Molecular Relationships among Obesity, Inflammation and Intervertebral Disc Degeneration: Are Adipokines the Common Link?

 ,
, 

 ,
,  ,
, 
Abstract
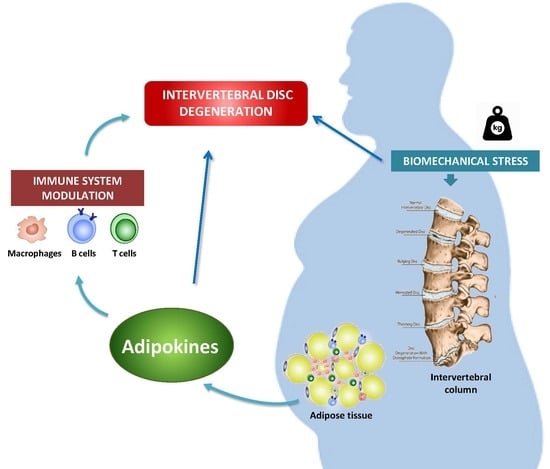
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
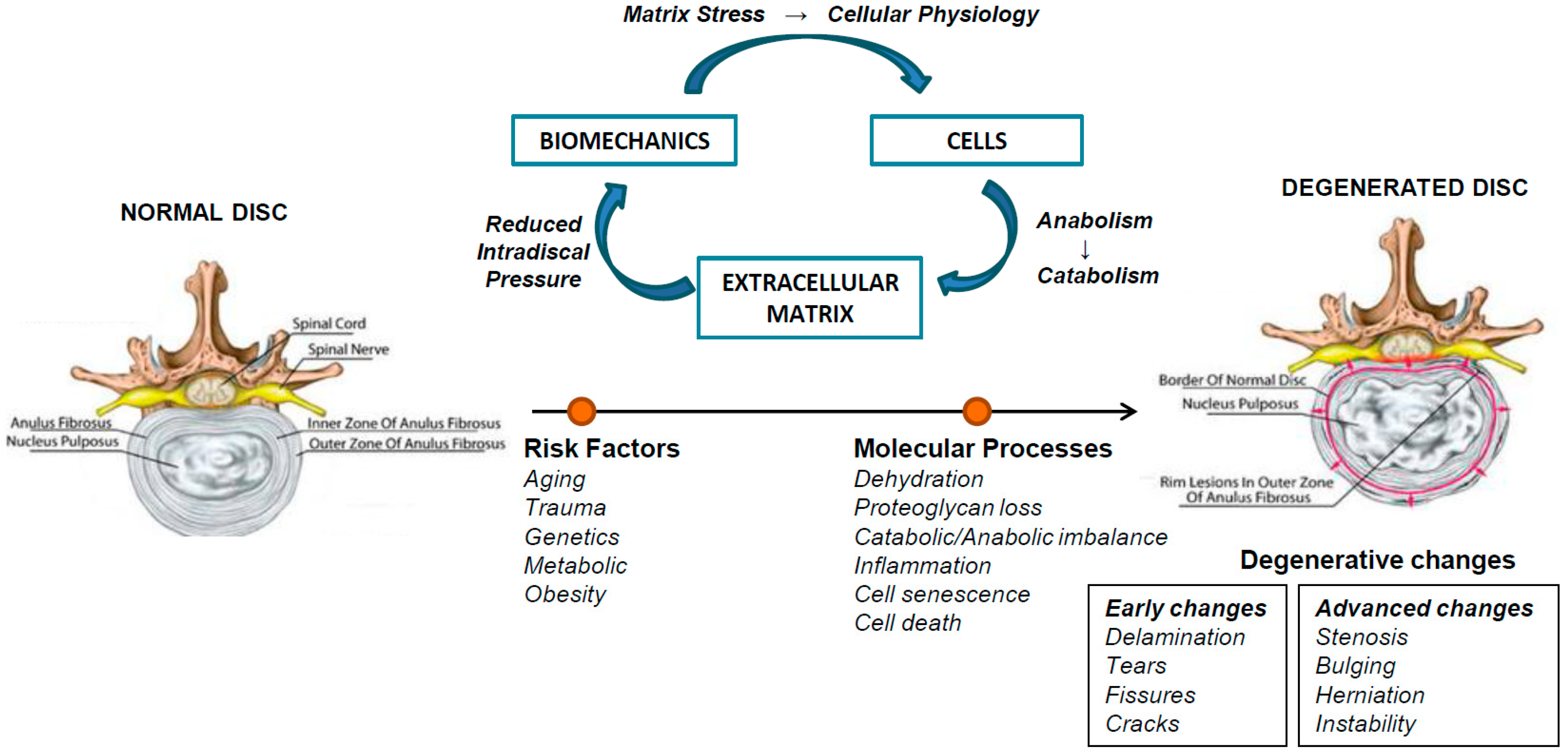
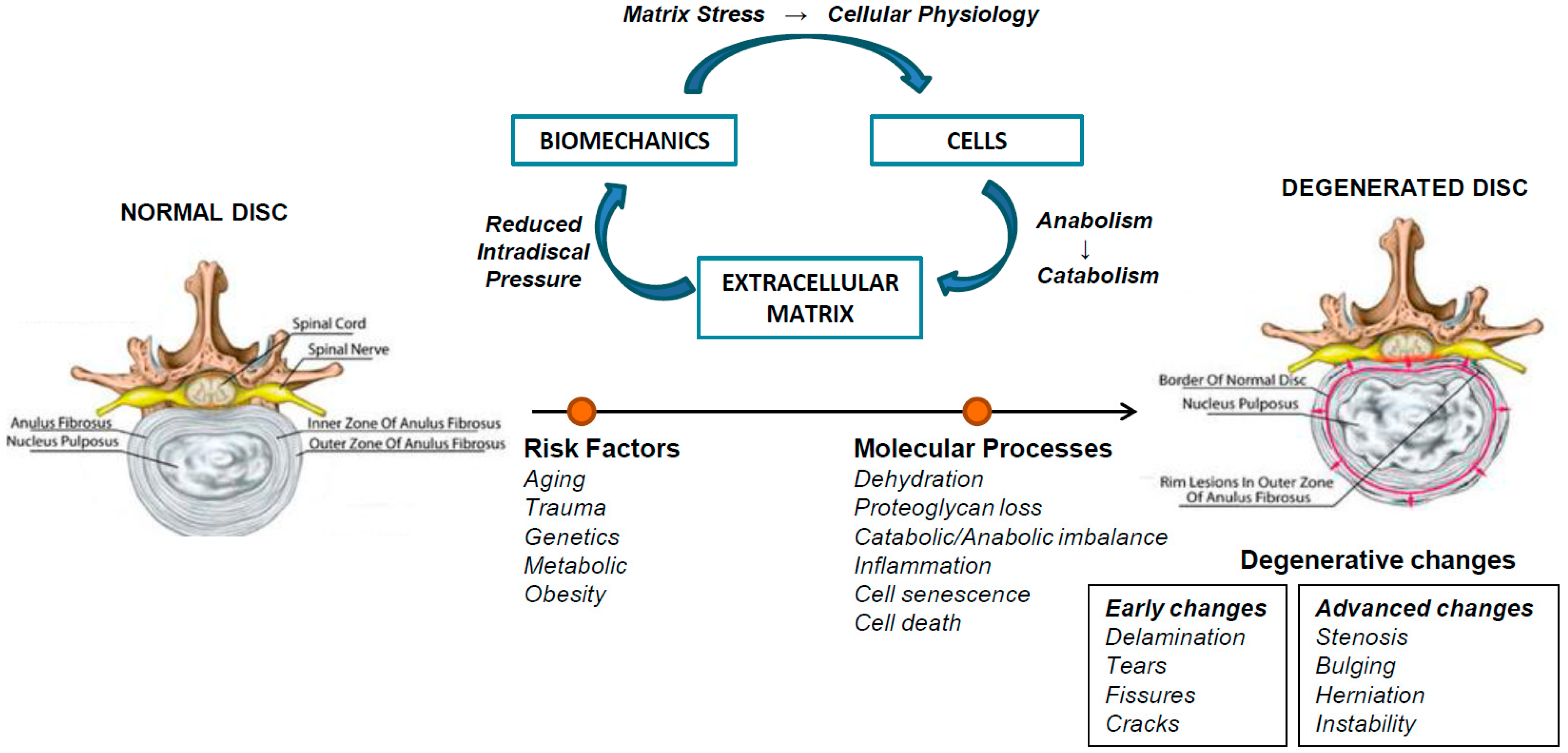
2. Intervertebral Disc Degeneration (IVDD)
3. Inflammation in IVDD
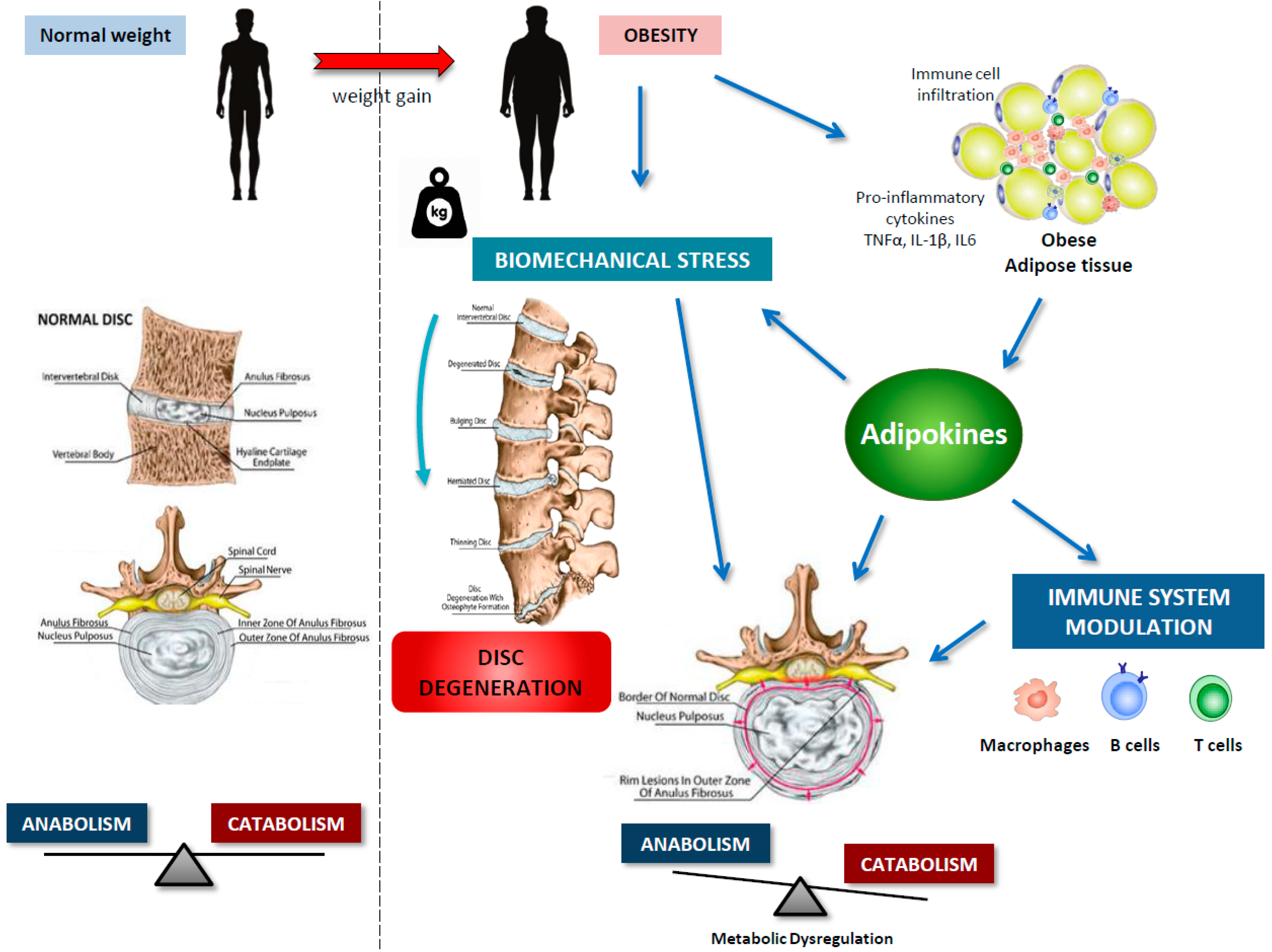
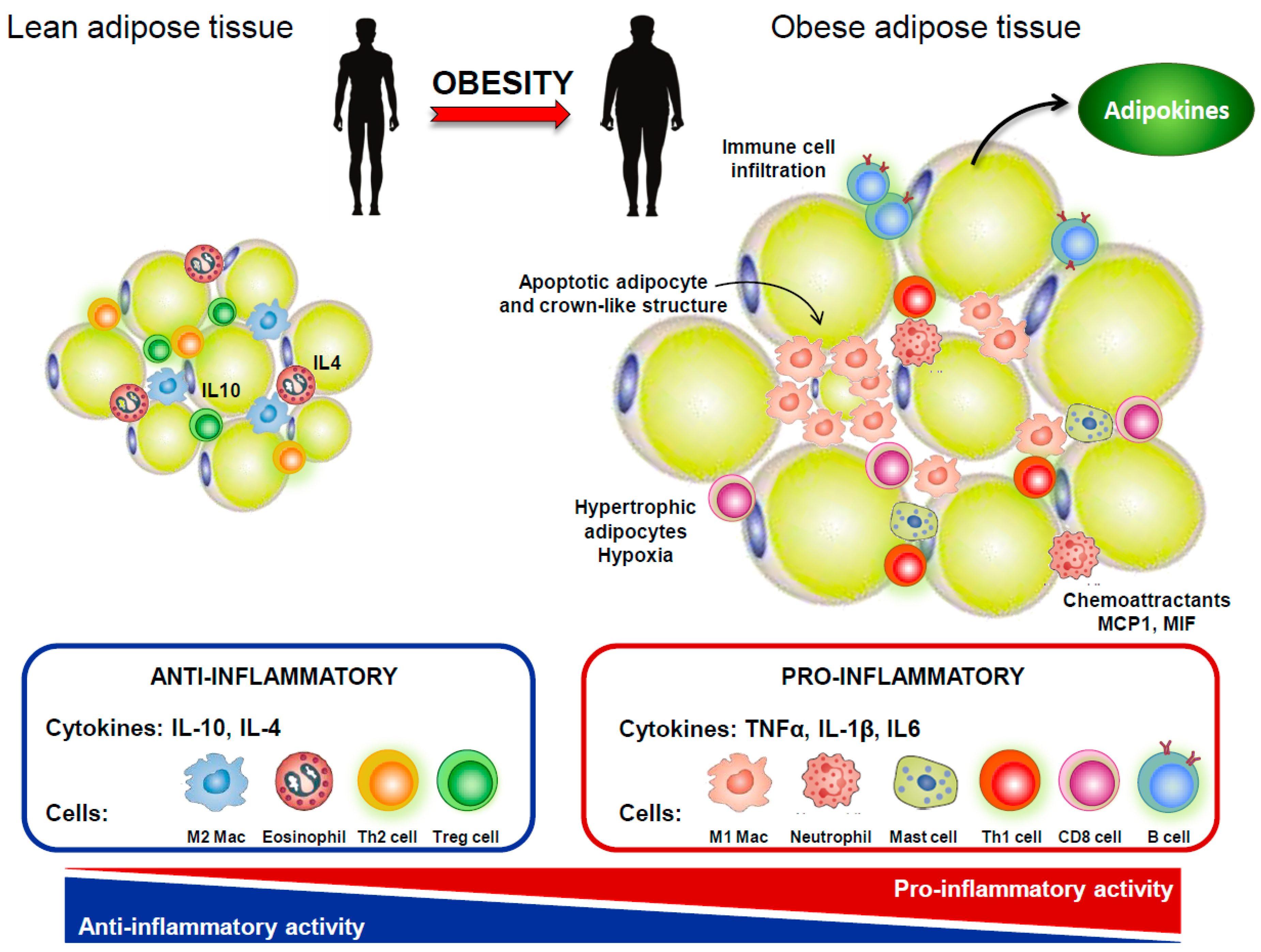
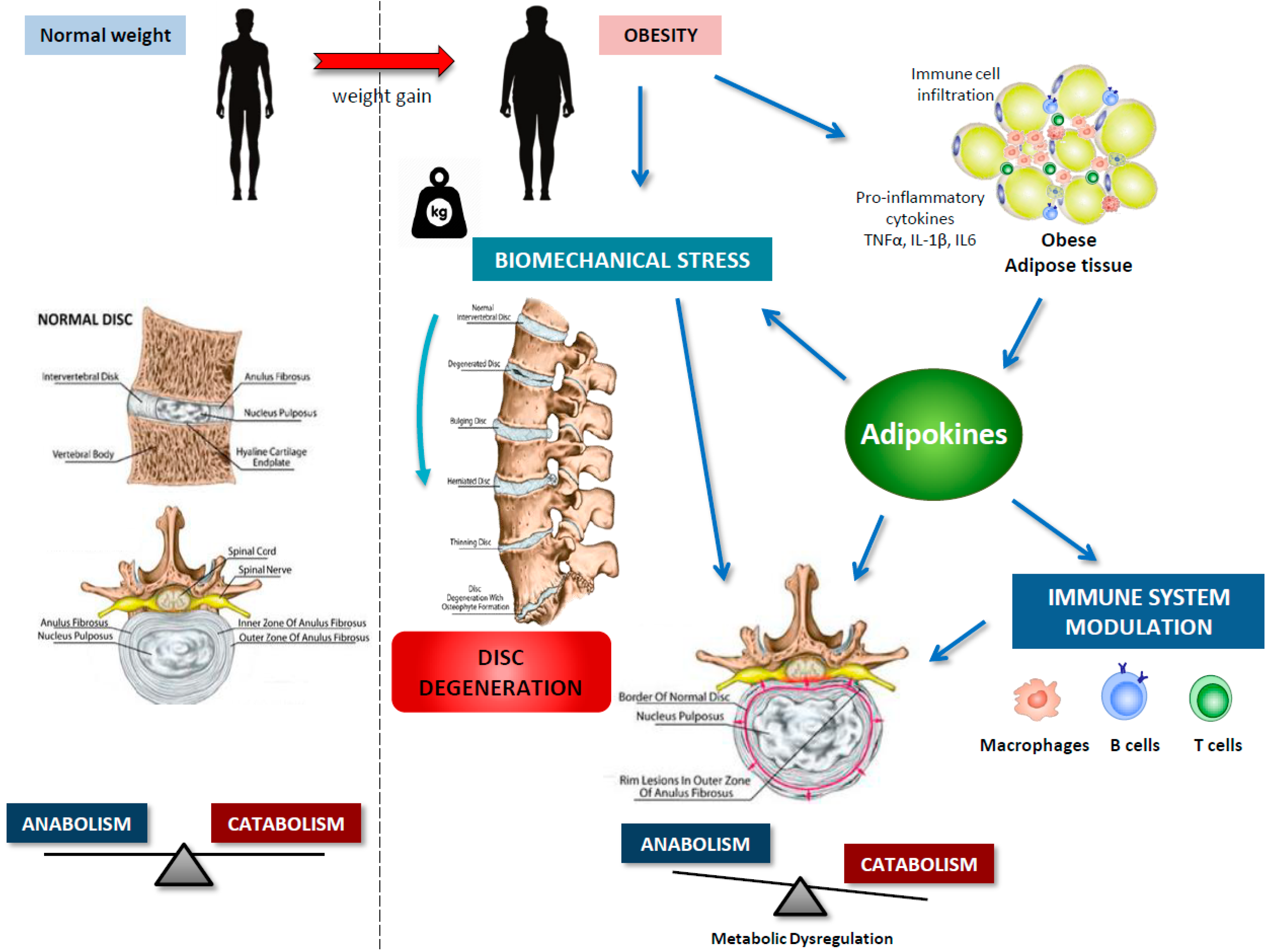
4. IVDD and Obesity
5. Adipokines in IVDD
5.1. Leptin
5.2. Adiponectin
5.3. Resistin
5.4. Progranulin
5.5. Visfatin
5.6. Lipocalin-2
5.7. Ghrelin
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| IVDD | Intervertebral disc (IVD) degeneration |
| OA | Osteoarthritis |
| RA | Rheumatoid arthritis |
| IL | Interleukin |
| TNF | Tumor necrosis factor |
| NP | Nucleus pulposus |
| AF | Annulus fibrosus |
| CEP | Cartilaginous end plate |
| ECM | Extracellular matrix |
| MMP | Matrix metalloprotease |
| ADAMTS | A disintegrin and metalloproteinase with thrombospondin motifs |
| NOS | Nitric oxide (NO) synthase |
| JAK | Janus kinase |
| STAT | Signal transducer and activator of transcription |
| ERK | Extracellular-signal-regulated kinase |
| JNK | c-Jun N-terminal kinase |
| MAPK | Mitogen Activated Protein Kinase |
| PI3K | Phosphoinositide 3-kinase |
| LEPR | Leptin receptor |
| PGRN | Progranulin |
| TNFR | TNF-α receptor |
| LCN2 | Lipocalin-2 |
References
- Vergroesen, P.-P.; Kingma, I.; Emanuel, K.S.; Hoogendoorn, R.J.; Welting, T.J.; Van Royen, B.J.; Van Dieën, J.H. Mechanics and biology in intervertebral disc degeneration: A vicious circle. Osteoarthr. Cartil. 2015, 23, 1057–1070. [Google Scholar] [CrossRef] [PubMed]
- Sampara, P.; Banala, R.R.; Vemuri, S.K.; Av, G.R.; Gpv, S. Understanding the molecular biology of intervertebral disc degeneration and potential gene therapy strategies for regeneration: A review. Gene Ther. 2018, 25, 67–82. [Google Scholar] [CrossRef] [PubMed]
- Kadow, T.; Sowa, G.; Vo, N.; Kang, J.D. Molecular Basis of Intervertebral Disc Degeneration and Herniations: What Are the Important Translational Questions? Clin. Orthop. Relat. Res. 2015, 473, 1903–1912. [Google Scholar] [CrossRef] [PubMed]
