Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications

 ,
, 


{kind=link}
{kind=link}
Abstract
1. Introduction
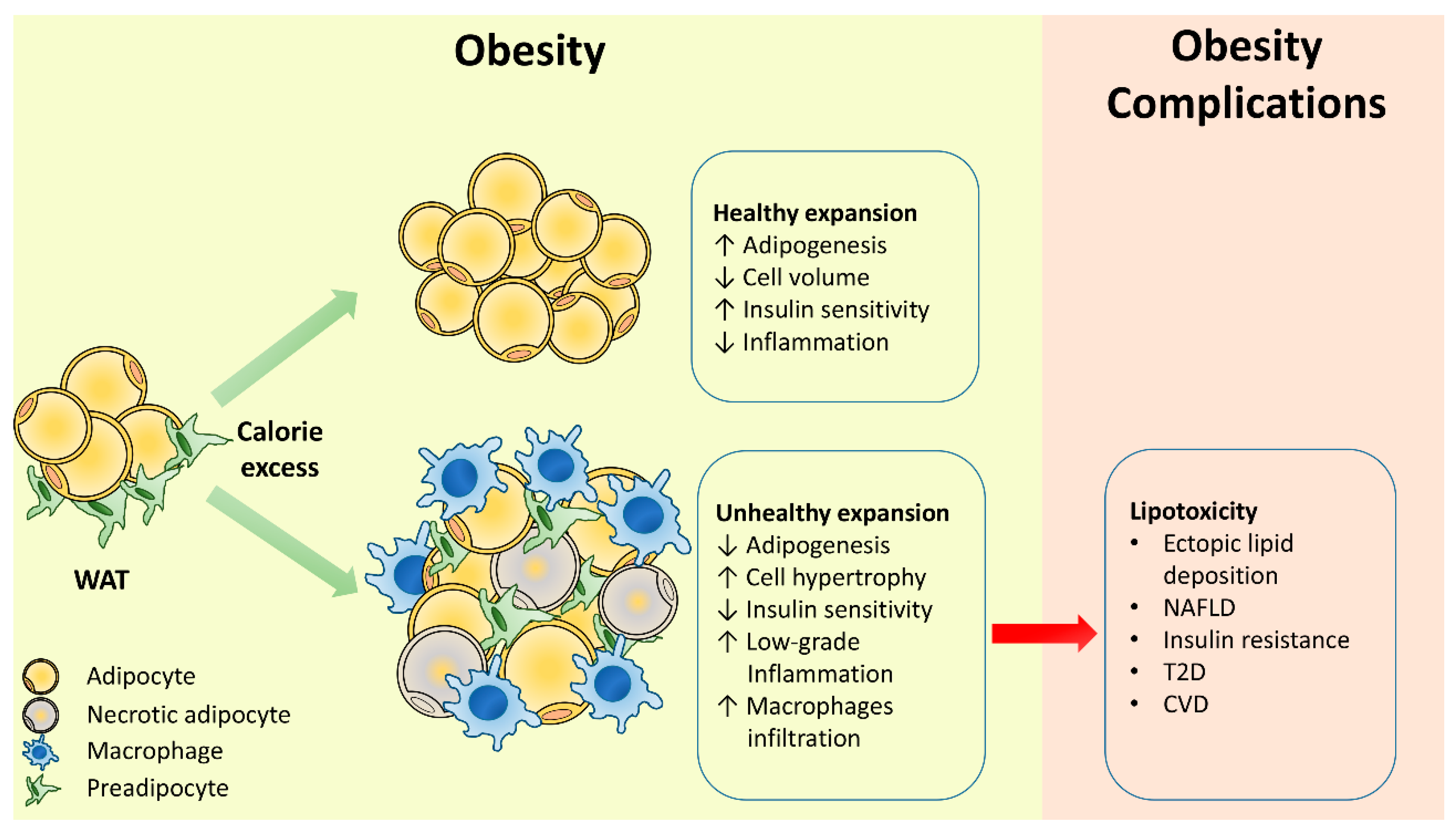
2. Adipose Tissue Remodeling in Obesity
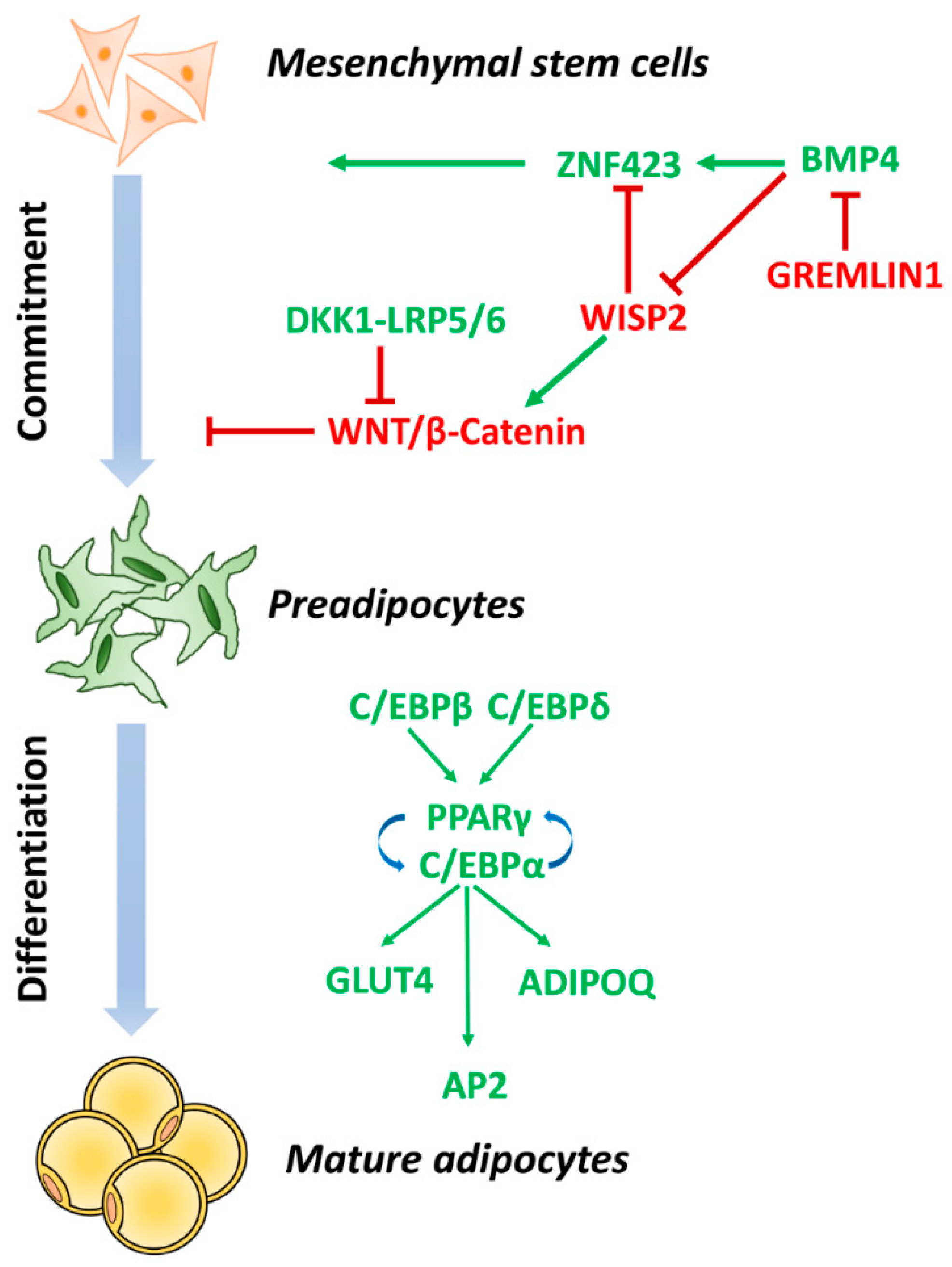
3. Impaired Adipogenesis and Insulin Resistance in Adipose Tissue Dysfunction
3.1. Adipocyte Commitment
3.2. Adipocyte Terminal Differentiation
4. Chronic Inflammation Links Obesity to Insulin Resistance
5. Ectopic Fat Accumulation and Insulin Resistance
5.1. Liver
5.2. Skeletal Muscle
5.3. Heart
6. Concluding Remarks
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| T2D | type 2 diabetes |
| WAT | white adipose tissue |
| SAT | subcutaneous adipose tissue |
| NAFLD | non-alcoholic fatty liver disease |
| BMI | body mass index |
| VAT | visceral adipose tissue |
| CVD | cardiovascular disease |
| MONW | metabolically obese normal-weight |
| GLUT4 | glucose transporter type 4 |
| TNF-α | tumor necrosis factor-α |
| IL-6 | interleukin-6 |
| IL-8 | interleukin-8 |
| MCP-1 | monocyte chemoattractant protein 1 |
| IRS | insulin receptor substrate |
| HIF | hypoxia-inducible factor |
| FFA | free fatty acid |
| MSC | pluripotent mesenchymal stem cell |
| WNT | wingless-type mouse mammary tumor virus integration site family |
| PPARγ | peroxisome proliferator-activated receptor-γ |
| C/EBP-α | CCAAT/enhancer-binding protein α |
| LRP5/6 | lipoprotein-receptor-related protein-5 or -6 |
| DKK 1 | proadipogenic factors Dickkopf 1 |
| BMP4 | bone morphogenetic protein 4 |
| WISP2 | WNT1-inducible signaling pathway protein 2 |
| TZD | thiazolidinediones |
| ATM | adipose tissue macrophage |
| NF-κB | nuclear factor-kappa B |
| JNK | c-Jun N-terminal kinase |
| PI3K | phosphatidylinositol 3-kinase |
| IκB | inhibitor of κB |
| ER | endoplasmic reticulum |
| PKCε | protein kinase Cε |
| DGAT2 | diacylglycerol acyl transferase 2 |
| EAT | epicardial adipose tissue |
| PVAT | perivascular adipose tissue |
References
- NCD Risk Factor Collaboration (NCD-RisC). Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 2016, 387, 1377–1396. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Seeley, R.J.; Zeltser, L.M.; Drewnowski, A.; Ravussin, E.; Redman, L.M.; Leibel, R.L. Obesity pathogenesis: An Endocrine Society scientific statement. Endocr. Rev. 2017, 38, 267–296. [Google Scholar] [CrossRef]
- Hopkins, M.; Blundell, J.E. Energy balance, body composition, sedentariness and appetite regulation: Pathways to obesity. Clin. Sci. 2016, 130, 1615–1628. [Google Scholar] [CrossRef]
- MacLean, P.S.; Blundell, J.E.; Mennella, J.A.; Batterham, R.L. Biological control of appetite: A daunting complexity. Obesity (Silver Spring) 2017, 25, S8–S16. [Google Scholar] [CrossRef]
- Albuquerque, D.; Stice, E.; Rodríguez-López, R.; Manco, L.; Nóbrega, C. Current review of genetics of human obesity: From molecular mechanisms to an evolutionary perspective. Mol. Genet. Genomics 2015, 290, 1191–1221. [Google Scholar] [CrossRef] [PubMed]
- Parrillo, L.; Costa, V.; Raciti, G.A.; Longo, M.; Spinelli, R.; Esposito, R.; Nigro, C.; Vastolo, V.; Desiderio, A.; Zatterale, F.; et al. Hoxa5 undergoes dynamic DNA methylation and transcriptional repression in the adipose tissue of mice exposed to high-fat diet. Int. J. Obes. 2016, 40, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Raciti, G.A.; Spinelli, R.; Desiderio, A.; Longo, M.; Parrillo, L.; Nigro, C.; D’Esposito, V.; Mirra, P.; Fiory, F.; Pilone, V.; et al. Specific CpG hyper-methylation leads to Ankrd26 gene down-regulation in white adipose tissue of a mouse model of diet-induced obesity. Sci. Rep. 2017, 7, 43526. [Google Scholar] [CrossRef] [PubMed]
- Magkos, F.; Fraterrigo, G.; Yoshino, J.; Luecking, C.; Kirbach, K.; Kelly, S.C.; de Las Fuentes, L.; He, S.; Okunade, A.L.; Patterson, B.W.; et al. Effects of Moderate and Subsequent Progressive Weight Loss on Metabolic Function and Adipose Tissue Biology in Humans with Obesity. Cell Metab. 2016, 23, 591–601. [Google Scholar] [CrossRef]
- Collaboration NRF. Worldwide trends in diabetes since 1980: A pooled analysis of 751 population-based studies with 4.4 million participants. Lancet 2016, 387, 1513–1530. [Google Scholar] [CrossRef]
- Kusminski, C.M.; Bickel, P.E.; Scherer, P.E. Targeting adipose tissue in the treatment of obesity-associated diabetes. Nat. Rev. Drug Discov. 2016, 15, 639–660. [Google Scholar] [CrossRef]
- Hall, J.E.; do Carmo, J.M.; da Silva, A.A.; Wang, Z.; Hall, M.E. Obesity-induced hypertension: Interaction of neurohumoral and renal mechanisms. Circ. Res. 2015, 116, 991–1006. [Google Scholar] [CrossRef] [PubMed]
- Van Gaal, L.F.; Mertens, I.L.; De Block, C.E. Mechanisms linking obesity with cardiovascular disease. Nature 2006, 444, 875–880. [Google Scholar] [CrossRef]
- Ye, J. Mechanisms of insulin resistance in obesity. Front. Med. 2013, 7, 14–24. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Bonadonna, R.C.; Ferrannini, E. Pathogenesis of NIDDM. A balanced overview. Diabetes Care 1992, 15, 318–368. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Reaven, G.M. Insulin resistance and hyperinsulinemia: You can’t have one without the other. Diabetes Care 2008, 31, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Czech, M.P. Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 2017, 23, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Scherer, P.E. Adipose tissue: From lipid storage compartment to endocrine organ. Diabetes 2006, 55, 1537–1545. [Google Scholar] [CrossRef]
