Pathophysiological, Molecular and Therapeutic Issues of Nonalcoholic Fatty Liver Disease: An Overview



 and
and
Abstract
1. Nonalcoholic Fatty Liver Disease: Definition and Diagnosis
Differential Diagnosis
2. Epidemiologic Features and Relevance of NAFLD
3. NAFLD Natural History
4. Pathophysiological Features
4.1. Genetic Factors
4.2. Insulin Resistance and Metabolic Factors
4.2.1. Does Insulin Resistance Cause NAFLD?
4.2.2. Does NAFLD Cause Insulin Resistance?
4.2.3. Other Metabolic Factors: The Role of Notch and The Skeletal Muscle
4.3. Diet
4.4. Overweight and Obesity
4.5. Gut Microbiota
4.6. Iron Deposits
4.7. Bile Acids
4.8. Circadian Clock
5. Therapy
5.1. Lifestyle Intervention
5.2. Antidiabetic Drugs
5.3. Antioxidants
5.4. Drugs acting on The Gut Microbiota
5.5. Drugs Acting on Bile Acids System
5.6. Lipid-Lowering Therapies
5.7. Novel Classes of Drugs
6. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NAFLD | Nonalcoholic Fatty Liver Disease |
| NASH | Nonalcoholic Steatohepatitis |
| CT | Computed Tomography |
| MRS | Magnetic Resonance Spectroscopy |
| MRI | Magnetic Resonance Imaging |
| GPT | Glutamic Pyruvic Transaminase |
| HCV | Hepatitis C Virus |
| OLT | Orthotopic Liver Transplantation |
| AASLD | American Association for the Study of Liver Disease |
| TE | Transient Elastography |
| BMI | Body Mass Index |
| MONW | Metabolically Obese but Normal Weight |
| TG | Triglycerides |
| LDL | Low Density Lipoprotein |
| sdLDL | Small dense Low Density Lipoprotein |
| HDL | High Density Lipoprotein |
| VLDL | Very Low Density Lipoprotein |
| HCC | Hepatocarcinoma |
| NAFL | Nonalcoholic Fatty Liver |
| FA | Fatty Acid |
| NEFA | Non-Esterified Fatty Acids |
| DNL | De Novo Lipogenesis |
| CPT1 | Carnitine-O-palmitoyltransferase 1 |
| ROS | Radical Oxygen Species |
| RE | Endoplasmic Reticulum |
| FATP5 | Fatty Acid Transport Protein |
| PKCε | Protein Kinase C-ε |
| SREBP1c | Sterol Regulatory Element Binding Protein 1c |
| LXR | Liver X Receptor |
| RXR | Retinoid X Receptor |
| LIRKO | Insulin Receptor Knockout |
| ChREBP | Carbohydrate Response Element Binding Protein |
| GM | Gut Microbiota |
| LPS | Lipopolysaccharide |
| PAMPs | Pathogen-Associated Molecular Patterns |
| HSC | Hepatic Stellate Cells |
| TLR4 | Toll-like Receptor 4 |
| DAMPs | Damage-Associated Molecular Patterns |
| PNPLA3 | Palatin-like Phospholipase Domain Containing 3 |
| iPLA2-ε | Calcium-Independent Phospholipase A2 Epsilon |
| TM6SF2 | Transmembrane 6 Superfamily Member 2 |
| GCKR | Glucokinase Regulator |
| CETP | Cholesteryl Ester Transfer Protein |
| SREBP-1c | Sterol Regulatory Element-Binding Protein 1c |
| MBOAT7 | Membrane Bound O-Acyltransferase Domain-Containing 7 |
| MTTP | Microsomal Triglyceride Transfer Protein |
| ApoB | Apolipoprotein B |
| SOD2 | Superoxide Dismutase 2 |
| GST | Glutathione S-Transferase |
| TNF-α | Tumor Necrosis Factor-alpha |
| PPARA | Peroxisome Proliferator-Activated Receptor α |
| APOC3 | APOLIPOPROTEIN C-III |
| IL6 | Interleukin-6 |
| BA | Bile acid |
| FXR | Farnesoid X Receptor |
| TGR5 | Takeda G-Protein-Coupled Receptor 5 |
| GLP1 | Glucagon-Like Peptide-1 |
| RCT | Randomized Controlled Trial |
References