- Samartzis, D.; Karppinen, J.; Chan, D.; Luk, K.D.K.; Cheung, K.M.C. The association of lumbar intervertebral disc degeneration on magnetic resonance imaging with body mass index in overweight and obese adults: A population-based study. Arthritis Rheum. 2012, 64, 1488–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieira-Potter, V.J. Inflammation and macrophage modulation in adipose tissues. Cell. Microbiol. 2014, 16, 1484–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Suhaimi, E.A.; Shehzad, A. Leptin, resistin and visfatin: The missing link between endocrine metabolic disorders and immunity. Eur. J. Med. Res. 2013, 18, 12. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Francisco, V.; Pérez, T.; Pino, J.; López, V.; Franco, E.; Alonso, A.; Gonzalez-Gay, M.A.; Mera, A.; Lago, F.; Gómez, R.; et al. Biomechanics, obesity, and osteoarthritis. The role of adipokines: When the levee breaks. J. Orthop. Res. 2017, 36, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Francisco, V.; Pino, J.; Gonzalez-Gay, M.A.; Mera, A.; Lago, F.; Gómez, R.; Mobasheri, A.; Gualillo, O. Adipokines and inflammation: Is it a question of weight? Br. J. Pharmacol. 2018, 175, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Risbud, M.V.; Shapiro, I.M. Role of cytokines in intervertebral disc degeneration: Pain and disc content. Nat. Rev. Rheumatol. 2014, 10, 44–56. [Google Scholar] [CrossRef]
- Johnson, Z.I.; Schoepflin, Z.R.; Choi, H.; Shapiro, I.M.; Risbud, M.V. Disc in flames: Roles of TNF-α and IL-1β in intervertebral disc degeneration. Eur. Cells Mater. 2015, 30, 104–117. [Google Scholar] [CrossRef]
- Gruber, H.E.; Ingram, J.A.; Hoelscher, G.L.; Hanley, E.N. Leptin expression by annulus cells in the human intervertebral disc. Spine J. 2007, 7, 437–443. [Google Scholar] [CrossRef]
- Zhao, C.Q.; Liu, D.; Li, H.; Jiang, L.S.; Dai, L.Y. Expression of leptin and its functional receptor on disc cells: Contribution to cell proliferation. Spine (Phila. Pa. 1976) 2008, 33, 858–864. [Google Scholar] [CrossRef]
- Daniels, J.; Binch, A.A.L.; Le Maitre, C.L. Inhibiting IL-1 signaling pathways to inhibit catabolic processes in disc degeneration. J. Orthop. Res. 2017, 35, 74–85. [Google Scholar] [CrossRef]
- De Geer, C.M. Cytokine Involvement in Biological Inflammation Related to Degenerative Disorders of the Intervertebral Disk: A Narrative Review. J. Chiropr. Med. 2018, 17, 54–62. [Google Scholar] [CrossRef]
- Kubaszewski, Ł.; Zioła-Frankowska, A.; Frankowski, M.; Rogala, P.; Gasik, Z.; Kaczmarczyk, J.; Nowakowski, A.; Dabrowski, M.; Labedz, W.; Miękisiak, G.; et al. Comparison of trace element concentration in bone and intervertebral disc tissue by atomic absorption spectrometry techniques. J. Orthop. Surg. Res. 2014, 9, 99. [Google Scholar] [CrossRef] [Green Version]
- Martirosyan, N.L.; Patel, A.A.; Carotenuto, A.; Kalani, M.Y.S.; Belykh, E.; Walker, C.T.; Preul, M.C.; Theodore, N. Genetic Alterations in Intervertebral Disc Disease. Front. Surg. 2016, 3. [Google Scholar] [CrossRef]