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef]
- Reilly, S.M.; Saltiel, A.R. Adapting to obesity with adipose tissue inflammation. Nat. Rev. Endocrinol. 2017, 13, 633–643. [Google Scholar] [CrossRef]
- Rutkowski, J.M.; Stern, J.H.; Scherer, P.E. The cell biology of fat expansion. J. Cell Biol. 2015, 208, 501–512. [Google Scholar] [CrossRef]
- Singh, R.G.; Yoon, H.D.; Wu, L.M.; Lu, J.; Plank, L.D.; Petrov, M.S. Ectopic fat accumulation in the pancreas and its clinical relevance: A systematic review, metaanalysis, and meta-regression. Metabolism 2017, 69, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gaborit, B.; Abdesselam, I.; Kober, F.; Jacquier, A.; Ronsin, O.; Emungania, O.; Lesavre, N.; Alessi, M.C.; Martin, J.C.; Bernard, M.; et al. Ectopic fat storage in the pancreas using 1H-MRS: Importance of diabetic status and modulation with bariatric surgery-induced weight loss. Int. J. Obes. 2015, 39, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Tchernof, A.; Després, J.P. Pathophysiology of human visceral obesity: An update. Physiol. Rev. 2013, 93, 359–404. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, T.; Craig, C.; Liu, L.F.; Perelman, D.; Allister, C.; Spielman, D.; Cushman, S.W. Adipose cell size and regional fat deposition as predictors of metabolic response to overfeeding in insulin resistant and insulin-sensitive humans. Diabetes 2016, 65, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Festuccia, W.T.; Laplante, M. A comparative perspective on lipid storage in animals. J. Cell. Sci. 2013, 126, 1541–1552. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Wang, Z.M.; Punyanita, M.; Lei, J.; Sinav, A.; Kral, J.G.; Imielinska, C.; Ross, R.; Heymsfield, S.B. Adipose tissue quantification by imaging methods: A proposed classification. Obes. Res. 2003, 11, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, A.; Heeren, J. Adipose tissue browning and metabolic health. Nat. Rev. Endocrinol. 2014, 10, 24–36. [Google Scholar] [CrossRef]
- Cinti, S. The adipose organ at a glance. Dis. Model. Mech. 2012, 5, 588–594. [Google Scholar] [CrossRef]
- Minchin, J.E.N.; Rawls, J.F. A classification system for zebrafish adipose tissues. Dis. Model. Mech. 2017, 10, 797–809. [Google Scholar] [CrossRef]
- Chen, L.; Dai, Y.M.; Ji, C.B.; Yang, L.; Shi, C.M.; Xu, G.F.; Pang, L.X.; Huang, F.Y.; Zhang, C.M.; Guo, X.R. MiR-146b is a regulator of human visceral preadipocyte proliferation and differentiation and its expression is altered in human obesity. Mol. Cell. Endocrinol. 2014, 393, 65–74. [Google Scholar] [CrossRef]
- Long, J.Z.; Svensson, K.J.; Tsai, L.; Zeng, X.; Roh, H.C.; Kong, X.; Rao, R.R.; Lou, J.; Lokurkar, I.; Baur, W.; et al. A smooth muscle-like origin for beige adipocytes. Cell Metab. 2014, 19, 810–820. [Google Scholar] [CrossRef]
- Fehlert, E.; Wagner, R.; Ketterer, C.; Böhm, A.; Machann, J.; Fritsche, L.; Machicao, F.; Schick, F.; Staiger, H.; Stefan, N.; et al. Genetic determination of body fat distribution and the attributive influence on metabolism. Obesity 2017, 25, 1277–1283. [Google Scholar] [CrossRef]
- Sanchez-Gurmaches, J.; Guertin, D.A. Adipocytes arise from multiple lineages that are heterogeneously and dynamically distributed. Nat. Commun. 2014, 5, 4099. [Google Scholar] [CrossRef]
- Lopes, H.F.; Corrêa-Giannella, M.L.; Consolim-Colombo, F.M.; Egan, B.M. Visceral adiposity syndrome. Diabetol. Metab. Syndr. 2016, 8, 40. [Google Scholar] [CrossRef]
- Zhang, M.; Hu, T.; Zhang, S.; Zhou, L. Associations of different adipose tissue depots with insulin resistance: A systematic review and meta-analysis of observational studies. Sci. Rep. 2015, 5, 18495. [Google Scholar] [CrossRef]
- Karpe, F.; Pinnick, K.E. Biology of upper-body and lower-body adipose tissue–link to whole-body phenotypes. Nat. Rev. Endocrinol. 2015, 11, 90–100. [Google Scholar] [CrossRef]
- Porter, S.A.; Massaro, J.M.; Hoffmann, U.; Vasan, R.S.; O’Donnel, C.J.; Fox, C.S. Abdominal subcutaneous adipose tissue: A protective fat depot? Diabetes Care 2009, 32, 1068–1075. [Google Scholar] [CrossRef]
- Pellegrinelli, V.; Carobbio, S.; Vidal-Puig, A. Adipose tissue plasticity: How fat depots respond differently to pathophysiological cues. Diabetologia 2016, 59, 1075–1088. [Google Scholar] [CrossRef]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose Tissue Remodeling: Its Role in Energy Metabolism and Metabolic Disorders. Front. Endocrinol. 2016, 7, 30. [Google Scholar] [CrossRef]
- Chawla, A.; Nguyen, K.D.; Goh, Y.P. Macrophage-mediated inflammation in metabolic disease. Nat. Rev. Immunol. 2011, 11, 738–749. [Google Scholar] [CrossRef]
- Huh, J.Y.; Park, Y.J.; Ham, M.; Kim, J.B. Crosstalk between adipocytes and immune cells in adipose tissue inflammation and metabolic dysregulation in obesity. Mol. Cells 2014, 37, 365–371. [Google Scholar] [CrossRef]
- Spalding, K.L.; Arner, E.; Westermark, P.O.; Bernard, S.; Buchholz, B.A.; Bergmann, O.; Blomqvist, L.; Hoffstedt, J.; Näslund, E.; Britton, T.; et al. Dynamics of fat cell turnover in humans. Nature 2008, 453, 783–787. [Google Scholar] [CrossRef]
- Blüher, M. Adipose tissue inflammation: A cause or consequence of obesity-related insulin resistance? Clin. Sci. 2016, 130, 1603–1614. [Google Scholar] [CrossRef]
- Wang, Q.A.; Tao, C.; Gupta, R.K.; Scherer, P.E. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat. Med. 2013, 19, 1338–1344. [Google Scholar] [CrossRef]
