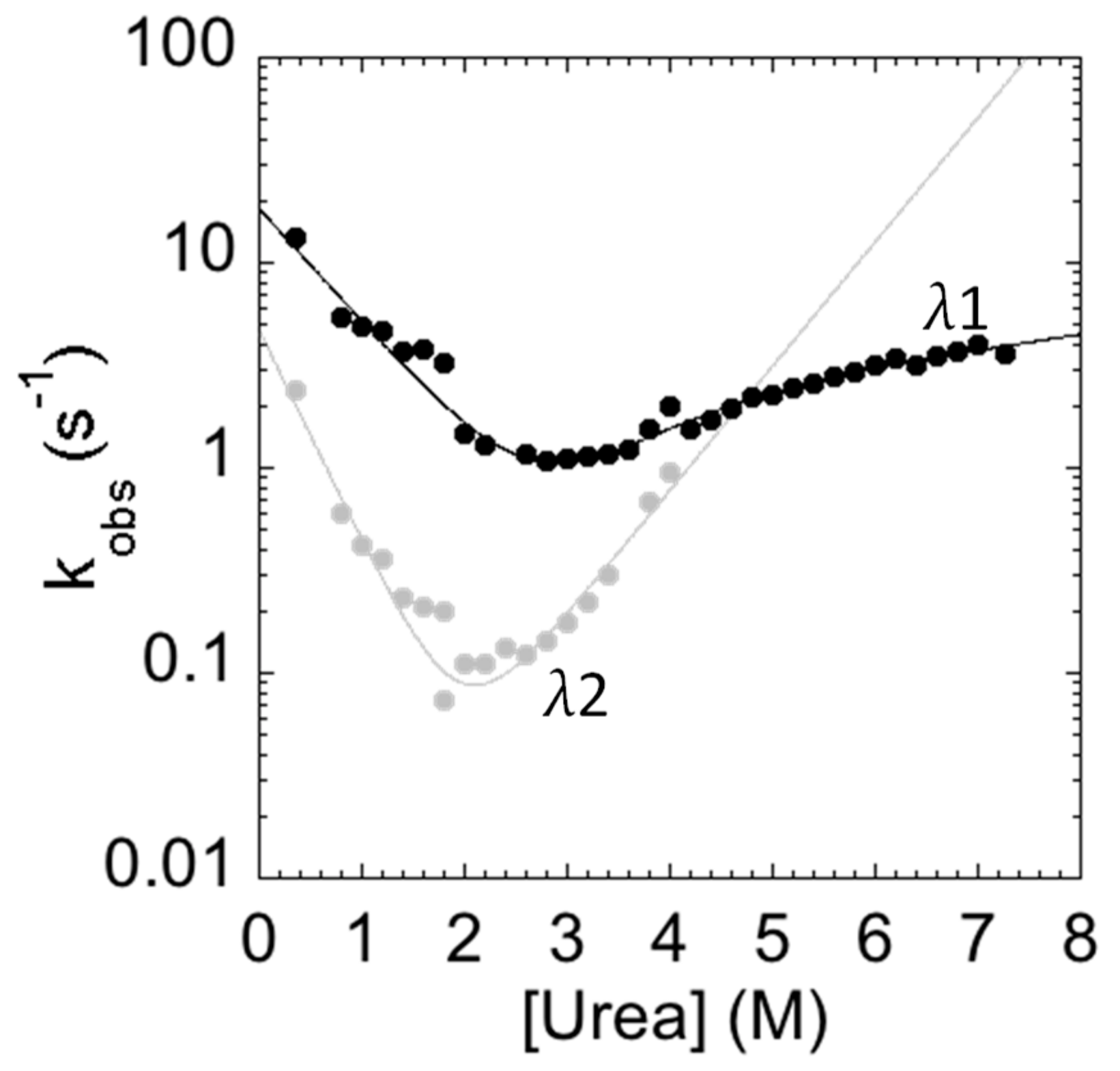
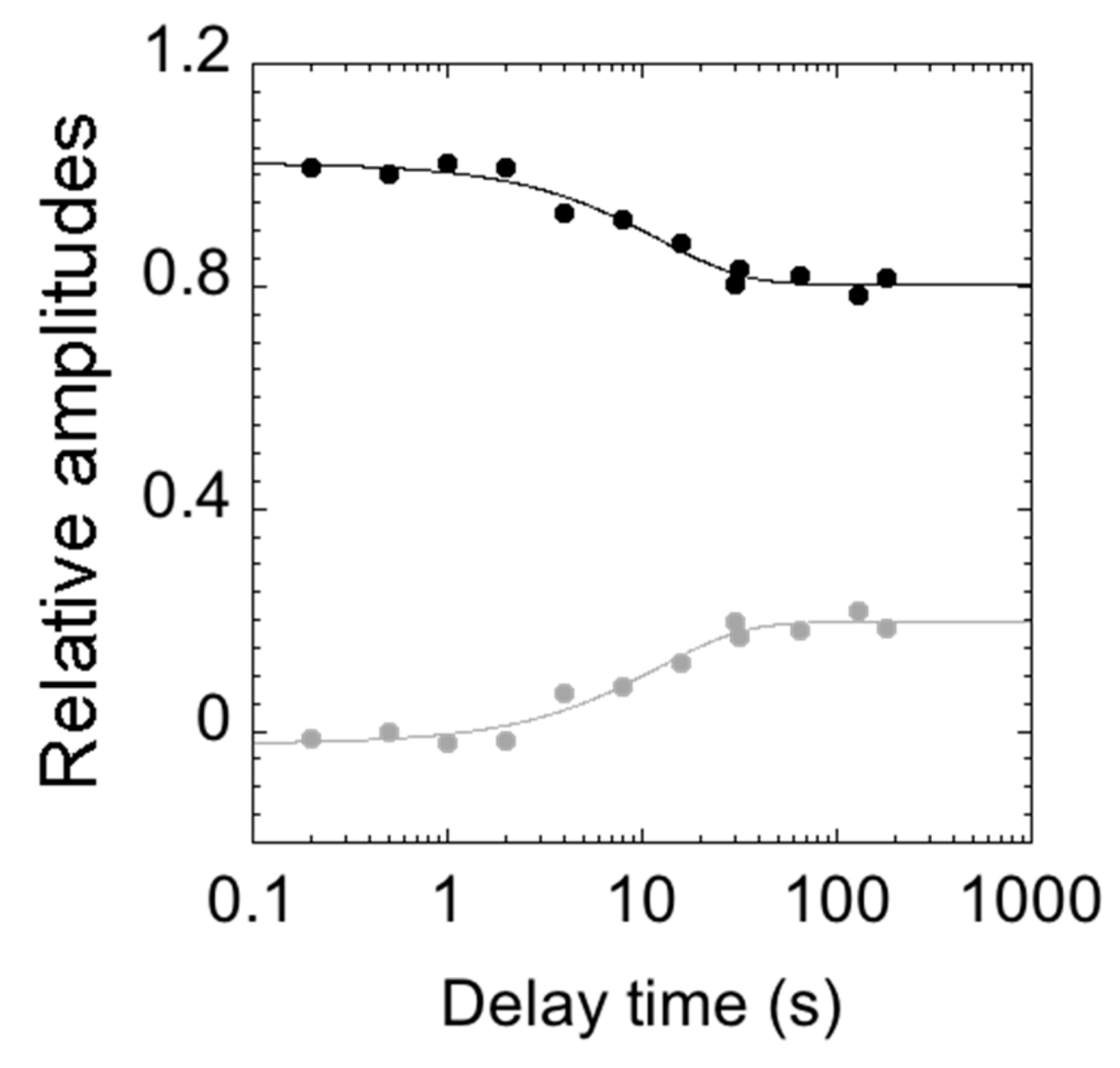
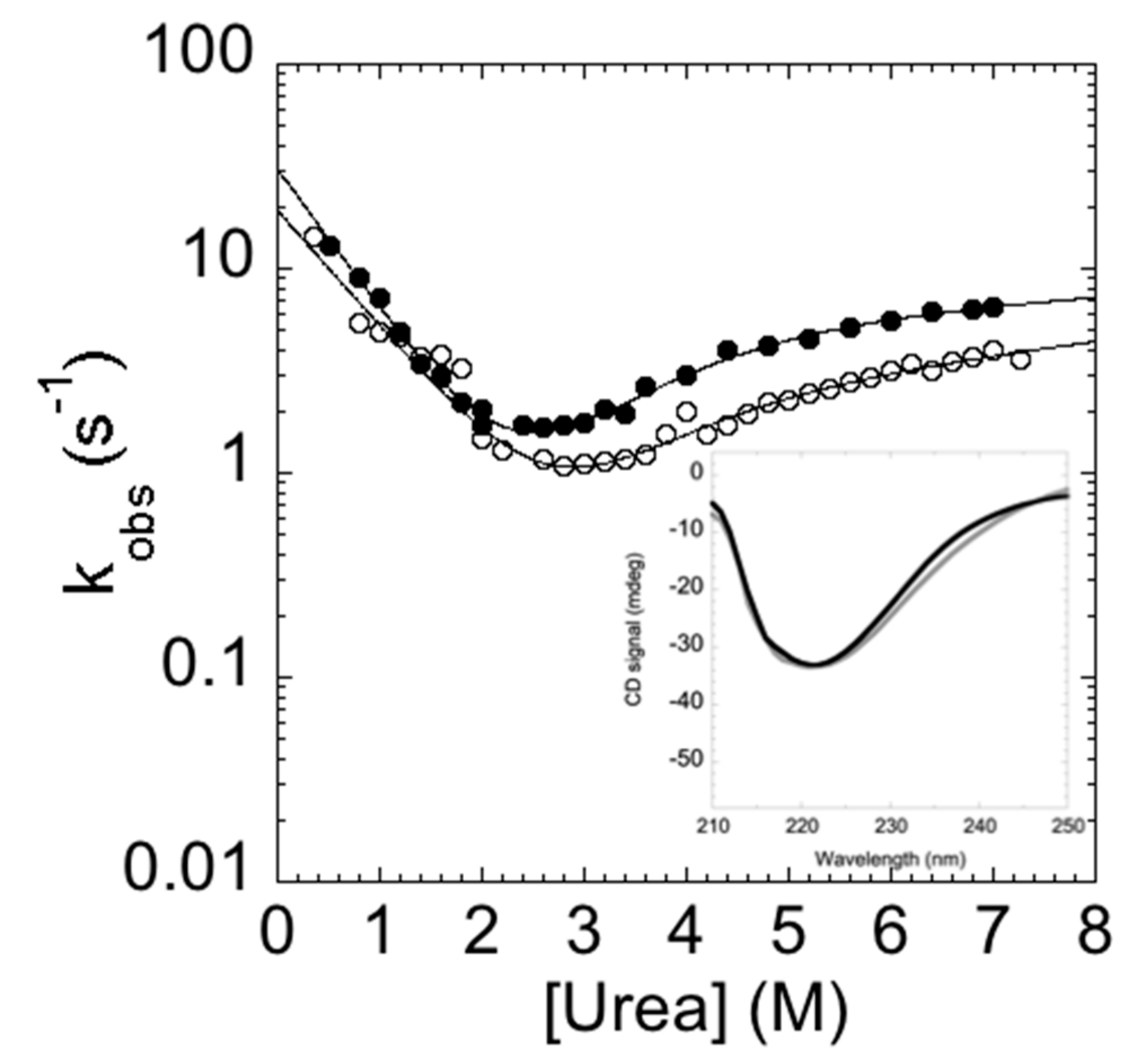
2. Results and Discussion
3. Methods
3.1. Site-Directed Mutagenesis
3.2. Protein Expression and Purification
3.3. Equilibrium Unfolding Experiments
3.4. Stopped-Flow Experiments
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bartlett, A.I.; Radford, S.E. An expanding arsenal of experimental methods yields an explosion of insights into protein folding mechanisms. Nat. Struct. Mol. Biol. 2009, 16, 582–588. [Google Scholar] [CrossRef]
- Chung, H.S.; McHale, K.; Louis, J.M.; Eaton, W.A. Single-molecule fluorescence experiments determine protein folding transition path times. Science 2012, 335, 981–984. [Google Scholar] [CrossRef]
- Daggett, V.; Fersht, A.R. Is there a unifying mechanism for protein folding? Trends Biochem. Sci. 2003, 28, 18–25. [Google Scholar] [CrossRef]
- Editorial. So much more to know Science. Science 2005, 309, 78–102. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Shaw, D.E. How fast-folding proteins fold. Science 2011, 334, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Schuler, B.; Eaton, W.A. Protein folding studied by single-molecule FRET. Curr. Opin. Struct. Biol. 2008, 18, 16–26. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Ann. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell. Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, J.; Antonelli, A.C.; Afridi, A.; Vatsia, S.; Joshi, G.; Romanov, V.; Murray, I.V.J.; Khan, S.A. Protein misfolding and aggregation in neurodegenerative diseases: A review of pathogeneses, novel detection strategies, and potential therapeutics. Rev. Neurosci. 2019, 30, 339–358. [Google Scholar] [CrossRef] [PubMed]