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL–EASD–EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Chang, Y.; Cho, Y.K.; Kim, Y.; Sung, E.; Ahn, J.; Jung, H.-S.S.; Yun, K.E.; Shin, H.; Ryu, S. Nonheavy Drinking and Worsening of Noninvasive Fibrosis Markers in Nonalcoholic Fatty Liver Disease: A Cohort Study. Hepatology 2019, 69, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Ballestri, S.; Nascimbeni, F.; Baldelli, E.; Marrazzo, A.; Romagnoli, D.; Targher, G.; Lonardo, A. Ultrasonographic fatty liver indicator detects mild steatosis and correlates with metabolic/histological parameters in various liver diseases. Metabolism 2017, 72, 57–65. [Google Scholar] [CrossRef]
- Castera, L.; Friedrich-Rust, M.; Loomba, R. Noninvasive Assessment of Liver Disease in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2019, 156, 1264–1281.e4. [Google Scholar] [CrossRef]
- Torres, D.M.; Harrison, S.A. Diagnosis and Therapy of Nonalcoholic Steatohepatitis. Gastroenterology 2008, 134, 1682–1698. [Google Scholar] [CrossRef]
- Obika, M.; Noguchi, H. Diagnosis and evaluation of nonalcoholic fatty liver disease. Exp. Diabetes Res. 2012, 2012, 145754. [Google Scholar] [CrossRef]
- Park, S.H.; Kim, P.N.; Kim, K.W.; Lee, S.W.; Yoon, S.E.; Park, S.W.; Ha, H.K.; Lee, M.-G.; Hwang, S.; Lee, S.-G.; et al. Macrovesicular Hepatic Steatosis in Living Liver Donors: Use of CT for Quantitative and Qualitative Assessment. Radiology 2006, 239, 105–112. [Google Scholar] [CrossRef]
- Charlton, M.; Sanyal, A.J.; Cusi, K.; Lavine, J.E.; Brunt, E.M.; Harrison, S.A.; Younossi, Z.; Rinella, M.; Chalasani, N.; Younossi, Z.; et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2017, 67, 328–357. [Google Scholar]
- Reeder, S.B.; Cruite, I.; Hamilton, G.; Sirlin, C.B. Quantitative assessment of liver fat with magnetic resonance imaging and spectroscopy. J. Magn. Reson. Imaging 2011, 34, 4. [Google Scholar] [CrossRef] [PubMed]
- Idilman, I.S.; Keskin, O.; Celik, A.; Savas, B.; Halil Elhan, A.; Idilman, R.; Karcaaltincaba, M. A comparison of liver fat content as determined by magnetic resonance imaging-proton density fat fraction and MRS versus liver histology in non-alcoholic fatty liver disease. Acta Radiol. 2016, 57, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.D.; Szczepaniak, L.S.; Dobbins, R.; Nuremberg, P.; Horton, J.D.; Cohen, J.C.; Grundy, S.M.; Hobbs, H.H. Prevalence of hepatic steatosis in an urban population in the United States: Impact of ethnicity. Hepatology 2004, 40, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Bedogni, G.; Miglioli, L.; Masutti, F.; Tiribelli, C.; Marchesini, G.; Bellentani, S. Prevalence of and risk factors for nonalcoholic fatty liver disease: The Dionysos nutrition and liver study. Hepatology 2005, 42, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Bedogni, G.; Bellentani, S.; Miglioli, L.; Masutti, F.; Passalacqua, M.; Castiglione, A.; Tiribelli, C. The Fatty Liver Index: A simple and accurate predictor of hepatic steatosis in the general population. BMC Gastroenterol. 2006, 6, 33. [Google Scholar] [CrossRef]
- Kotronen, A.; Peltonen, M.; Hakkarainen, A.; Sevastianova, K.; Bergholm, R.; Johansson, L.M.; Lundbom, N.; Rissanen, A.; Ridderstråle, M.; Groop, L.; et al. Prediction of Non-Alcoholic Fatty Liver Disease and Liver Fat Using Metabolic and Genetic Factors. Gastroenterology 2009, 137, 865–872. [Google Scholar] [CrossRef]