- Kim, S.M.; Lun, M.; Wang, M.; Senyo, S.E.; Guillermier, C.; Patwari, P.; Steinhauser, M.L. Loss of white adipose hyperplastic potential is associated with enhanced susceptibility to insulin resistance. Cell Metab. 2014, 20, 1049–1058. [Google Scholar] [CrossRef]
- Guillermier, C.; Fazeli, P.K.; Kim, S.; Lun, M.; Zuflacht, J.P.; Milian, J.; Lee, H.; Francois-Saint-Cyr, H.; Horreard, F.; Larson, D.; et al. Imaging mass spectrometry demonstrates age-related decline in human adipose plasticity. JCI Insight 2017, 2, e90349. [Google Scholar] [CrossRef]
- Wildman, R.P.; Muntner, P.; Reynolds, K.; McGinn, A.P.; Rajpathak, S.; Wylie-Rosett, J.; Sowers, M.R. The obese without cardiometabolic risk factor clustering and the normal weight with cardiometabolic risk factor clustering: Prevalence and correlates of 2 phenotypes among the US population (NHANES 1999–2004). Arch. Intern. Med. 2008, 168, 1617–1624. [Google Scholar] [CrossRef]
- Naukkarinen, J.; Heinonen, S.; Hakkarainen, A.; Lundbom, J.; Vuolteenaho, K.; Saarinen, L.; Hautaniemi, S.; Rodriguez, A.; Frühbeck, G.; Pajunen, P.; et al. Characterising metabolically healthy obesity in weight-discordant monozygotic twins. Diabetologia 2014, 57, 167–176. [Google Scholar] [CrossRef]
- Rey-López, J.; Rezende, L.; Pastor-Valero, M.; Tess, B. The prevalence of metabolically healthy obesity: A systematic review and critical evaluation of the definitions used. Obes. Rev. 2014, 15, 781–790. [Google Scholar] [CrossRef]
- Blüher, M. The distinction of metabolically ‘healthy’ from ‘unhealthy’ obese individuals. Curr. Opin. Lipidol. 2010, 21, 38–43. [Google Scholar] [CrossRef]
- Rey-López, J.P.; de Rezende, L.F.; de Sá, T.H.; Stamatakis, E. Is the metabolically healthy obesity phenotype an irrelevant artifact for public health? Am. J. Epidemiol. 2015, 182, 737–741. [Google Scholar] [CrossRef]
- Bell, J.A.; Kivimaki, M.; Hamer, M. Metabolically healthy obesity and risk of incident type 2 diabetes: A meta-analysis of prospective cohort studies. Obes. Rev. 2014, 15, 504–515. [Google Scholar] [CrossRef]
- Du, T.; Yu, X.; Zhang, J.; Sun, X. Lipid accumulation product and visceral adiposity index are effective markers for identifying the metabolically obese normal-weight phenotype. Acta Diabetol. 2015, 52, 855–863. [Google Scholar] [CrossRef]
- Yoo, H.J.; Hwang, S.Y.; Hong, H.C.; Choi, H.Y.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Choi, D.S.; Baik, S.H.; Choi, K.M. Association of metabolically abnormal but normal weight (MANW) and metabolically healthy but obese (MHO) individuals with arterial stiffness and carotid atherosclerosis. Atherosclerosis 2014, 234, 218–223. [Google Scholar] [CrossRef]
- Ruderman, N.B.; Schneider, S.H.; Berchtold, P. The “metabolically- obese”, normal–weight individual. Am. J. Clin. Nutr. 1981, 34, 1617–1621. [Google Scholar] [CrossRef]
- Lee, S.H.; Han, K.; Yang, H.K.; Kim, H.S.; Cho, J.H.; Kwon, H.S.; Park, Y.M.; Cha, B.Y.; Yoon, K.H. A novel criterion for identifying metabolically obese but normal weight individuals using the product of triglycerides and glucose. Nutr. Diabetes 2015, 5, e149. [Google Scholar] [CrossRef]
- Fang, L.; Guo, F.; Zhou, L.; Stahl, R.; Grams, J. The cell size and distribution of adipocytes from subcutaneous and visceral fat is associated with type 2 diabetes mellitus in humans. Adipocyte 2015, 4, 273–279. [Google Scholar] [CrossRef]
- Cotillard, A.; Poitou, C.; Torcivia, A.; Bouillot, J.L.; Dietrich, A.; Kloting, N.; Grégoire, C.; Lolmede, K.; Blüher, M.; Clément, K. Adipocyte size threshold matters: Link with risk of type 2 diabetes and improved insulin resistance after gastric bypass. J. Clin. Endocrinol. Metab. 2014, 99, E1466–E1470. [Google Scholar] [CrossRef]
- Kim, J.I.; Huh, J.Y.; Sohn, J.H.; Choe, S.S.; Lee, Y.S.; Lim, C.Y.; Jo, A.; Park, S.B.; Han, W.; Kim, J.B. Lipid-overloaded enlarged adipocytes provoke insulin resistance independent of inflammation. Mol. Cell. Biol. 2015, 35, 1686–1699. [Google Scholar] [CrossRef]
- Jernas, M.; Palming, J.; Sjoholm, K.; Jennische, E.; Svensson, P.A.; Gabrielsson, B.G.; Levin, M.; Sjögren, A.; Rudemo, M.; Lystig, T.C.; et al. Separation of human adipocytes by size: Hypertrophic fat cells display distinct gene expression. FASEB J. 2006, 20, 1540–1542. [Google Scholar] [CrossRef]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Gorgun, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, P. The role of adipokines in chronic inflammation. Immunotargets Ther. 2016, 5, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Rasouli, N. Adipose tissue hypoxia and insulin resistance. J. Investig. Med. 2016, 64, 830–832. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, J.W.; Osborne, O.; Oh, D.Y.; Sasik, R.; Schenk, S.; Chen, A.; Chung, H.; Murphy, A.; Watkins, S.M.; et al. Increased adipocyte O2 consumption triggers HIF-1alpha, causing inflammation and insulin resistance in obesity. Cell 2014, 157, 1339–1352. [Google Scholar] [CrossRef] [PubMed]
- Wueest, S.; Rapold, R.A.; Rytka, J.M.; Schoenle, E.J.; Konrad, D. Basal lipolysis, not the degree of insulin resistance, differentiates large from small isolated adipocytes in high-fat fed mice. Diabetologia 2009, 52, 541–546. [Google Scholar] [CrossRef]