- Le Maitre, C.; Freemont, A.J.; Hoyland, J.; Luoma, K.; Riihimaki, H.; Luukkonen, R.; Raininko, R.; Viikari-Juntura, E.; Lamminen, A.; Freemont, A.; et al. The role of interleukin-1 in the pathogenesis of human Intervertebral disc degeneration. Arthritis Res. Ther. 2005, 7, R732. [Google Scholar] [CrossRef]
- Phillips, K.L.E.; Jordan-Mahy, N.; Nicklin, M.J.H.; Le Maitre, C.L. Interleukin-1 receptor antagonist deficient mice provide insights into pathogenesis of human intervertebral disc degeneration. Ann. Rheum. Dis. 2013, 72, 1860–1867. [Google Scholar] [CrossRef]
- Hoyland, J.A.; Le Maitre, C.; Freemont, A.J. Investigation of the role of IL-1 and TNF in matrix degradation in the intervertebral disc. Rheumatology (Oxford) 2008, 47, 809–814. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Yin, Z.; Liu, C.; Liang, H.; Jiang, M.; Tian, J. IL-1β promotes ADAMTS enzyme-mediated aggrecan degradation through NF-κB in human intervertebral disc. J. Orthop. Surg. Res. 2015, 10. [Google Scholar] [CrossRef]
- Hu, B.; Shi, C.; Xu, C.; Cao, P.; Tian, Y.; Zhang, Y.; Deng, L.; Chen, H.; Yuan, W. Heme oxygenase-1 attenuates IL-1β induced alteration of anabolic and catabolic activities in intervertebral disc degeneration. Sci. Rep. 2016, 6, 21190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krupkova, O.; Hlavna, M.; Tahmasseb, J.A.; Zvick, J.; Kunz, D.; Ito, K.; Ferguson, S.J.; Wuertz-Kozak, K. An inflammatory nucleus pulposus tissue culture model to test molecular regenerative therapies: Validation with epigallocatechin 3-gallate. Int. J. Mol. Sci. 2016, 17, 1640. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tian, Y.; Phillips, K.L.E.; Chiverton, N.; Haddock, G.; Bunning, R.A.; Cross, A.K.; Shapiro, I.M.; Le Maitre, C.L.; Risbud, M.V. Tumor necrosis factor α- And interleukin-1β-dependent induction of CCL3 expression by nucleus pulposus cells promotes macrophage migration through CCR1. Arthritis Rheum. 2013, 65, 832–842. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, J.; Yao, J.; Ji, G.; Qian, L.; Wang, J.; Zhang, G.; Tian, J.; Nie, Y.; Zhang, Y.E.; et al. Obesity: Pathophysiology and intervention. Nutrients 2014, 6, 5153–5183. [Google Scholar] [CrossRef] [PubMed]
- Liuke, M.; Solovieva, S.; Lamminen, A.; Luoma, K.; Leino-Arjas, P.; Luukkonen, R.; Riihimäki, H. Disc degeneration of the lumbar spine in relation to overweight. Int. J. Obes. 2005, 29, 903–908. [Google Scholar] [CrossRef] [Green Version]