- Poynard, T.; Lassailly, G.; Diaz, E.; Clement, K.; Caïazzo, R.; Tordjman, J.; Munteanu, M.; Perazzo, H.; Demol, B.; Callafe, R.; et al. Performance of Biomarkers FibroTest, ActiTest, SteatoTest, and NashTest in Patients with Severe Obesity: Meta Analysis of Individual Patient Data. PLoS ONE 2012, 7, e30325. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Nascimbeni, F.; Targher, G.; Bernardi, M.; Bonino, F.; Bugianesi, E.; Casini, A.; Gastaldelli, A.; Marchesini, G.; Marra, F.; et al. AISF position paper on nonalcoholic fatty liver disease (NAFLD): Updates and future directions. Dig. Liver Dis. 2017, 49, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; Hui, J.M.; Marchesini, G.; Bugianesi, E.; George, J.; Farrell, G.C.; Enders, F.; Saksena, S.; Burt, A.D.; Bida, J.P.; et al. The NAFLD fibrosis score: A noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology 2007, 45, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.G.; Lydecker, A.; Murray, K.; Tetri, B.N.; Contos, M.J.; Sanyal, A.J.; Nash Clinical Research Network. Comparison of Noninvasive Markers of Fibrosis in Patients With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2009, 7, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Vanni, E.; Bugianesi, E.; Di Marco, V.; Cammà, C.; Cabibi, D.; Mezzabotta, L.; Craxì, A. The combination of liver stiffness measurement and NAFLD fibrosis score improves the noninvasive diagnostic accuracy for severe liver fibrosis in patients with nonalcoholic fatty liver disease. Liver Int. 2015, 35, 1566–1573. [Google Scholar] [CrossRef]
- Petta, S.; Wong, V.W.-S.; Cammà, C.; Hiriart, J.-B.; Wong, G.L.-H.; Vergniol, J.; Chan, A.W.-H.; Di Marco, V.; Merrouche, W.; Chan, H.L.-Y.; et al. Serial combination of non-invasive tools improves the diagnostic accuracy of severe liver fibrosis in patients with NAFLD. Aliment. Pharmacol. Ther. 2017, 46, 617–627. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Meroni, M.; Longo, M.; Fargion, S.; Fracanzani, A.L. miRNA Signature in NAFLD: A Turning Point for a Non-Invasive Diagnosis. Int. J. Mol. Sci. 2018, 19, 3966. [Google Scholar] [CrossRef] [PubMed]
- Rasineni, K.; Casey, C.A. Molecular mechanism of alcoholic fatty liver. Indian J. Pharmacol. 2012, 44, 299–303. [Google Scholar] [CrossRef]
- Modaresi Esfeh, J.; Ansari-Gilani, K. Steatosis and hepatitis C. Gastroenterol. Rep. 2016, 4, 24–29. [Google Scholar] [CrossRef][Green Version]
- Amacher, D.; Chalasani, N. Drug-Induced Hepatic Steatosis. Semin. Liver Dis. 2014, 34, 205–214. [Google Scholar]
- Whitfield, A.J. Liver Dysfunction and Steatosis in Familial Hypobetalipoproteinemia. Clin. Chem. 2004, 51, 266–269. [Google Scholar] [CrossRef][Green Version]
- A-Kader, H.H. Lysosomal acid lipase deficiency: A form of non-obese fatty liver disease (NOFLD). Expert Rev. Gastroenterol. Hepatol. 2017, 11, 911–924. [Google Scholar] [CrossRef]
- Liggi, M.; Murgia, D.; Civolani, A.; Demelia, E.; Sorbello, O.; Demelia, L. The relationship between copper and steatosis in Wilson’s disease. Clin. Res. Hepatol. Gastroenterol. 2013, 37, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Arinaga-Hino, T.; Ohira, H.; Abe, K.; Torimura, T.; Zeniya, M.; Abe, M.; Yoshizawa, K.; Takaki, A.; Suzuki, Y.; et al. Non-alcoholic fatty liver disease in patients with autoimmune hepatitis. JGH Open 2018, 2, 54–58. [Google Scholar] [CrossRef]