- Roberts, R.; Hodson, L.; Dennis, A.L.; Neville, M.J.; Humphreys, S.M.; Harnden, K.E.; Micklem, K.J.; Frayn, K.N. Markers of de novo lipogenesis in adipose tissue: Associations with small adipocytes and insulin sensitivity in humans. Diabetologia 2009, 52, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Slawik, M.; Vidal-Puig, A.J. Lipotoxicity, overnutrition and energy metabolism in aging. Ageing Res. Rev. 2006, 5, 144–164. [Google Scholar] [CrossRef] [PubMed]
- Haczeyni, F.; Bell-Anderson, K.S.; Farrell, G.C. Causes and mechanisms of adipocyte enlargement and adipose expansion. Obes. Rev. 2018, 19, 406–420. [Google Scholar] [CrossRef]
- Arner, E.; Westermark, P.O.; Spalding, K.L.; Britton, T.; Ryden, M.; Frisen, J.; Bernard, S.; Arner, P. Adipocyte turnover: Relevance to human adipose tissue morphology. Diabetes 2010, 59, 105–109. [Google Scholar] [CrossRef]
- McLaughlin, T.; Lamendola, C.; Coghlan, N.; Liu, T.C.; Lerner, K.; Sherman, A.; Cushman, S.W. Subcutaneous adipose cell size and distribution: Relationship to insulin resistance and body fat. Obesity 2014, 22, 673–680. [Google Scholar] [CrossRef]
- Gustafson, B.; Hedjazifar, S.; Gogg, S.; Hammarstedt, A.; Smith, U. Insulin resistance and impaired adipogenesis. Trends Endocrinol. Metab. 2015, 26, 193–200. [Google Scholar] [CrossRef]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and metabolic health. Nat. Rev. Mol. Cell Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef]
- Tang, Q.Q.; Lane, M.D. Adipogenesis: From stem cell to adipocyte. Annu. Rev. Biochem. 2012, 81, 715–736. [Google Scholar] [CrossRef]
- Rydén, M.; Uzunel, M.; Hård, J.L.; Borgström, E.; Mold, J.E.; Arner, E.; Mejhert, N.; Andersson, D.P.; Widlund, Y.; Hassan, M.; et al. Transplanted bone marrow-derived cells contribute to human adipogenesis. Cell Metab. 2015, 22, 408–417. [Google Scholar] [CrossRef]
- Farmer, S.R. Transcriptional control of adipocyte formation. Cell Metab. 2006, 4, 263–273. [Google Scholar] [CrossRef]
- Rosen, E.D.; MacDougald, O.A. Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 2006, 7, 885–896. [Google Scholar] [CrossRef]
- Christodoulides, C.; Lagathu, C.; Sethi, J.K.; Vidal-Puig, A. Adipogenesis and WNT signalling. Trends Endocrinol. Metab. 2009, 20, 16–24. [Google Scholar] [CrossRef]
- Ross, S.E.; Hemati, N.; Longo, K.A.; Bennett, C.N.; Lucas, P.C.; Erickson, R.L.; MacDougald, O.A. Inhibition of adipogenesis by Wnt signaling. Science 2000, 289, 950–953. [Google Scholar] [CrossRef]
- Grünberg, J.R.; Hammarstedt, A.; Hedjazifar, S.; Smith, U. The novel secreted adipokines WNT1-inducible signaling pathway protein 2 (WISP2) is a mesenchymal cell activator of canonical WNT. J. Biol. Chem. 2014, 289, 6899–6907. [Google Scholar] [CrossRef]
- Gustafson, B.; Smith, U. The WNT inhibitor Dickkopf 1 and bone morphogenetic protein 4 rescue adipogenesis in hypertrophic obesity in humans. Diabetes 2012, 61, 1217–1224. [Google Scholar] [CrossRef]
- Huang, H.; Song, T.J.; Li, X.; Hu, L.; He, Q.; Liu, M.; Lane, M.D.; Tang, Q.Q. BMP signaling pathway is required for commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc. Natl. Acad. Sci. USA 2009, 106, 12670–12675. [Google Scholar] [CrossRef]
- Gustafson, B.; Hammarstedt, A.; Hedjazifar, S.; Hoffmann, J.M.; Svensson, P.A.; Grimsby, J.; Rondinone, C.; Smith, U. BMP4 and BMP antagonists regulate human white and beige adipogenesis. Diabetes 2015, 64, 1670–1681. [Google Scholar] [CrossRef] [PubMed]
- Hammarstedt, A.; Hedjazifar, S.; Jenndahl, L.; Gogg, S.; Grünberg, J.; Gustafson, B.; Klimcakova, E.; Stich, V.; Langin, D.; Laakso, M.; et al. WISP2 regulates preadipocyte commitment and PPARgamma activation by BMP4. Proc. Natl. Acad. Sci. USA 2013, 110, 2563–2568. [Google Scholar] [CrossRef] [PubMed]
- Hammarstedt, A.; Gogg, S.; Hedjazifar, S.; Nerstedt, A.; Smith, U. Impaired Adipogenesis and Dysfunctional Adipose Tissue in Human Hypertrophic Obesity. Physiol. Rev. 2018, 98, 1911–1941. [Google Scholar] [CrossRef] [PubMed]
- Grünberg, J.R.; Hoffmann, J.M.; Hedjazifar, S.; Nerstedt, A.; Jenndahl, L.; Elvin, J.; Castellot, J.; Wei, L.; Movérare-Skrtic, S.; Ohlsson, C.; et al. Overexpressing the novel autocrine/endocrine adipokine WISP2 induces hyperplasia of the heart, white and brown adipose tissues and prevents insulin resistance. Sci. Rep. 2017, 7, 43515. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Arany, Z.; Seale, P.; Mepani, R.J.; Ye, L.; Conroe, H.M.; Roby, Y.A.; Kulaga, H.; Reed, R.R.; Spiegelman, B.M. Transcriptional control of preadipocyte determination by Zfp423. Nature 2010, 464, 619–623. [Google Scholar] [CrossRef]
- Shao, M.; Ishibashi, J.; Kusminski, C.M.; Wang, Q.A.; Hepler, C.; Vishvanath, L.; MacPherson, K.A.; Spurgin, S.B.; Sun, K.; Holland, W.L.; et al. Zfp423 maintains white adipocyte identity through suppression of the beige cell thermogenic gene program. Cell Metab. 2016, 23, 1167–1184. [Google Scholar] [CrossRef] [PubMed]
- Longo, M.; Raciti, G.A.; Zatterale, F.; Parrillo, L.; Desiderio, A.; Spinelli, R.; Hammarstedt, A.; Hedjazifar, S.; Hoffmann, J.M.; Nigro, C.; et al. Epigenetic modifications of the Zfp/ZNF423 gene control murine adipogenic commitment and are dysregulated in human hypertrophic obesity. Diabetologia 2018, 61, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Lefterova, M.I.; Zhang, Y.; Steger, D.J.; Schupp, M.; Schug, J.; Cristancho, A.; Feng, D.; Zhuo, D.; Stoeckert, C.J., Jr.; Liu, X.S.; et al. PPARγ and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome- wide scale. Genes Dev. 2008, 22, 2941–2952. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Waki, H.; Murakami, K.; Motojima, K.; Komeda, K.; Ide, T.; Kubota, N.; Terauchi, Y.; Tobe, K.; et al. The mechanisms by which both heterozygous peroxisome proliferator-activated receptor gamma (PPARgamma) deficiency and PPARgamma agonist improve insulin resistance. J. Biol. Chem. 2001, 276, 41245–41254. [Google Scholar] [CrossRef]
