Hepatoprotective Effect of Kombucha Tea in Rodent Model of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis

 ,
, 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
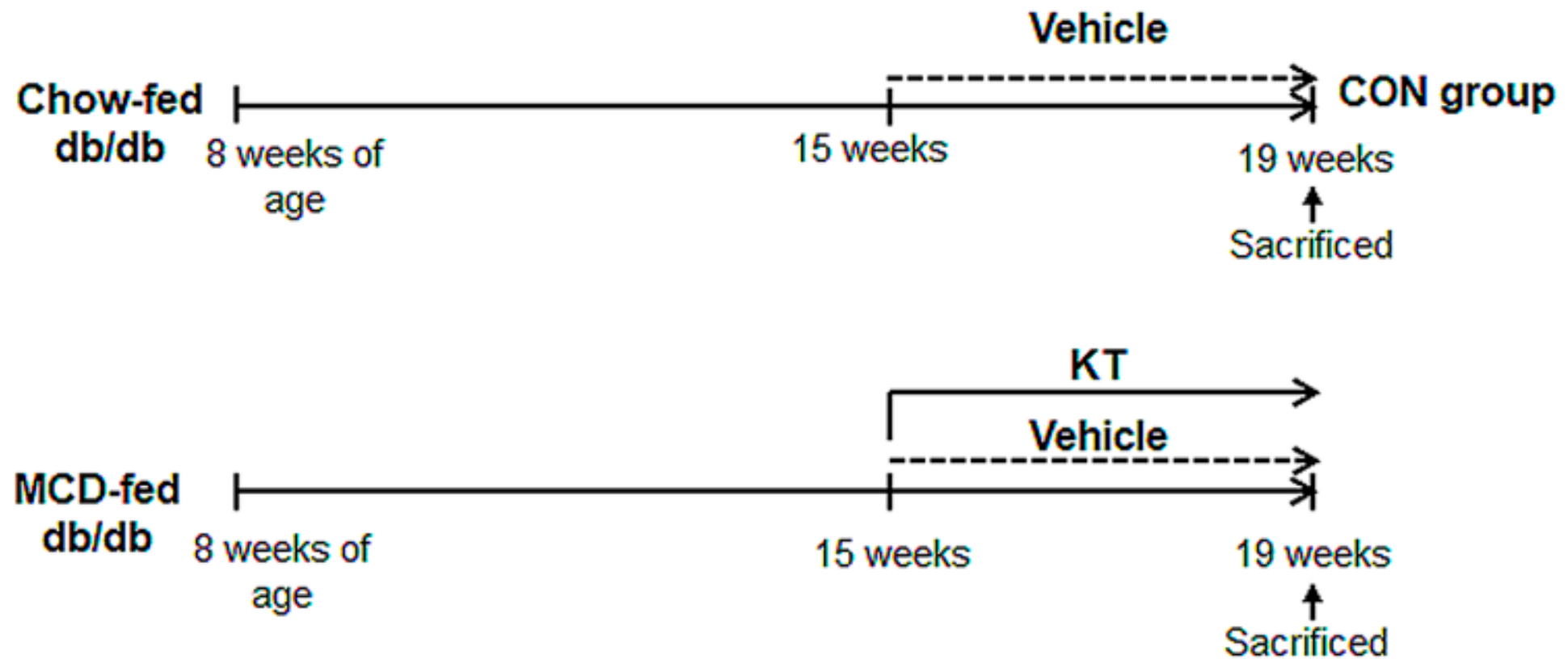
2.1. KT Attenuates Hepatic Damage Caused by MCD Diet in db/db Mice
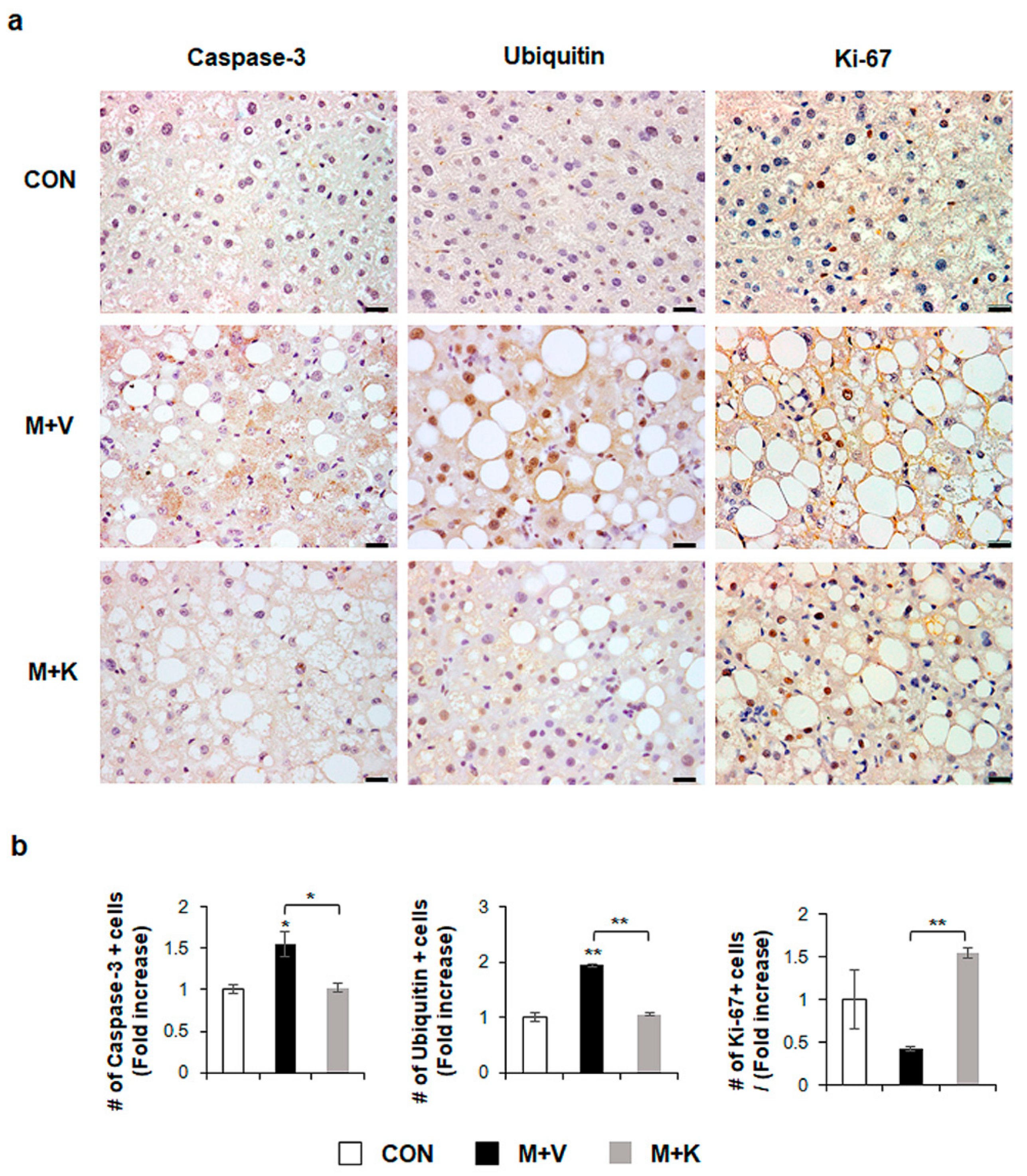
2.2. KT Improves Hepatocyte Survival
2.3. KT Reduces Lipotoxicity by Decreasing Hepatic FFA Level in MCD-Treated db/db Mice
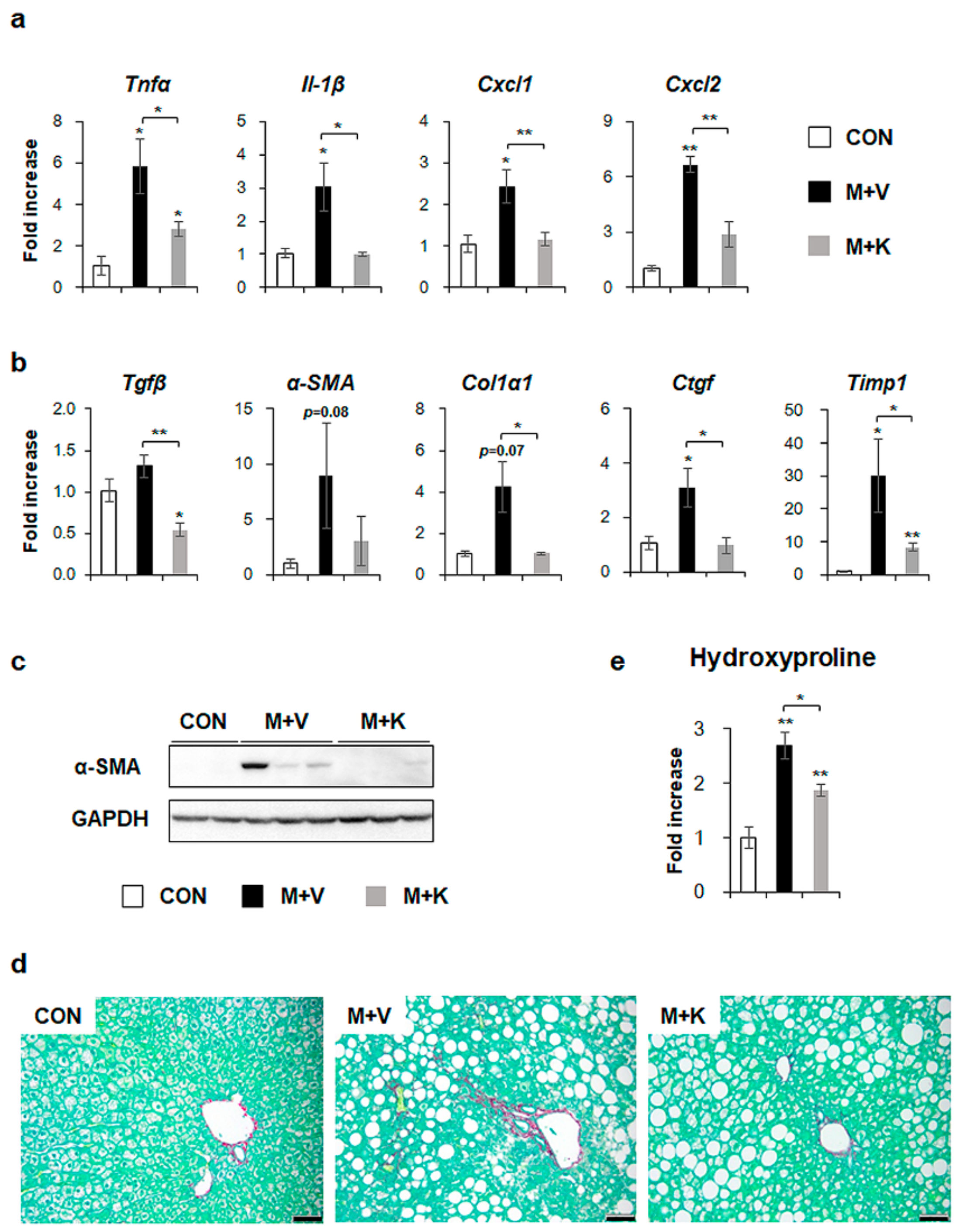
2.4. KT Reduces Inflammation and Fibrosis in the Liver of db/db Mice
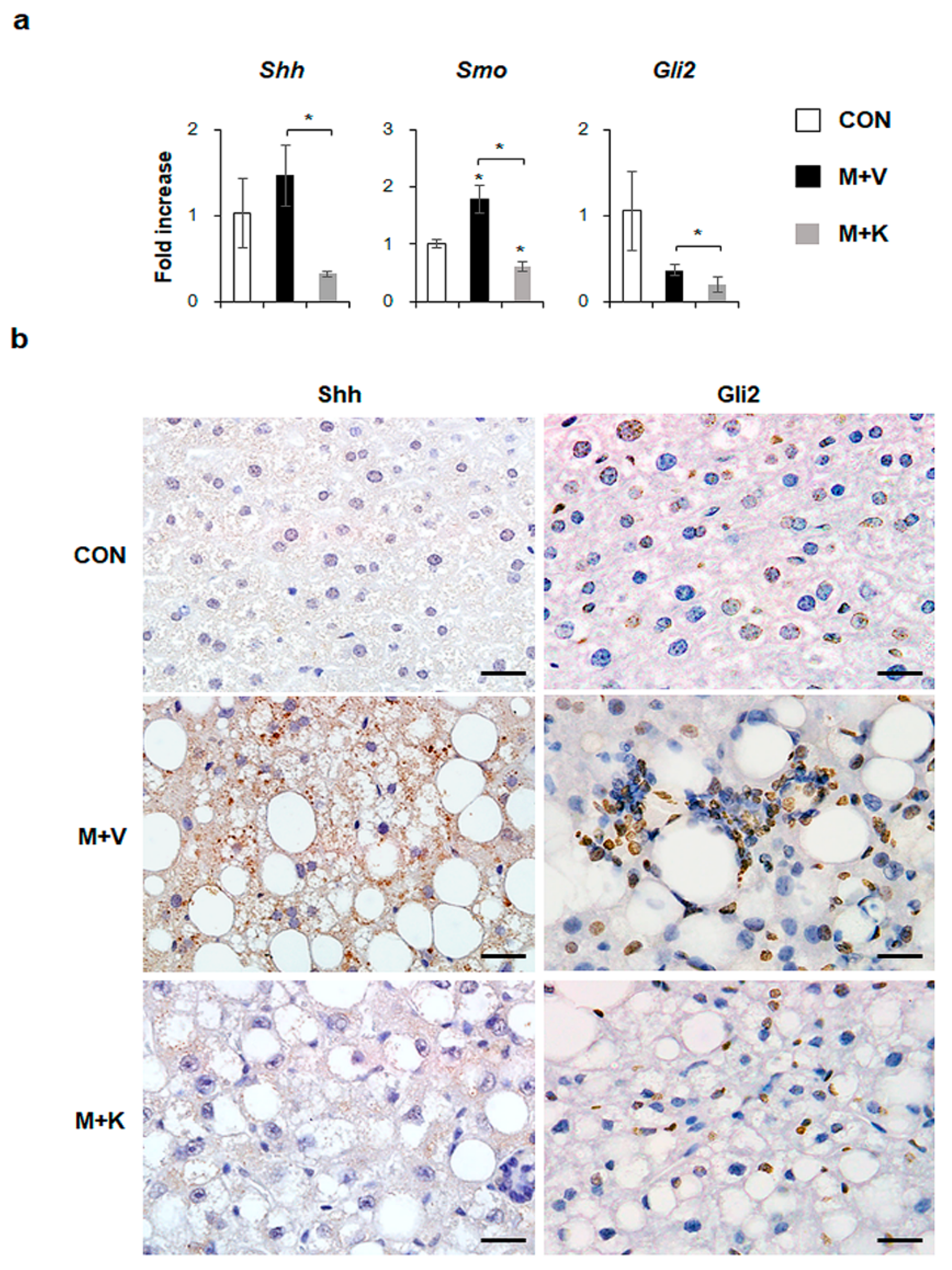
2.5. KT Suppresses Activation of Hh Signaling Pathways in Diet-Induced NASH
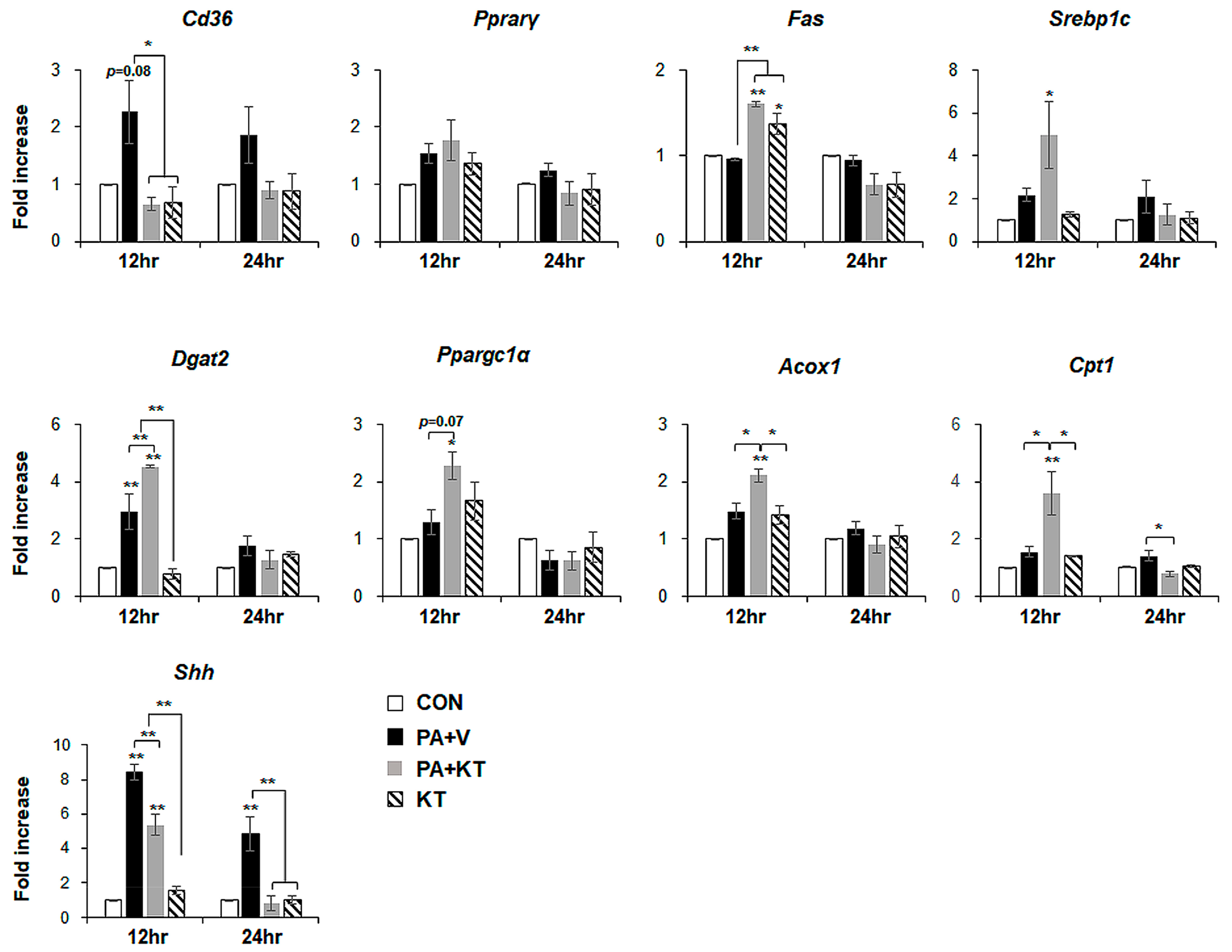
2.6. KT Prevents Palmitate-Induced Steatosis in Primary Hepatocytes
3. Discussion
4. Materials and Methods
4.1. Preparation of KT Extract
4.2. Animal Experiments
4.3. Isolation of Primary Hepatocytes and Cell Experiments
4.4. Liver Histology and Immunohistochemistry
4.5. RNA Isolation and Quantitative RT-PCR
4.6. Western Blot
4.7. Cell Proliferation Assay
4.8. Measurement of Triglyceride Level
4.9. Hydroxyproline Assay
4.10. Measurement of AST/ALT
4.11. Cell Quantification
4.12. Oil Red O Staining
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MCD | Methionine/Choline-deficient |
| KT | Kombucha Tea |
| NAFLD | Nonalcoholic Fatty liver Disease |
| NASH | Nonalcoholic Steatpohepatitis |
| ALT | Alanine Transaminase |
| AST | Aspartate Transaminase |
| TG | Triglyceride |
| G6pc | Glucose-6-phosphatase catalytic subunit |
| FFA | Free Fatty Acid |
| Cd36 | Cluster of differentiation 36 |
| Pparγ | Peroxisome proliferator-activated receptor gamma |
| Dgat2 | Diacylglycerol-O-acyltransferase 2 |
| Fas | Fatty acid synthase |
| Srebp1c | Sterol regulatory element binding protein 1c |
| Ppargc1α | Peroxisome proliferator-activated receptor gamma coactivator 1 alpha |
| Fabp1 | Fatty acid binding protein 1 |
| Acox1 | Acyl coenzyme A oxidase 1 |
| Cpt1 | Carnitine Palmitoyltansferase 1 |
| Pparα | Peroxisome proliferator-activated receptor alpha |
| Mcad | Medium-chain-coenzyme A dehydrogenase |