- Abkevich, V.I.; Gutin, A.M.; Shakhnovich, E.I. Specific nucleus as the transition state for protein folding: Evidence from the lattice model. Biochemistry 1994, 33, 10026–10036. [Google Scholar] [CrossRef] [PubMed]
- Fersht, A.R. From the first protein structures to our current knowledge of protein folding: Delights and scepticisms. Nat. Rev. Mol. Cell. Biol. 2008, 9, 650–654. [Google Scholar] [CrossRef]
- Gianni, S.; Jemth, P. Conserved nucleation sites reinforce the significance of Phi value analysis in protein-folding studies. IUBMB Life 2014, 66, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, L.S.; Otzen, D.E.; Fersht, A.R. The structure of the transition state for folding of chymotrypsin inhibitor 2 analysed by protein engineering methods: Evidence for a nucleation-condensation mechanism for protein folding. J. Mol. Biol. 1995, 254, 260–288. [Google Scholar] [CrossRef]
- Jackson, S.E.; Fersht, A.R. Folding of chymotrypsin inhibitor 2.1. Evidence for a two-state transition. Biochemistry 1991, 30, 10428–10435. [Google Scholar] [CrossRef]
- Gianni, S.; Ivarsson, Y.; De Simone, A.; Travaglini-Allocatelli, C.; Brunori, M.; Vendruscolo, M. Structural characterization of a misfolded intermediate populated during the folding process of a PDZ domain. Nat. Struct. Mol. Biol. 2010, 17, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Capaldi, A.P.; Shastry, M.C.; Kleanthous, C.; Roder, H.; Radford, S.E. Ultrarapid mixing experiments reveal that Im7 folds via an on-pathway intermediate. Nat. Struct. Biol. 2001, 8, 68–72. [Google Scholar] [PubMed]