- Corrales, P.; Vidal-Puig, A.; Medina-Gómez, G. PPARs and Metabolic Disorders Associated with Challenged Adipose Tissue Plasticity. Int. J. Mol. Sci. 2018, 19, 2124. [Google Scholar] [CrossRef]
- Majithia, A.R.; Flannick, J.; Shahinian, P.; Guo, M.; Bray, M.A.; Fontanillas, P.; Gabriel, S.B.; Rosen, E.D.; Altshuler, D.; GoT2D Consortium; et al. Rare variants in PPARG with decreased activity in adipocyte differentiation are associated with increased risk of type 2 diabetes. Proc. Natl. Acad Sci. USA 2014, 111, 13127–13132. [Google Scholar] [CrossRef]
- Aprile, M.; Cataldi, S.; Ambrosio, M.R.; D’Esposito, V.; Lim, K.; Dietrich, A.; Bluher, M.; Savage, D.B.; Formisano, P.; Ciccodicola, A.; et al. PPARgD5, a naturally occurring dominant-negative splice isoform, impairs PPARg function and adipocyte differentiation. Cell Rep. 2018, 25, 1577–1592.e6. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Ivanov, S.; Merlin, J.; Lee, M.K.S.; Murphy, A.J.; Guinamard, R.R. Biology and function of adipose tissue macrophages, dendritic cells and B cells. Atherosclerosis 2018, 71, 102–110. [Google Scholar] [CrossRef]
- Miller, N.E.; Michel, C.C.; Nanjee, M.N.; Olszewski, W.L.; Miller, I.P.; Hazell, M.; Olivecrona, G.; Sutton, P.; Humphreys, S.M.; Frayn, K.N. Secretion of adipokines by human adipose tissue in vivo: Partitioning between capillary and lymphatic transport. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E659–E667. [Google Scholar] [CrossRef]
- Boumelhem, B.B.; Assinder, S.J.; Bell-Anderson, K.S.; Fraser, S.T. Flow cytometric single cell analysis reveals heterogeneity between adipose depots. Adipocyte 2017, 3, 112–123. [Google Scholar] [CrossRef]
- Alcalá, M.; Calderon-Dominguez, M.; Bustos, E.; Ramos, P.; Casals, N.; Serra, D.; Viana, M.; Herrero, L. Increased inflammation, oxidative stress and mitochondrial respiration in brown adipose tissue from obese mice. Sci. Rep. 2017, 7, 16082. [Google Scholar] [CrossRef]
- Lawler, H.M.; Underkofler, C.M.; Kern, P.A.; Erickson, C.; Bredbeck, B.; Rasouli, N. Adipose Tissue Hypoxia, Inflammation, and Fibrosis in Obese Insulin-Sensitive and Obese Insulin-Resistant Subjects. J. Clin. Endocrinol. Metab. 2016, 101, 1422–1428. [Google Scholar] [CrossRef]
- Schmitz, J.; Evers, N.; Awazawa, M.; Nicholls, H.T.; Brönneke, H.S.; Dietrich, A.M.J.; Blüher, M.; Brüning, J.C. Obesogenic memory can confer long-term increases in adipose tissue but not liver inflammation and insulin resistance after weight loss. Mol. Metab. 2016, 5, 328–339. [Google Scholar] [CrossRef]
- Henninger, A.M.; Eliasson, B.; Jenndahl, L.E.; Hammarstedt, A. Adipocyte hypertrophy, inflammation and fibrosis characterize subcutaneous adipose tissue of healthy, non-obese subjects predisposed to type 2 diabetes. PLoS ONE 2014, 9, e105262. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Castoldi, A.; Naffah de Souza, C.; Câmara, N.O.; Moraes-Vieira, P.M. The Macrophage Switch in Obesity Development. Front. Immunol. 2016, 6, 637. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef]
- Boulenouar, S.; Michelet, X.; Duquette, D.; Alvarez, D.; Hogan, A.E.; Dold, C.; O’Connor, D.; Stutte, S.; Tavakkoli, A.; Winters, D.; et al. Adipose Type One Innate Lymphoid Cells Regulate Macrophage Homeostasis through Targeted Cytotoxicity. Immunity 2017, 46, 273–286. [Google Scholar] [CrossRef]
- Wensveen, F.M.; Jelenčić, V.; Valentić, S.; Šestan, M.; Wensveen, T.T.; Theurich, S.; Glasner, A.; Mendrila, D.; Štimac, D.; Wunderlich, F.T.; et al. NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat. Immunol. 2015, 16, 376–385. [Google Scholar] [CrossRef]
- Cinti, S.; Mitchell, G.; Barbatelli, G.; Murano, I.; Ceresi, E.; Faloia, E.; Wang, S.; Fortier, M.; Greenberg, A.S.; Obin, M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid. Res. 2005, 46, 2347–2355. [Google Scholar] [CrossRef]
- Haase, J.; Weyer, U.; Immig, K.; Klöting, N.; Blüher, M.; Eilers, J.; Bechmann, I.; Gericke, M. Local proliferation of macrophages in adipose tissue during obesity-induced inflammation. Diabetologia 2014, 57, 562–571. [Google Scholar] [CrossRef]
- Cohen, P.; Levy, J.D.; Zhang, Y.; Frontini, A.; Kolodin, D.P.; Svensson, K.J.; Lo, J.C.; Zeng, X.Y.L.; Khandekar, M.J.; Wu, J.; et al. Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell 2014, 156, 304–316. [Google Scholar] [CrossRef]
- Pasarica, M.; Sereda, O.R.; Redman, L.M.; Albarado, D.C.; Hymel, D.T.; Roan, L.E.; Rood, J.C.; Burk, D.H.; Smith, S.R. Reduced Adipose Tissue Oxygenation in Human Obesity. Evidence for Rarefaction, Macrophage Chemotaxis, and Inflammation Without an Angiogenic Response. Diabetes 2009, 58, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Elson, D.A.; Thurston, G.; Huang, L.E.; Ginzinger, D.G.; McDonald, D.M.; Johnson, R.S.; Arbeit, J.M. Induction of hypervascularity without leakage or inflammation in transgenic mice overexpressing hypoxia-inducible factor-1α. Genes. Dev. 2001, 15, 2520–2532. [Google Scholar] [CrossRef]
- Wang, B.; Wood, I.S.; Trayhurn, P. Dysregulation of the expression and secretion of inflammation-related adipokines by hypoxia in human adipocytes. Pflugers. Arch. 2007, 455, 479–492. [Google Scholar] [CrossRef]