| Tnfα | Tumor necrosis factor alpha |
| Il-1β | Interleukin-1β |
| Cxc1 | Chemokine (C-X-C motif) ligand 1 |
| Cxcl2 | Chemokine (C-X-C motif) ligand 2 |
| Tgfβ | Transforming growth factor β |
| α-SMA | Alpha-smooth muscle actin |
| Col1α1 | Collagen type 1 alpha 1 |
| Ctgf | Connective tissue growth factor |
| Timp1 | Tissue inhibitor of metalloproteinase 1 |
| Shh | Sonic hedgehog |
| Smo | Smoothened |
| Gli2 | GLI-Kruppel family zinc finger 2 |
References
- Yu, J.; Shen, J.; Sun, T.T.; Zhang, X.; Wong, N. Obesity, insulin resistance, NASH and hepatocellular carcinoma. Semin. Cancer Biol. 2013, 23, 483–491. [Google Scholar]
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar]
- Fon Tacer, K.; Rozman, D. Nonalcoholic Fatty liver disease: Focus on lipoprotein and lipid deregulation. J. Lipids 2011, 2011, 783976. [Google Scholar]
- Brunt, E.M. Nonalcoholic steatohepatitis: Definition and pathology. Semin. Liver Dis. 2001, 21, 3–16. [Google Scholar]
- Musso, G.; Gambino, R.; Cassader, M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD). Prog. Lipid Res. 2009, 48, 1–26. [Google Scholar]
- Chitturi, S.; Farrell, G.C. Etiopathogenesis of nonalcoholic steatohepatitis. Semin. Liver Dis. 2001, 21, 27–41. [Google Scholar]
- Puri, P.; Bailie, R.A.; Weist, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar]
- Alkhouri, N.; Dixon, L.J.; Feldstein, A.E. Lipotoxicity in nonalcoholic fatty liver disease: Not all lipids are created equal. Expert. Rev. Gastroenterol. Hepatol. 2009, 3, 445–451. [Google Scholar]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar]
- Serviddio, G.; Bellanti, F.; Tamborra, R.; Rollo, T.; Capitanio, N.; Romano, A.D.; Sastre, J.; Vendemiale, G.; Altomare, E. Uncoupling protein-2 (UCP2) induces mitochondrial proton leak and increases susceptibility of non-alcoholic steatohepatitis (NASH) liver to ischaemia–reperfusion injury. Gut 2008, 57, 957–965. [Google Scholar]
- Feldstein, A.E.; Werneburg, N.W.; Canbay, A.; Guicciardi, M.E.; Bronk, S.F.; Rydzewski, R.; Burgart, L.J.; Gores, G.J. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-α expression via a lysosomal pathway. Hepatology 2004, 40, 185–194. [Google Scholar]