- Dowdell, J.; Erwin, M.; Choma, T.; Vaccaro, A.; Iatridis, J.; Cho, S.K. Intervertebral disk degeneration and repair. Clin. Neurosurg. 2017, 80, S46–S54. [Google Scholar] [CrossRef] [PubMed]
- Dario, A.B.; Ferreira, M.L.; Refshauge, K.M.; Lima, T.S.; Ordoñana, J.R.; Ferreira, P.H. The relationship between obesity, low back pain, and lumbar disc degeneration when genetics and the environment are considered: A systematic review of twin studies. Spine J. 2015, 15, 1106–1117. [Google Scholar] [CrossRef]
- Videman, T.; Gibbons, L.E.; Kaprio, J.; Battié, M.C. Challenging the cumulative injury model: Positive effects of greater body mass on disc degeneration. Spine J. 2010, 10, 26–31. [Google Scholar] [CrossRef]
- Sharma, A. The Role of Adipokines in Intervertebral Disc Degeneration. Med. Sci. 2018, 6, 34. [Google Scholar] [CrossRef]
- Segar, A.H.; Fairbank, J.C.T.; Urban, J. Leptin and the intervertebral disc: A biochemical link exists between obesity, intervertebral disc degeneration and low back pain—an in vitro study in a bovine model. Eur. Spine J. 2019, 28, 214–223. [Google Scholar] [CrossRef]
- Huh, J.Y.; Park, Y.J.; Ham, M.; Kim, J.B. Crosstalk between adipocytes and immune cells in adipose tissue inflammation and metabolic dysregulation in obesity. Mol Cells 2014, 37, 365–371. [Google Scholar] [CrossRef]
- Vrselja, Z.; Curic, G. Vertebral marrow adipose tissue adipokines as a possible cause of intervertebral disc inflammation. Jt. Bone Spine 2018, 85, 143–146. [Google Scholar] [CrossRef]
- Francisco, V.; Pino, J.; Campos-Cabaleiro, V.; Ruiz-Fernández, C.; Mera, A.; Gonzalez-Gay, M.A.; Gómez, R.; Gualillo, O. Obesity, fat mass and immune system: Role for leptin. Front. Physiol. 2018, 9, 640. [Google Scholar] [CrossRef]
- Zhou, Y.; Rui, L. Leptin signaling and leptin resistance. Front. Med. 2014, 7, 207–222. [Google Scholar] [CrossRef]
- Koerner, J.D.; Markova, D.Z.; Yadla, S.; Mendelis, J.; Hilibrand, A.; Vaccaro, A.R.; Risbud, M.V.; Albert, T.J.; Anderson, D.G.; Kepler, C.K. Differential Gene Expression in Anterior and Posterior Annulus Fibrosus. Spine (Phila. Pa. 1976) 2014, 39, 1917–1923. [Google Scholar] [CrossRef]
- Li, Z.; Shen, J.; Wu, W.K.K.; Yu, X.; Liang, J.; Qiu, G.; Liu, J. Leptin Induces Cyclin D1 Expression and Proliferation of Human Nucleus Pulposus Cells via JAK/STAT, PI3K/Akt and MEK/ERK Pathways. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Li, Z.; Shen, J.; Wu, W.K.K.; Yu, X.; Liang, J.; Qiu, G.; Liu, J. The role of leptin on the organization and expression of cytoskeleton elements in nucleus pulposus cells. J. Orthop. Res. 2013, 31, 847–857. [Google Scholar] [CrossRef]