- Jackson, S.E. How do small single-domain proteins fold? Fold. Des. 1998, 3, 81–91. [Google Scholar] [CrossRef]
- Schmid, F.X. Proline isomerization in unfolded ribonuclease A. The equilibrium between fast-folding and slow-folding species is independent of temperature. Eur. J. Biochem. 1992, 128, 77–80. [Google Scholar] [CrossRef]
- Kim, P.S.; Baldwin, R.L. Structural intermediates trapped during the folding of ribonuclease A by amide proton exchange. Biochemistry 1980, 19, 6124–6129. [Google Scholar] [CrossRef] [PubMed]
- Hurle, M.R.; Matthews, C.R. Proline isomerization and the slow folding reactions of the alpha subunit of tryptophan synthase from Escherichia coli. Biochim. Biophys. Acta 1987, 913, 179–184. [Google Scholar] [CrossRef]
- Kiefhaber, T.; Kohler, H.H.; Schmid, F.X. Kinetic coupling between protein folding and prolyl isomerization. I. Theoretical models. J. Mol. Biol. 1992, 224, 217–229. [Google Scholar] [CrossRef]
- Schultz, D.A.; Schmid, F.X.; Baldwin, R.L. Cis proline mutants of ribonuclease A. II. Elimination of the slow-folding forms by mutation. Protein Sci. 1992, 1, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, S.S.; Scheraga, H.A. Stability of cis, trans, and nonplanar peptide groups. Macromolecules 1976, 9, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Payne, G.; Stolz, L.A.; Pei, D.; Band, H.; Shoelson, S.E.; Walsh, C.T. The phosphopeptide-binding specificity of Src family SH2 domains. Chem. Biol. 1994, 1, 99–105. [Google Scholar] [CrossRef]