- Choe, S.S.; Shin, K.C.; Ka, S.; Lee, Y.K.; Chun, J.S.; Kim, J.B. Macrophage HIF-2α ameliorates adipose tissue inflammation and insulin resistance in obesity. Diabetes 2014, 63, 3359–3371. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.J.; Barnard, R.J. Effect of diet on fat cell size and hormone-sensitive lipase activity. J. Appl. Physiol. 1999, 87, 227–232. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Favelyukis, S.; Nguyen, A.K.; Reichart, D.; Scott, P.A.; Jenn, A.; Liu-Bryan, R.; Glass, C.K.; Neels, J.G.; Olefsky, J.M. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J. Biol. Chem. 2007, 282, 35279–35292. [Google Scholar] [CrossRef] [PubMed]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef]
- Aguirre, V.; Werner, E.D.; Giraud, J.; Lee, Y.H.; Shoelson, S.E.; White, M.F. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J. Biol. Chem. 2002, 277, 1531–1537. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Gao, Y.; Zhang, Y. Inhibition of JNK suppresses autophagy and attenuates insulin resistance in a rat model of nonalcoholic fatty liver disease. Mol. Med. Rep. 2017, 15, 180–186. [Google Scholar] [CrossRef]
- Park, M.H.; Hong, J.T. Roles of NF-κB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef]
- Panahi, G.; Pasalar, P.; Zare, M.; Rizzuto, R.; Meshkani, R. High glucose induces inflammatory responses in HepG2 cells via the oxidative stress-mediated activation of NF-κB, and MAPK pathways in HepG2 cells. Arch. Physiol. Biochem. 2018, 124, 468–474. [Google Scholar] [CrossRef]
- Yekollu, S.K.; Thomas, R.; O’Sullivan, B. Targeting curcusomes to inflammatory dendritic cells inhibits NF-κB and improves insulin resistance in obese mice. Diabetes 2011, 60, 2928–2938. [Google Scholar] [CrossRef]
- Zhang, N.; Valentine, J.M.; Zhou, Y.; Li, M.E.; Zhang, Y.; Bhattacharya, A.; Walsh, M.E.; Fischer, K.E.; Austad, S.N.; Osmulski, P.; et al. Sustained NFκB inhibition improves insulin sensitivity but is detrimental to muscle health. Aging Cell 2017, 16, 847–858. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef]
- Longo, M.; Spinelli, R.; D’Esposito, V.; Zatterale, F.; Fiory, F.; Nigro, C.; Raciti, G.A.; Miele, C.; Formisano, P.; Beguinot, F.; et al. Pathologic endoplasmic reticulum stress induced by glucotoxic insults inhibits adipocyte differentiation and induces an inflammatory phenotype. Biochim. Biophys. Acta 2016, 1863, 1146–1156. [Google Scholar] [CrossRef]
- Gustafson, B.; Smith, U. Cytokines promote Wnt signaling and inflammation and impair the normal differentiation and lipid accumulation in 3T3-L1 preadipocytes. J. Biol. Chem. 2006, 281, 9507–9516. [Google Scholar] [CrossRef]
- Samad, F.; Yamamoto, K.; Pandey, M.; Loskutoff, D.J. Elevated expression of transforming growth factor-beta in adipose tissue from obese mice. Mol. Med. 1997, 3, 37–48. [Google Scholar] [CrossRef]
- Spalding, K.L.; Bernard, S.; Näslund, E.; Salehpour, M.; Possnert, G.; Appelsved, L.; Fu, K.Y.; Alkass, K.; Druid, H.; Thorell, A.; et al. Impact of fat mass and distribution on lipid turnover in human adipose tissue. Nat. Commun. 2017, 8, 15253. [Google Scholar] [CrossRef]
- Serra, M.C.; Ryan, A.S.; Goldberg, A.P. Reduced LPL and subcutaneous lipid storage capacity are associated with metabolic syndrome in postmenopausal women with obesity. Obes. Sci. Pract. 2017, 3, 106–114. [Google Scholar] [CrossRef]
- Muir, L.A.; Neeley, C.K.; Meyer, K.A.; Baker, N.A.; Brosius, A.M.; Washabaugh, A.R.; Varban, O.A.; Finks, J.F.; Zamarron, B.F.; Flesher, C.G.; et al. Adipose tissue fibrosis, hypertrophy, and hyperplasia: Correlations with diabetes in human obesity. Obesity 2016, 24, 597–605. [Google Scholar] [CrossRef]
- Goossens, G.H. The Metabolic Phenotype in Obesity: Fat Mass, Body Fat Distribution, and Adipose Tissue Function. Obes. Facts 2017, 10, 207–215. [Google Scholar] [CrossRef]
- Cornier, M.A.; Després, J.P.; Davis, N.; Grossniklaus, D.A.; Klein, S.; Lamarche, B.; Lopez-Jimenez, F.; Rao, G.; St-Onge, M.P.; Towfighi, A.; et al. Assessing adiposity: A scientific statement from the American Heart Association. Circulation 2011, 124, 1996–2019. [Google Scholar] [CrossRef]
- Mollard, R.C.; Senechal, M.; MacIntosh, A.C.; Hay, J.; Wicklow, B.A.; Wittmeier, K.D.; Sellers, E.A.; Dean, H.J.; Ryner, L.; Berard, L.; et al. Dietary determinants of hepatic steatosis and visceral adiposity in overweight and obese youth at risk of type 2 diabetes. Am. J. Clin. Nutr. 2014, 99, 804–812. [Google Scholar] [CrossRef]
- Perfilyev, A.; Dahlman, I.; Gillberg, L.; Rosqvist, F.; Iggman, D.; Volkov, P.; Nilsson, E.; Risérus, U.; Ling, C. Impact of polyunsaturated and saturated fat overfeeding on the DNA-methylation pattern in human adipose tissue: A randomized controlled trial. Am. J. Clin. Nutr. 2017, 105, 991–1000. [Google Scholar] [CrossRef]
- Haufe, S.; Engeli, S.; Kast, P.; Böhnke, J.; Utz, W.; Haas, V.; Hermsdorf, M.; Mähler, A.; Wiesner, S.; Birkenfeld, A.L.; et al. Randomised comparison of reduced fat and reduced carbohydrate hypocaloric diets on intrahepatic fat in overweight and obese human subjects. Hepatology 2011, 53, 1504–1514. [Google Scholar] [CrossRef]
- Parry, S.A.; Hodson, L.J. Influence of dietary macronutrients on liver fat accumulation and metabolism. Investig. Med. 2017, 65, 1102–1115. [Google Scholar] [CrossRef]