- Fujii, H.; Kawada, N. Inflammation and fibrogenesis in steatohepatitis. J. Gastroenterol. 2012, 47, 215–225. [Google Scholar]
- Jung, Y.; Brown, K.D.; Witek, R.P.; Omenetti, A.; Yang, L.; Vandongen, M.; Milton, R.J.; Hines, I.N.; Rippe, R.A.; Spahr, L.; et al. Accumulation of hedgehog-responsive progenitors parallels alcoholic liver disease severity in mice and humans. Gastroenterology 2008, 134, 1532–1543. [Google Scholar]
- Verdelho Machado, M.; Diehl, A.M. Role of Hedgehog Signaling Pathway in NASH. Int. J. Mol. Sci. 2016, 17, 857. [Google Scholar]
- Hirsova, P.; Gores, G.J. Ballooned hepatocytes, undead cells, sonic hedgehog, and vitamin E: Therapeutic implications for nonalcoholic steatohepatitis. Hepatology 2015, 61, 15–17. [Google Scholar]
- Rangwala, F.; Guy, C.D.; Lu, J.; Suzuki, A.; Burchette, J.L.; Abdelmalek, M.F.; Chen, W.; Diehl, A.M. Increased production of sonic hedgehog by ballooned hepatocytes. J. Pathol. 2011, 224, 401–410. [Google Scholar]
- Guy, C.D.; Suzuki, A.; Zdanowicz, M.; Abdelmalek, M.F.; Burchette, J.; Unalp, A.; Diehl, A.M. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology 2012, 55, 1711–1721. [Google Scholar]
- Machado, M.V.; Diehl, A.M. Pathogenesis of Nonalcoholic Steatohepatitis. Gastroenterology 2016, 150, 1769–1777. [Google Scholar]
- Dufresne, C.; Farnworth, E. Tea, Kombucha, and health: A review. Food Res. Int. 2000, 33, 409–421. [Google Scholar]
- Abshenas, J.; Derakhshanfar, A.; Ferdosi, M.H.; Hasanzadeh, S. Protective effect of kombucha tea against acetaminophen-induced hepatotoxicity in mice: A biochemical and histopathological study. Comp. Clin. Path. 2012, 21, 1243–1248. [Google Scholar]
- Aloulou, A.; Hamden, K.; Elloumi, D.; Ali, M.B.; Hargafi, K.; Jaouadi, B.; Ayadi, F.; Elfeki, A.; Ammar, E. Hypoglycemic and antilipidemic properties of kombucha tea in alloxan-induced diabetic rats. BMC Complement. Altern. Med. 2012, 12, 63–71. [Google Scholar]