- Songyang, Z.; Shoelson, S.E.; Chaudhuri, M.; Gish, G.; Pawson, T.; Haser, W.G.; King, F.; Roberts, T.; Ratnofsky, S.; Lechleider, R.J.; et al. SH2 domains recognize specific phosphopeptide sequences. Cell 1993, 72, 767–778. [Google Scholar]
- Waksman, G.; Shoelson, S.E.; Pant, N.; Cowburn, D.; Kuriyan, J. Binding of a high affinity phosphotyrosyl peptide to the Src SH2 domain: Crystal structures of the complexed and peptide-free forms. Cell 1993, 72, 779–790. [Google Scholar] [CrossRef]
- Visconti, L.; Malagrinò, F.; Toto, A.; Gianni, S. The kinetics of folding of the NSH2 domain from p85. Sci. Rep. 2019, 9, 4058. [Google Scholar] [CrossRef]
- Myers, J.K.; Pace, C.N.; Scholtz, J.M. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995, 4, 2138–2148. [Google Scholar] [CrossRef]
- Dogan, J.; Toto, A.; Andersson, E.; Gianni, S.; Jemth, P. Activation barrier-limited folding and conformational sampling of a dynamic protein domain. Biochemistry 2016, 55, 5289–5295. [Google Scholar] [CrossRef]
- Ivarsson, Y.; Travaglini-Allocatelli, C.; Jemth, P.; Malatesta, F.; Brunori, M.; Gianni, S. An on-pathway intermediate in the folding of a PDZ domain. J. Biol. Chem. 2007, 282, 8568–8572. [Google Scholar] [CrossRef]
- Parker, M.J.; Spencer, J.; Clarke, A.R. An integrated kinetic analysis of intermediates and transition states in protein folding reactions. J. Mol. Biol. 1995, 253, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Shastry, M.C.; Roder, H. Evidence for barrier-limited protein folding kinetics on the microsecond time scale. Nat. Struct. Biol. 1998, 5, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Travaglini-Allocatelli, C.; Gianni, S.; Morea, V.; Tramontano, A.; Soulimane, T.; Brunori, M. Exploring the cytochrome c folding mechanism: Cytochrome c552 from thermus thermophilus folds through an on-pathway intermediate. J. Biol. Chem. 2003, 278, 41136–41140. [Google Scholar] [CrossRef] [PubMed]