- Freeman, H.J. Hepatic manifestations of celiac disease. Clin. Exp. Gastroenterol. 2010, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Marino, L.; Jornayvaz, F.R. Endocrine causes of nonalcoholic fatty liver disease. World J. Gastroenterol. 2015, 21, 11053–11076. [Google Scholar] [CrossRef]
- Ferrandino, G.; Kaspari, R.R.; Spadaro, O.; Reyna-Neyra, A.; Perry, R.J.; Cardone, R.; Kibbey, R.G.; Shulman, G.I.; Dixit, V.D.; Carrasco, N. Pathogenesis of hypothyroidism-induced NAFLD is driven by intra- and extrahepatic mechanisms. Proc. Natl. Acad. Sci. 2017, 114, E9172–E9180. [Google Scholar] [CrossRef]
- Mantovani, A.; Nascimbeni, F.; Lonardo, A.; Zoppini, G.; Bonora, E.; Mantzoros, C.S.; Targher, G. Association Between Primary Hypothyroidism and Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Thyroid 2018, 28, 1270–1284. [Google Scholar] [CrossRef]
- Petta, S.; Ciresi, A.; Bianco, J.; Geraci, V.; Boemi, R.; Galvano, L.; Magliozzo, F.; Merlino, G.; Craxì, A.; Giordano, C. Insulin resistance and hyperandrogenism drive steatosis and fibrosis risk in young females with PCOS. PLoS ONE 2017, 12, e0186136. [Google Scholar] [CrossRef] [PubMed]
- Kneeman, J.M.; Misdraji, J.; Corey, K.E. Secondary causes of nonalcoholic fatty liver disease. Therap. Adv. Gastroenterol. 2012, 5, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Safar Zadeh, E.; Lungu, A.O.; Cochran, E.K.; Brown, R.J.; Ghany, M.G.; Heller, T.; Kleiner, D.E.; Gorden, P. The liver diseases of lipodystrophy: The long-term effect of leptin treatment. J. Hepatol. 2013, 59, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Stepanova, M.; Afendy, M.; Fang, Y.; Younossi, Y.; Mir, H.; Srishord, M. Changes in the Prevalence of the Most Common Causes of Chronic Liver Diseases in the United States From 1988 to 2008. Clin. Gastroenterol. Hepatol. 2011, 9, 524–530. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Seto, W.-K.; Yuen, M.-F. Nonalcoholic fatty liver disease in Asia: Emerging perspectives. J. Gastroenterol. 2017, 52, 164–174. [Google Scholar] [CrossRef]
- Bellentani, S.; Tiribelli, C.; Saccoccio, G.; Sodde, M.; Fratti, N.; De Martin, C.; Cristianini, G. Prevalence of chronic liver disease in the general population of northern Italy: The Dionysos Study. Hepatology 1994, 20, 1442–1449. [Google Scholar] [CrossRef]
- Bedogni, G.; Bellentani, S. Fatty liver: How frequent is it and why? Ann. Hepatol. 2004, 3, 63–65. [Google Scholar]
- Lonardo, A.; Lugari, S.; Ballestri, S.; Nascimbeni, F.; Baldelli, E.; Maurantonio, M. A round trip from nonalcoholic fatty liver disease to diabetes: Molecular targets to the rescue? Acta Diabetol. 2018, 385–396. [Google Scholar] [CrossRef]
- Estes, C.; Anstee, Q.M.; Arias-Loste, M.T.; Bantel, H.; Bellentani, S.; Caballeria, J.; Colombo, M.; Craxi, A.; Crespo, J.; Day, C.P.; et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Charlton, M.R.; Burns, J.M.; Pedersen, R.A.; Watt, K.D.; Heimbach, J.K.; Dierkhising, R.A. Frequency and Outcomes of Liver Transplantation for Nonalcoholic Steatohepatitis in the United States. Gastroenterology 2011, 141, 1249–1253. [Google Scholar] [CrossRef]
- Wong, R.J.; Cheung, R.; Ahmed, A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the U.S. Hepatology 2014, 59, 2188–2195. [Google Scholar] [CrossRef]