- Hyun, J.; Lee, Y.; Wang, S.; Kim, J.; Kim, J.; Cha, J.; Seo, Y.-S.; Jung, Y. Kombucha tea prevents obese mice from developing hepatic steatosis and liver damage. Food Sci. Biotechnol. 2016, 25, 861–866. [Google Scholar]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar]
- Machado, M.V.; Michelotti, G.A.; Pereira, T. d. A.; Boursier, J.; Kruger, L.; Swiderska-Syn, M.; Karaca, G.; Xie, G.; Guy, C.D.; Bohinc, B.; et al. Reduced lipoapoptosis, hedgehog pathway activation and fibrosis in caspase-2 deficient mice with non-alcoholic steatohepatitis. Gut 2014, 64, 1148–1157. [Google Scholar]
- Lackner, C.; Gogg-Kamerer, M.; Zatloukal, K.; Stumptner, C.; Brunt, E.M.; Denk, H. Ballooned hepatocytes in steatohepatitis: The value of keratin immunohistochemistry for diagnosis. J. Hepatol. 2008, 48, 821–828. [Google Scholar]
- Guy, C.D.; Suzuki, A.; Burchette, J.L.; Brunt, E.M.; Abdelmalek, M.F.; Cardona, D.; McCall, S.J.; Unalp, A.; Belt, P.; Ferrell, L.D.; et al. Costaining for keratins 8/18 plus ubiquitin improves detection of hepatocyte injury in nonalcoholic fatty liver disease. Hum. Pathol. 2012, 43, 790–800. [Google Scholar]
- Thomas, S.; Johannes, G. The Ki-67 protein: From the known and the unknown. J. Cell Physiol. 2000, 182, 311–322. [Google Scholar]
- Varga, T.; Nagy, L. Nuclear receptors, transcription factors linking lipid metabolism and immunity: The case of peroxisome proliferator-activated receptor gamma. Eur. J. Clin. Investig. 2008, 38, 695–707. [Google Scholar]
- Zhou, J.; Febbraio, M.; Wada, T.; Zhai, Y.; Kuruba, R.; He, J.; Lee, J.H.; Khadem, S.; Ren, S.; Li, S.; et al. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology 2008, 134, 556–567. [Google Scholar]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar]
- Hellerstein, M.K. De novo lipogenesis in humans: Metabolic and regulatory aspects. Eur. J. Clin. Nutr. 1999, 53 (Suppl. 1), S53–S65. [Google Scholar]
- Dorn, C.; Riener, M.O.; Kirovski, G.; Saugspier, M.; Steib, K.; Weiss, T.S.; Gabele, E.; Kristiansen, G.; Hartmann, A.; Hellerbrand, C. Expression of fatty acid synthase in nonalcoholic fatty liver disease. Int. J. Clin. Exp. Pathol. 2010, 3, 505–514. [Google Scholar]