- Bonetti, D.; Troilo, F.; Toto, A.; Travaglini-Allocatelli, C.; Brunori, M.; Gianni, S. Mechanism of Folding and Binding of the N-Terminal SH2 Domain from SHP2. J. Phys. Chem. B 2016, 122, 11108–11114. [Google Scholar] [CrossRef] [PubMed]
- Gianni, S.; Camilloni, C.; Giri, R.; Toto, A.; Bonetti, D.; Morrone, A.; Sormanni, P.; Brunori, M.; Vendruscolo, M. Understanding the frustration arising from the competition between function, misfolding, and aggregation in a globular protein. Proc. Natl. Acad. Sci. USA 2014, 111, 14141–14146. [Google Scholar] [CrossRef] [PubMed]
- Englander, S.W. Protein folding intermediates and pathways studied by hydrogen exchange. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 213–238. [Google Scholar] [CrossRef] [PubMed]
- Khorasanizadeh, S.; Peters, I.D.; Roder, H. Evidence for a three-state model of protein folding from kinetic analysis of ubiquitin variants with altered core residues. Nat. Struct. Biol. 1996, 3, 193–205. [Google Scholar] [CrossRef]
- Matouschek, A.; Kellis, J.T., Jr.; Serrano, L.; Bycroft, M.; Fersht, A.R. Transient folding intermediates characterized by protein engineering. Nature 1990, 346, 440–445. [Google Scholar] [CrossRef]