- Gealekman, O.; Guseva, N.; Hartigan, C.; Apotheker, S.; Gorgoglione, M.; Gurav, K.; Tran, K.V.; Straubhaar, J.; Nicoloro, S.; Czech, M.P.; et al. Depot-specific differences and insufficient subcutaneous adipose tissue angiogenesis in human obesity. Circulation 2011, 123, 186–194. [Google Scholar] [CrossRef]
- Britton, K.A.; Fox, C.S. Ectopic fat depots and cardiovascular disease. Circulation 2011, 124, e837–e841. [Google Scholar] [CrossRef]
- Nguyen, P.; Leray, V.; Diez, M.; Serisier, S.; Le Bloc’h, J.; Siliart, B.; Dumon, H. Liver lipid metabolism. J. Anim. Physiol. Anim. Nutr. 2008, 92, 272–283. [Google Scholar] [CrossRef]
- Yaligar, J.; Gopalan, V.; Kiat, O.W.; Sugii, S.; Shui, G.; Lam, B.D.; Henry, C.J.; Wenk, M.R.; Tai, E.S.; Velan, S.S. Evaluation of dietary effects on hepatic lipids in high fat and placebo diet fed rats by in vivo MRS and LC-MS techniques. PLoS ONE 2014, 9, e91436. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Koska, J.; Stefan, N.; Permana, P.A.; Weyer, C.; Sonoda, M.; Bogardus, C.; Smith, S.R.; Joanisse, D.R.; Funahashi, T.; Krakoff, J.; et al. Increased fat accumulation in liver may link insulin resistance with subcutaneous abdominal adipocyte enlargement, visceral adiposity, and hypoadiponectinemia in obese individuals. Am. J. Clin. Nutr. 2008, 87, 295–302. [Google Scholar] [CrossRef]
- Kabir, M.; Catalano, K.J.; Ananthnarayan, S.; Kim, S.P.; Van Citters, G.W.; Dea, M.K.; Bergman, R.N. Molecular evidence supporting the portal theory: A causative link between visceral adiposity and hepatic insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E454–E461. [Google Scholar] [CrossRef]
- Carr, R.M.; Peralta, G.; Yin, X.; Ahima, R.S. Absence of perilipin 2 prevents hepatic steatosis, glucose intolerance and ceramide accumulation in alcohol-fed mice. PLoS ONE 2014, 9, e97118. [Google Scholar] [CrossRef] [PubMed]
- Gassaway, B.M.; Petersen, M.C.; Surovtseva, Y.V.; Barber, K.W.; Sheetz, J.B.; Aerni, H.R.; Merkel, J.S.; Samuel, V.T.; Shulman, G.I.; Rinehart, J. PKCε contributes to lipid-induced insulin resistance through cross talk with p70S6K and through previously unknown regulators of insulin signaling. Proc. Natl. Acad. Sci. USA 2018, 115, E8996–E9005. [Google Scholar] [CrossRef]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef]
- Xia, J.; Holland, W.L.; Kusminski, C.M.; Sun, K.; Sharma, A.X.; Pearson, M.J.; Sifuentes, A.J.; McDonald, J.G.; Gordillo, R.; Scherer, P.E. Targeted Induction of Ceramide Degradation Leads to Improved Systemic Metabolism and Reduced Hepatic Steatosis. Cell Metab. 2015, 22, 266–278. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Roles of Diacylglycerols and Ceramides in Hepatic Insulin Resistance. Trends Pharmacol. Sci. 2017, 38, 649–665. [Google Scholar] [CrossRef]
- Samuel, V.T.; Liu, Z.X.; Wang, A.; Beddow, S.A.; Geisler, J.G.; Kahn, M.; Zhang, X.M.; Monia, B.P.; Bhanot, S.; Shulman, G.I. Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J. Clin. Investig. 2007, 117, 739–745. [Google Scholar] [CrossRef]
- Liu, L.; Mei, M.; Yang, S.; Li, Q. Roles of chronic low-grade inflammation in the development of ectopic fat deposition. Mediators Inflamm. 2014, 2014, 418185. [Google Scholar] [CrossRef]
- Jin, Y.; McFie, P.J.; Banman, S.L.; Brandt, C.; Stone, S.J. Diacylglycerol acyltransferase-2 (DGAT2) and monoacylglycerol acyltransferase-2 (MGAT2) interact to promote triacylglycerol synthesis. J. Biol. Chem. 2014, 289, 28237–28248. [Google Scholar] [CrossRef]
- Jornayvaz, F.R.; Birkenfeld, A.L.; Jurczak, M.J.; Kanda, S.; Guigni, B.A.; Jiang, D.C.; Zhang, D.; Lee, H.Y.; Samuel, V.T.; Shulman, G.I. Hepatic insulin resistance in mice with hepatic overexpression of diacylglycerol acyltransferase 2. Proc. Natl. Acad. Sci. USA 2011, 108, 5748–5752. [Google Scholar] [CrossRef]
- Choi, C.S.; Befroy, D.E.; Codella, R.; Kim, S.; Reznick, R.M.; Hwang, Y.J.; Liu, Z.X.; Lee, H.Y.; Distefano, A.; Samuel, V.T.; et al. Paradoxical effects of increased expression of PGC-1alpha on muscle mitochondrial function and insulin-stimulated muscle glucose metabolism. Proc. Natl. Acad. Sci. USA 2008, 105, 19926–19931. [Google Scholar] [CrossRef]
- Brøns, C.; Grunnet, L.G. MECHANISMS IN ENDOCRINOLOGY: Skeletal muscle lipotoxicity in insulin resistance and type 2 diabetes: A causal mechanism or an innocent bystander? Eur. J. Endocrinol. 2017, 176, R67–R78. [Google Scholar] [CrossRef]
- Itani, S.I.; Zhou, Q.; Pories, W.J.; MacDonald, K.G.; Dohm, G.L. Involvement of protein kinase C in human skeletal muscle insulin resistance and obesity. Diabetes 2000, 49, 1353–1358. [Google Scholar] [CrossRef]
- Szendroedi, J.; Yoshimura, T.; Phielix, E.; Koliaki, C.; Marcucci, M.; Zhang, D.; Jelenik, T.; Müller, J.; Herder, C.; Nowotny, P.; et al. Role of diacylglycerol activation of PKCθ in lipid-induced muscle insulin resistance in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 9597–9602. [Google Scholar] [CrossRef]
- Wang, H.; Knaub, L.A.; Jensen, D.R.; Young Jung, D.; Hong, E.G.; Ko, H.J.; Coates, A.M.; Goldberg, I.J.; de la Houssaye, B.A.; Janssen, R.C.; et al. Skeletal muscle-specific deletion of lipoprotein lipase enhances insulin signaling in skeletal muscle but causes insulin resistance in liver and other tissues. Diabetes 2009, 58, 116–124. [Google Scholar] [CrossRef]
- Goodpaster, B.H.; He, J.; Watkins, S.; Kelley, D.E. Skeletal muscle lipid content and insulin resistance: Evidence for a paradox in endurance-trained athletes. J. Clin. Endocrinol. Metab. 2001, 86, 5755–5761. [Google Scholar] [CrossRef]