- Li, Z.; Liang, J.; Wu, W.K.K.; Yu, X.; Yu, J.; Weng, X.; Shen, J. Leptin activates RhoA/ROCK pathway to induce cytoskeleton remodeling in nucleus pulposus cells. Int. J. Mol. Sci. 2014, 15, 1176–1188. [Google Scholar] [CrossRef]
- Miao, D.; Zhang, L. Leptin modulates the expression of catabolic genes in rat nucleus pulposus cells through the mitogen-activated protein kinase and Janus kinase 2/signal transducer and activator of transcription 3 pathways. Mol. Med. Rep. 2015, 12, 1761–1768. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Yu, X.; Liang, J.; Ka, W.; Wu, K.; Yu, J.; Shen, J.; Li, Z.; Liang, Y.X.; Wkk, W.; et al. Leptin Downregulates Aggrecan through the p38-ADAMST Pathway in Human Nucleus Pulposus Cells. PLoS ONE 2014, 9, e109595. [Google Scholar] [CrossRef]
- Han, Y.C.; Ma, B.; Guo, S.; Yang, M.; Li, L.J.; Wang, S.J.; Tan, J. Leptin regulates disc cartilage endplate degeneration and ossification through activation of the MAPK-ERK signalling pathway in vivo and in vitro. J. Cell. Mol. Med. 2018, 22, 2098–2109. [Google Scholar] [CrossRef]
- Ding, W.; Zhao, C.; Cao, L.; Zhang, K.; Sun, W.; Xie, Y.; Li, H.; Zhao, J. Leptin induces terminal differentiation of rat annulus fibrosus cells via activation of MAPK signaling. Anat. Rec. 2013, 296, 1806–1812. [Google Scholar] [CrossRef]
- Sun, C.; Wang, Z.; Tian, J.-W.; Wang, Y.-H. Leptin-induced inflammation by activating IL-6 expression contributes to the fibrosis and hypertrophy of ligamentum flavum in lumbar spinal canal stenosis. Biosci. Rep. 2018, 38, 20171214. [Google Scholar] [CrossRef]
- Sun, Y.; Xun, K.; Wang, C.; Zhao, H.; Bi, H.; Chen, X.; Wang, Y. Adiponectin, an unlocking adipocytokine. Cardiovasc. Ther. 2009, 27, 59–75. [Google Scholar] [CrossRef]
- Liu, M.; Liu, F. Regulation of adiponectin multimerization, signaling and function. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Kadowaki, T.; Yamauchi, T. Adiponectin and adiponectin receptors. Endocr. Rev. 2005, 26, 439–451. [Google Scholar] [CrossRef]
- Khabour, O.F.; Abu-Rumeh, L.; Al-Jarrah, M.; Jamous, M.; Alhashimi, F. Association of adiponectin protein and ADIPOQ gene variants with lumbar disc degeneration. Exp. Ther. Med. 2014, 8, 1340–1344. [Google Scholar] [CrossRef] [Green Version]
- Yuan, B.; Huang, L.; Yan, M.; Zhang, S.; Zhang, Y.; Jin, B.; Ma, Y.; Luo, Z. Adiponectin Downregulates TNF-α Expression in Degenerated Intervertebral Discs. Spine (Phila. Pa. 1976) 2018, 43, E381–E389. [Google Scholar] [CrossRef]