- Siegal, G.; Davis, B.; Kristensen, S.M.; Sankar, A.; Linacre, J.; Stein, R.C.; Panayotou, G.; Waterfield, M.D.; Driscoll, P.C. Solution structure of the C-terminal SH2 domain of the p85 alpha regulatory subunit of phosphoinositide 3-kinase. J. Mol. Biol. 1998, 276, 461–478. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | kf (s−1) | mf (kcal·M−1·mol−1) | ku (s−1) | mu (kcal·M−1·mol−1) | Kp (s−1) | mu (kcal·M−1·mol−1) |
|---|---|---|---|---|---|---|
| λ1 | 19.24 ± 2.15 | 0.81 ±0.07 | 0.030 ± 0.003 | 0.76 ± 0.33 | 0.020 ± 0.008 | 0.66 ± 0.26 |
| λ2 | 4.70 ± 0.60 | 1.47 ± 0.27 | 0.0030 ± 0.0008 | 0.86 ± 0.1 | ||
| P684A | 30.74 ± 4.12 | 0.98 ± 0.09 | 0.080 ± 0.008 | 0.70 ± 0.30 | 0.020 ± 0.003 | 0.64 ± 0.21 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Troilo, F.; Malagrinò, F.; Visconti, L.; Toto, A.; Gianni, S. The Effect of Proline cis-trans Isomerization on the Folding of the C-Terminal SH2 Domain from p85. Int. J. Mol. Sci. 2020, 21, 125. https://doi.org/10.3390/ijms21010125
Troilo F, Malagrinò F, Visconti L, Toto A, Gianni S. The Effect of Proline cis-trans Isomerization on the Folding of the C-Terminal SH2 Domain from p85. International Journal of Molecular Sciences. 2020; 21(1):125. https://doi.org/10.3390/ijms21010125
Chicago/Turabian StyleTroilo, Francesca, Francesca Malagrinò, Lorenzo Visconti, Angelo Toto, and Stefano Gianni. 2020. "The Effect of Proline cis-trans Isomerization on the Folding of the C-Terminal SH2 Domain from p85" International Journal of Molecular Sciences 21, no. 1: 125. https://doi.org/10.3390/ijms21010125
APA StyleTroilo, F., Malagrinò, F., Visconti, L., Toto, A., & Gianni, S. (2020). The Effect of Proline cis-trans Isomerization on the Folding of the C-Terminal SH2 Domain from p85. International Journal of Molecular Sciences, 21(1), 125. https://doi.org/10.3390/ijms21010125