- Dubé, J.J.; Amati, F.; Stefanovic-Racic, M.; Toledo, F.G.; Sauers, S.E.; Goodpaster, B.H. Exercise-induced alterations in intramyocellular lipids and insulin resistance: The athlete’s paradox revisited. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E882–E888. [Google Scholar] [CrossRef]
- Daemen, S.; van Polanen, N.; Hesselink, M.K.C. The effect of diet and exercise on lipid droplet dynamics in human muscle tissue. J. Exp. Biol. 2018, 221, jeb167015. [Google Scholar] [CrossRef]
- Sharma, S.; Adrogue, J.V.; Golfman, L.; Uray, I.; Lemm, J.; Youker, K.; Noon, G.P.; Frazier, O.H.; Taegtmeyer, H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004, 18, 1692–1700. [Google Scholar] [CrossRef] [PubMed]
- Schulze, P.C.; Drosatos, K.; Goldberg, I.J. Lipid Use and Misuse by the Heart. Circ. Res. 2016, 118, 1736–1751. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Folmes, C.D.; Stanley, W.C. Cardiac energy metabolism in obesity. Circ. Res. 2007, 101, 335–347. [Google Scholar] [CrossRef]
- Ji, R.; Akashi, H.; Drosatos, K.; Liao, X.; Jiang, H.; Kennel, P.J.; Brunjes, D.L.; Castillero, E.; Zhang, X.; Deng, L.Y.; et al. Increased de novo ceramide synthesis and accumulation in failing myocardium. JCI Insight 2017, 2, e82922. [Google Scholar] [CrossRef]
- Warfel, J.D.; Bermudez, E.M.; Mendoza, T.M.; Ghosh, S.; Zhang, J.; Elks, C.M.; Mynatt, R.; Vandanmagsar, B. Mitochondrial fat oxidation is essential for lipid-induced inflammation in skeletal muscle in mice. Sci. Rep. 2016, 6, 37941. [Google Scholar] [CrossRef] [PubMed]
- Mori, J.; Patel, V.B.; Abo Alrob, O.; Basu, R.; Altamimi, T.; Desaulniers, J.; Wagg, C.S.; Kassiri, Z.; Lopaschuk, G.D.; Oudit, G.Y. Angiotensin 1-7 ameliorates diabetic cardiomyopathy and diastolic dysfunction in db/db mice by reducing lipotoxicity and inflammation. Circ. Heart. Fail. 2014, 7, 327–339. [Google Scholar] [CrossRef]
- Reali, F.; Morine, M.J.; Kahramanoğulları, O.; Raichur, S.; Schneider, H.C.; Crowther, D.; Priami, C. Mechanistic interplay between ceramide and insulin resistance. Sci. Rep. 2017, 7, 41231. [Google Scholar] [CrossRef]
- Iozzo, P. Myocardial, perivascular, and epicardial fat. Diabetes Care 2011, 34, S371–S379. [Google Scholar] [CrossRef]
- Greulich, S.; Maxhera, B.; Vandenplas, G.; de Wiza, D.H.; Smiris, K.; Mueller, H.; Heinrichs, J.; Blumensatt, M.; Cuvelier, C.; Akhyari, P.; et al. Secretory products from epicardial adipose tissue of patients with type 2 diabetes mellitus induce cardiomyocyte dysfunction. Circulation 2012, 126, 2324–2334. [Google Scholar] [CrossRef]
- Koliaki, C.; Liatis, S.; Kokkinos, A. Obesity and cardiovascular disease: Revisiting an old relationship. Metabolism 2019, 92, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.M.; Neves, K.B.; Tostes, R.C.; Lobato, N.S. Perivascular Adipose Tissue as a Relevant Fat Depot for Cardiovascular Risk in Obesity. Front. Physiol. 2018, 9, 253. [Google Scholar] [CrossRef] [PubMed]
- Xia, N.; Weisenburger, S.; Koch, E.; Burkart, M.; Reifenberg, G.; Förstermann, U.; Li, H. Restoration of perivascular adipose tissue function in diet-induced obese mice without changing bodyweight. Br. J. Pharmacol. 2017, 174, 3443–3453. [Google Scholar] [CrossRef]
- Murai, T.; Takebe, N.; Nagasawa, K.; Todate, Y.; Nakagawa, R.; Nakano, R.; Hangai, M.; Hasegawa, Y.; Takahashi, Y.; Yoshioka, K.; et al. Association of epicardial adipose tissue with serum level of cystatin C in type 2 diabetes. PLoS ONE 2017, 12, e0184723. [Google Scholar] [CrossRef] [PubMed]
- Gaborit, B.; Venteclef, N.; Ancel, P.; Pelloux, V.; Gariboldi, V.; Leprince, P.; Amour, J.; Hatem, S.N.; Jouve, E.; Dutour, A.; et al. Human epicardial adipose tissue has a specific transcriptomic signature depending on its anatomical peri-atrial, peri-ventricular, or peri-coronary location. Cardiovasc. Res. 2015, 108, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Lambadiari, V.; Kadoglou, N.P.; Stasinos, V.; Maratou, E.; Antoniadis, A.; Kolokathis, F.; Parissis, J.; Hatziagelaki, E.; Iliodromitis, E.K.; Dimitriadis, G. Serum levels of retinol-binding protein-4 are associated with the presence and severity of coronary artery disease. Cardiovasc. Diabetol. 2014, 13, 121. [Google Scholar] [CrossRef] [PubMed]
- Soucek, F.; Covassin, N.; Singh, P.; Ruzek, L.; Kara, T.; Suleiman, M.; Lerman, A.; Koestler, C.; Friedman, P.A.; Lopez-Jimenez, F.; et al. Effects of Atorvastatin (80 mg) Theray on Quantity of Epicardial Adipose Tissue in Patients Undergoing Pulmonary Vein Isolation for Atrial Fibrillation. Am. J. Cardiol. 2015, 116, 1443–1446. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358. https://doi.org/10.3390/ijms20092358
Longo M, Zatterale F, Naderi J, Parrillo L, Formisano P, Raciti GA, Beguinot F, Miele C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. International Journal of Molecular Sciences. 2019; 20(9):2358. https://doi.org/10.3390/ijms20092358
Chicago/Turabian StyleLongo, Michele, Federica Zatterale, Jamal Naderi, Luca Parrillo, Pietro Formisano, Gregory Alexander Raciti, Francesco Beguinot, and Claudia Miele. 2019. "Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications" International Journal of Molecular Sciences 20, no. 9: 2358. https://doi.org/10.3390/ijms20092358
APA StyleLongo, M., Zatterale, F., Naderi, J., Parrillo, L., Formisano, P., Raciti, G. A., Beguinot, F., & Miele, C. (2019). Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. International Journal of Molecular Sciences, 20(9), 2358. https://doi.org/10.3390/ijms20092358