- Terashima, Y.; Kakutani, K.; Yurube, T.; Takada, T.; Maeno, K.; Hirata, H.; Miyazaki, S.; Ito, M.; Kakiuchi, Y.; Takeoka, Y.; et al. Expression of adiponectin receptors in human and rat intervertebral disc cells and changes in receptor expression during disc degeneration using a rat tail temporary static compression model. J. Orthop. Surg. Res. 2016, 11, 1–9. [Google Scholar] [CrossRef]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nature 2001, 409, 307–312. [Google Scholar] [CrossRef]
- Tarkowski, A.; Bjersing, J.; Shestakov, A.; Bokarewa, M.I. Resistin competes with lipopolysaccharide for binding to toll-like receptor 4. J. Cell. Mol. Med. 2010, 14, 1419–1431. [Google Scholar] [CrossRef]
- De Boer, T.N.; van Spil, W.E.; Huisman, A.M.; Polak, A.A.; Bijlsma, J.W.J.; Lafeber, F.P.J.G.; Mastbergen, S.C. Serum adipokines in osteoarthritis; comparison with controls and relationship with local parameters of synovial inflammation and cartilage damage. Osteoarthr. Cartil. 2012, 20, 846–853. [Google Scholar] [CrossRef] [Green Version]
- Presle, N.; Pottie, P.; Dumond, H.; Guillaume, C.; Lapicque, F.; Pallu, S.; Mainard, D.; Netter, P.; Terlain, B. Differential distribution of adipokines between serum and synovial fluid in patients with osteoarthritis. Contribution of joint tissues to their articular production. Osteoarthr. Cartil. 2006, 14, 690–695. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.Q.; Zhang, Q.; Peng, Y.B.; Chen, M.; Lin, X.P.; Wu, J.H.; Cai, C.H.; Mei, Y.F.; Jin, H. Resistin level is positively correlated with thrombotic complications in Southern Chinese metabolic syndrome patients. J. Endocrinol. Invest. 2011, 34, e36–e42. [Google Scholar] [CrossRef]
- Tarkowski, A.; Bokarewa, M.; Nagaev, I.; Dahlberg, L.; Smith, U. Proinflammatory Properties Resistin, an Adipokine with Potent. J. Immunol. Ref. 2005, 174, 5789–5795. [Google Scholar]
- Su, C.-M.; Hsu, C.-J.; Tsai, C.-H.; Huang, C.-Y.; Wang, S.-W.; Tang, C.-H. Resistin Promotes Angiogenesis in Endothelial Progenitor Cells Through Inhibition of MicroRNA206: Potential Implications for Rheumatoid Arthritis. Stem Cells 2015, 33, 2243–2255. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wang, X.; Pan, H.; Yang, H.; Li, X.; Zhang, K.; Wang, H.; Zheng, Z.; Liu, H.; Wang, J. Resistin promotes CCL4 expression through toll-like receptor-4 and activation of the p38-MAPK and NF-κB signaling pathways: Implications for intervertebral disc degeneration. Osteoarthr. Cartil. 2017, 25, 341–350. [Google Scholar] [CrossRef]
- Liu, C.; Yang, H.; Gao, F.; Li, X.; An, Y.; Wang, J.; Jin, A. Resistin promotes intervertebral disc degeneration by upregulation of ADAMTS-5 through p38 MAPK signaling pathway. Spine (Phila. Pa. 1976) 2016, 41, 1414–1420. [Google Scholar] [CrossRef]