- Nagaya, T.; Tanaka, N.; Suzuki, T.; Sano, K.; Horiuchi, A.; Komatsu, M.; Nakajima, T.; Nishizawa, T.; Joshita, S.; Umemura, T.; et al. Down-regulation of SREBP-1c is associated with the development of burned-out NASH. J. Hepatol. 2010, 53, 724–731. [Google Scholar]
- Farrell, G.C.; van Rooyen, D.; Gan, L.; Chitturi, S. NASH is an Inflammatory Disorder: Pathogenic, Prognostic and Therapeutic Implications. Gut Liver 2012, 6, 149–171. [Google Scholar]
- Joshi-Barve, S.; Barve, S.S.; Amancherla, K.; Gobejishvili, L.; Hill, D.; Cave, M.; Hote, P.; McClain, C.J. Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes. Hepatology 2007, 46, 823–830. [Google Scholar]
- Egnatchik, R.A.; Leamy, A.K.; Noguchi, Y.; Shiota, M.; Young, J.D. Palmitate-induced activation of mitochondrial metabolism promotes oxidative stress and apoptosis in H4IIEC3 rat hepatocytes. Metabolism 2014, 63, 283–295. [Google Scholar]
- Anstee, Q.M.; Goldin, R.D. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int. J. Exp. Pathol. 2006, 87, 1–16. [Google Scholar]
- Sahai, A.; Malladi, P.; Pan, X.; Paul, R.; Melin-Aldana, H.; Green, R.M.; Whitington, P.F. Obese and diabetic db/db mice develop marked liver fibrosis in a model of nonalcoholic steatohepatitis: Role of short-form leptin receptors and osteopontin. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1035–G1043. [Google Scholar]
- Gyamfi, M.A.; Damjanov, I.; French, S.; Wan, Y.J. The pathogenesis of ethanol versus methionine and choline deficient diet-induced liver injury. Biochem. Pharmacol. 2008, 75, 981–995. [Google Scholar]
- Sommerfeld, A.; Reinehr, R.; Haussinger, D. Free fatty acids shift insulin-induced hepatocyte proliferation towards CD95-dependent apoptosis. J. Biol. Chem. 2015, 290, 4398–4409. [Google Scholar]
- Schreurs, M.; Kuipers, F.; van der Leij, F.R. Regulatory enzymes of mitochondrial β-oxidation as targets for treatment of the metabolic syndrome. Obes. Rev. 2010, 11, 380–388. [Google Scholar]
- Perez-Carreras, M.; Del Hoyo, P.; Martin, M.A.; Rubio, J.C.; Martin, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar]
- Nakamuta, M.; Kohjima, M.; Morizono, S.; Kotoh, K.; Yoshimoto, T.; Miyagi, I.; Enjoji, M. Evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2005, 16, 631–635. [Google Scholar]
- Nakade, Y.; Sakamoto, K.; Yamauchi, T.; Inoue, T.; Kobayashi, Y.; Yamamoto, T.; Ishii, N.; Ohashi, T.; Sumida, Y.; Ito, K.; et al. Conophylline inhibits non-alcoholic steatohepatitis in mice. PLoS ONE 2017, 12, e0178436. [Google Scholar]
- Wortham, M.; He, L.; Gyamfi, M.; Copple, B.L.; Wan, Y.J. The transition from fatty liver to NASH associates with SAMe depletion in db/db mice fed a methionine choline-deficient diet. Dig. Dis. Sci. 2008, 53, 2761–2774. [Google Scholar]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389–397. [Google Scholar]
- Richardson, M.M.; Jonsson, J.R.; Powell, E.E.; Brunt, E.M.; Neuschwander-Tetri, B.A.; Bhathal, P.S.; Dixon, J.B.; Weltman, M.D.; Tilg, H.; Moschen, A.R.; et al. Progressive fibrosis in nonalcoholic steatohepatitis: Association with altered regeneration and a ductular reaction. Gastroenterology 2007, 133, 80–90. [Google Scholar]
- Strable, M.S.; Ntambi, J.M. Genetic control of de novo lipogenesis: Role in diet-induced obesity. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 199–214. [Google Scholar]
- Jayabalan, R.; Malbaša, R.V.; Lončar, E.S.; Vitas, J.S.; Sathishkumar, M. A Review on Kombucha Tea—Microbiology, Composition, Fermentation, Beneficial Effects, Toxicity, and Tea Fungus. Compr. Rev. Food Sci. Food Saf. 2014, 13, 538–550. [Google Scholar]
- Greenwalt, C.J.; Steinkraus, K.H.; Ledford, R.A. Kombucha, the fermented tea: Microbiology, composition, and claimed health effects. J. Food Prot. 2000, 63, 976–981. [Google Scholar]
- Kumar, V.; Joshi, V. Kombucha: Technology, microbiology, production, composition and therapeutic value. Int. J. Food Ferment. Technol. 2016, 6, 13. [Google Scholar]
- Tumanov, S.; Bulusu, V.; Kamphorst, J.J. Analysis of Fatty Acid Metabolism Using Stable Isotope Tracers and Mass Spectrometry. Methods Enzymol. 2015, 561, 197–217. [Google Scholar]