- Rajan, N.E.; Bloom, O.; Maidhof, R.; Stetson, N.; Sherry, B.; Levine, M.; Chahine, N.O. Toll-Like Receptor 4 (TLR4) expression and stimulation in a model of intervertebral disc inflammation and degeneration. Spine (Phila. Pa. 1976) 2013, 38, 1343–1351. [Google Scholar] [CrossRef]
- Wei, J.; Hettinghouse, A.; Liu, C. The role of progranulin in arthritis. Ann. N. Y. Acad. Sci. 2016, 1383, 5–20. [Google Scholar] [CrossRef]
- Jian, J.; Li, G.; Hettinghouse, A.; Liu, C. Progranulin: A key player in autoimmune diseases. Cytokine 2018, 101, 48–55. [Google Scholar] [CrossRef]
- Wang, S.; Wei, J.; Fan, Y.; Ding, H.; Tian, H.; Zhou, X.; Cheng, L. Progranulin Is Positively Associated with Intervertebral Disc Degeneration by Interaction with IL-10 and IL-17 Through TNF Pathways. Inflammation 2018, 41, 1852–1863. [Google Scholar] [CrossRef]
- Naphade, S.B.; Kigerl, K.A.; Jakeman, L.B.; Kostyk, S.K.; Popovich, P.G.; Kuret, J. Progranulin expression is upregulated after spinal contusion in mice. Acta Neuropathol. 2010, 119, 123–133. [Google Scholar] [CrossRef]
- Zhao, Y.P.; Tian, Q.Y.; Liu, B.; Cuellar, J.; Richbourgh, B.; Jia, T.H.; Liu, C.J. Progranulin Knockout Accelerates Intervertebral Disc Degeneration in Aging Mice. Sci. Rep. 2015, 5, 9102. [Google Scholar] [CrossRef] [Green Version]
- Ding, H.; Wei, J.; Zhao, Y.; Liu, Y.; Liu, L.; Cheng, L. Progranulin derived engineered protein Atsttrin suppresses TNF-α-mediated inflammation in intervertebral disc degenerative disease. Oncotarget 2017, 8, 109692–109702. [Google Scholar] [CrossRef] [Green Version]
- Samal, B.; Sun, Y.; Stearns, G.; Xie, C.; Suggs, S.; McNiece, I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol. Cell. Biol. 1994, 14, 1431–1437. [Google Scholar] [CrossRef]
- Shi, C.; Wu, H.; Du, D.; Im, H.J.; Zhang, Y.; Hu, B.; Chen, H.; Wang, X.; Liu, Y.; Cao, P.; et al. Nicotinamide Phosphoribosyltransferase Inhibitor APO866 Prevents IL-1β-Induced Human Nucleus Pulposus Cell Degeneration via Autophagy. Cell. Physiol. Biochem. 2018, 49, 2463–2482. [Google Scholar] [CrossRef]
- Kao, T.-H.; Peng, Y.-J.; Salter, D.M.; Lee, H.-S.; Kao, T.-H.; Lee, H.-S.; Peng, Y.-J.; Salter, D.M. Nerve growth factor increases MMP9 activity in annulus fibrosus cells by upregulating lipocalin 2 expression. Eur. Spine J. 2015, 24, 1959–1968. [Google Scholar] [CrossRef]
- Kao, T.-H.; Peng, Y.-J.; Tsou, H.-K.; Salter, D.M.; Lee, H.-S. Nerve growth factor promotes expression of novel genes in intervertebral disc cells that regulate tissue degradation. J. Neurosurg. Spine 2014, 21, 653–661. [Google Scholar] [CrossRef] [Green Version]