- Vacca, M.; Allison, M.; Griffin, J.L.; Vidal-Puig, A. Fatty Acid and Glucose Sensors in Hepatic Lipid Metabolism: Implications in NAFLD. Semin. Liver Dis. 2015, 35, 250–261. [Google Scholar]
- Korenblat, K.M.; Fabbrini, E.; Mohammed, B.S.; Klein, S. Liver, muscle, and adipose tissue insulin action is directly related to intrahepatic triglyceride content in obese subjects. Gastroenterology 2008, 134, 1369–1375. [Google Scholar]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783. [Google Scholar]
- Omenetti, A.; Popov, Y.; Jung, Y.; Choi, S.S.; Witek, R.P.; Yang, L.; Brown, K.D.; Schuppan, D.; Diehl, A.M. The hedgehog pathway regulates remodelling responses to biliary obstruction in rats. Gut 2008, 57, 1275–1282. [Google Scholar]
- Kodama, T.; Takehara, T.; Hikita, H.; Shimizu, S.; Shigekawa, M.; Tsunematsu, H.; Li, W.; Miyagi, T.; Hosui, A.; Tatsumi, T.; et al. Increases in p53 expression induce CTGF synthesis by mouse and human hepatocytes and result in liver fibrosis in mice. J. Clin. Investig. 2011, 121, 3343–3356. [Google Scholar]
- Yang, L.; Roh, Y.S.; Song, J.; Zhang, B.; Liu, C.; Loomba, R.; Seki, E. Transforming growth factor beta signaling in hepatocytes participates in steatohepatitis through regulation of cell death and lipid metabolism in mice. Hepatology 2014, 59, 483–495. [Google Scholar]
- Jung, Y.; Witek, R.P.; Syn, W.K.; Choi, S.S.; Omenetti, A.; Premont, R.; Guy, C.D.; Diehl, A.M. Signals from dying hepatocytes trigger growth of liver progenitors. Gut 2010, 59, 655–665. [Google Scholar]
- Omenetti, A.; Choi, S.; Michelotti, G.; Diehl, A.M. Hedgehog signaling in the liver. J. Hepatol. 2011, 54, 366–373. [Google Scholar]
- Chen, Y.; Choi, S.S.; Michelotti, G.A.; Chan, I.S.; Swiderska-Syn, M.; Karaca, G.F.; Xie, G.; Moylan, C.A.; Garibaldi, F.; Premont, R.; et al. Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology 2012, 143, 1319–1329. [Google Scholar]
- Hooper, J.E.; Scott, M.P. Communicating with Hedgehogs. Nat. Rev. Mol. Cell Biol. 2005, 6, 306–317. [Google Scholar]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar]
- Ip, E.; Farrell, G.C.; Robertson, G.; Hall, P.; Kirsch, R.; Leclercq, I. Central role of PPARalpha-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology 2003, 38, 123–132. [Google Scholar]
- Horiguchi, N.; Wang, L.; Mukhopadhyay, P.; Park, O.; Jeong, W.I.; Lafdil, F.; Osei-Hyiaman, D.; Moh, A.; Fu, X.Y.; Pacher, P.; et al. Cell type-dependent pro- and anti-inflammatory role of signal transducer and activator of transcription 3 in alcoholic liver injury. Gastroenterology 2008, 134, 1148–1158. [Google Scholar]
- Hyun, J.; Wang, S.; Kim, J.; Rao, K.M.; Park, S.Y.; Chung, I.; Ha, C.S.; Kim, S.W.; Yun, Y.H.; Jung, Y. MicroRNA-378 limits activation of hepatic stellate cells and liver fibrosis by suppressing Gli3 expression. Nat. Commun. 2016, 7, 10993. [Google Scholar]
- Hyun, J.; Wang, S.; Kim, J.; Kim, G.J.; Jung, Y. MicroRNA125b-mediated Hedgehog signaling influences liver regeneration by chorionic plate-derived mesenchymal stem cells. Sci. Rep. 2015, 5, 14135. [Google Scholar]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.; Kim, J.; Wang, S.; Sung, S.; Kim, N.; Lee, H.-H.; Seo, Y.-S.; Jung, Y. Hepatoprotective Effect of Kombucha Tea in Rodent Model of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2019, 20, 2369. https://doi.org/10.3390/ijms20092369
Lee C, Kim J, Wang S, Sung S, Kim N, Lee H-H, Seo Y-S, Jung Y. Hepatoprotective Effect of Kombucha Tea in Rodent Model of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. International Journal of Molecular Sciences. 2019; 20(9):2369. https://doi.org/10.3390/ijms20092369
Chicago/Turabian StyleLee, Chanbin, Jieun Kim, Sihyung Wang, Sumi Sung, Namgyu Kim, Hyun-Hee Lee, Young-Su Seo, and Youngmi Jung. 2019. "Hepatoprotective Effect of Kombucha Tea in Rodent Model of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis" International Journal of Molecular Sciences 20, no. 9: 2369. https://doi.org/10.3390/ijms20092369
APA StyleLee, C., Kim, J., Wang, S., Sung, S., Kim, N., Lee, H.-H., Seo, Y.-S., & Jung, Y. (2019). Hepatoprotective Effect of Kombucha Tea in Rodent Model of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. International Journal of Molecular Sciences, 20(9), 2369. https://doi.org/10.3390/ijms20092369