- Lorenzi, T.; Meli, R.; Marzioni, D.; Morroni, M.; Baragli, A.; Castellucci, M.; Gualillo, O.; Muccioli, G. Ghrelin: A metabolic signal affecting the reproductive system. Cytokine Growth Factor Rev. 2009, 20, 137–152. [Google Scholar] [CrossRef]
- Colldén, G.; Tschöp, M.H.; Müller, T.D. Molecular Sciences Therapeutic Potential of Targeting the Ghrelin Pathway. Int. J. Mol. Sci. 2017, 18, 798. [Google Scholar] [CrossRef]
- Caminos, J.E.; Gualillo, O.; Lago, F.; Otero, M.; Blanco, M.; Gallego, R.; Garcia-Caballero, T.; Goldring, M.B.; Casanueva, F.F.; Gomez-Reino, J.J.; et al. The endogenous growth hormone secretagogue (ghrelin) is synthesized and secreted by chondrocytes. Endocrinology 2005, 146, 1285–1292. [Google Scholar] [CrossRef]
- Pereira, J.A.D.S.; da Silva, F.C.; de Moraes-Vieira, P.M.M. The Impact of Ghrelin in Metabolic Diseases: An Immune Perspective. J. Diabetes Res. 2017, 2017, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Wu, X.; Qu, R.; Wang, W.; Chen, X.; Cheng, L. Ghrelin protects against nucleus pulposus degeneration through inhibition of NF-κB signaling pathway and activation of Akt signaling pathway. Oncotarget 2017, 8, 91887–91901. [Google Scholar] [Green Version]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz-Fernández, C.; Francisco, V.; Pino, J.; Mera, A.; González-Gay, M.A.; Gómez, R.; Lago, F.; Gualillo, O. Molecular Relationships among Obesity, Inflammation and Intervertebral Disc Degeneration: Are Adipokines the Common Link? Int. J. Mol. Sci. 2019, 20, 2030. https://doi.org/10.3390/ijms20082030
Ruiz-Fernández C, Francisco V, Pino J, Mera A, González-Gay MA, Gómez R, Lago F, Gualillo O. Molecular Relationships among Obesity, Inflammation and Intervertebral Disc Degeneration: Are Adipokines the Common Link? International Journal of Molecular Sciences. 2019; 20(8):2030. https://doi.org/10.3390/ijms20082030
Chicago/Turabian StyleRuiz-Fernández, Clara, Vera Francisco, Jesus Pino, Antonio Mera, Miguel Angel González-Gay, Rodolfo Gómez, Francisca Lago, and Oreste Gualillo. 2019. "Molecular Relationships among Obesity, Inflammation and Intervertebral Disc Degeneration: Are Adipokines the Common Link?" International Journal of Molecular Sciences 20, no. 8: 2030. https://doi.org/10.3390/ijms20082030
APA StyleRuiz-Fernández, C., Francisco, V., Pino, J., Mera, A., González-Gay, M. A., Gómez, R., Lago, F., & Gualillo, O. (2019). Molecular Relationships among Obesity, Inflammation and Intervertebral Disc Degeneration: Are Adipokines the Common Link? International Journal of Molecular Sciences, 20(8), 2030. https://doi.org/10.3390/ijms20082030