- Adam, R.; Karam, V.; Delvart, V.; O’Grady, J.; Mirza, D.; Klempnauer, J.; Castaing, D.; Neuhaus, P.; Jamieson, N.; Salizzoni, M.; et al. Evolution of indications and results of liver transplantation in Europe. A report from the European Liver Transplant Registry (ELTR). J. Hepatol. 2012, 57, 675–688. [Google Scholar] [CrossRef]
- Pais, R.; Barritt, A.S.; Calmus, Y.; Scatton, O.; Runge, T.; Lebray, P.; Poynard, T.; Ratziu, V.; Conti, F.; Conti, F. NAFLD and liver transplantation: Current burden and expected challenges. J. Hepatol. 2016, 65, 1245–1257. [Google Scholar] [CrossRef]
- Gitto, S.; Vukotic, R.; Vitale, G.; Pirillo, M.; Villa, E.; Andreone, P. Non-alcoholic steatohepatitis and liver transplantation. Dig. Liver Dis. 2016, 48, 587–591. [Google Scholar] [CrossRef]
- Adam, R.; Karam, V.; Cailliez, V.; O Grady, J.G.; Mirza, D.; Cherqui, D.; Klempnauer, J.; Salizzoni, M.; Pratschke, J.; Jamieson, N.; et al. 2018 Annual Report of the European Liver Transplant Registry (ELTR)—50-year evolution of liver transplantation. Transpl. Int. 2018, 31, 1293–1317. [Google Scholar] [CrossRef]
- Dare, A.J.; Plank, L.D.; Phillips, A.R.J.; Gane, E.J.; Harrison, B.; Orr, D.; Jiang, Y.; Bartlett, A.S.J.R. Additive effect of pretransplant obesity, diabetes, and cardiovascular risk factors on outcomes after liver transplantation. Liver Transplant. 2014, 20, 281–290. [Google Scholar] [CrossRef]
- Charlton, M. Evolving aspects of liver transplantation for nonalcoholic steatohepatitis. Curr. Opin. Organ Transplant. 2013, 18, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Wang, W.L. Risk factors of metabolic syndrome after liver transplantation. Hepatobiliary Pancreat. Dis. Int. 2015, 14, 582–587. [Google Scholar] [CrossRef]
- Koehler, E.M.; Leebeek, F.W.G.; Stricker, B.H.; Schouten, J.N.L.; Janssen, H.L.A.; Hansen, B.E.; Darwish Murad, S.; Castera, L.; Taimr, P.; Plompen, E.P.C.; et al. Presence of diabetes mellitus and steatosis is associated with liver stiffness in a general population: The Rotterdam study. Hepatology 2015, 63, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Noureddin, M.; Yates, K.P.; Vaughn, I.A.; Neuschwander-Tetri, B.A.; Sanyal, A.J.; McCullough, A.; Merriman, R.; Hameed, B.; Doo, E.; Kleiner, D.E.; et al. Clinical and histological determinants of nonalcoholic steatohepatitis and advanced fibrosis in elderly patients. Hepatology 2013, 58, 1644–1654. [Google Scholar] [CrossRef]
- Gagliano, N.; Arosio, B.; Grizzi, F.; Masson, S.; Tagliabue, J.; Dioguardi, N.; Vergani, C.; Annoni, G. Reduced collagenolytic activity of matrix metalloproteinases and development of liver fibrosis in the aging rat. Mech. Ageing Dev. 2002, 123, 413–425. [Google Scholar] [CrossRef]
- Kim, I.H.; Kisseleva, T.; Brenner, D.A. Aging and liver disease. Curr. Opin. Gastroenterol. 2015, 31, 184–191. [Google Scholar] [CrossRef]
- Collins, B.H.; Holzknecht, Z.E.; Lynn, K.A.; Sempowski, G.D.; Smith, C.C.; Liu, S.; Parker, W.; Rockey, D.C. Association of age-dependent liver injury and fibrosis with immune cell populations. Liver Int. 2013, 33, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.P. Paediatric NAFLD: More than just small adults. Lancet Gastroenterol. Hepatol. 2018, 3, 222. [Google Scholar] [CrossRef]
- Yerian, L.; Lopez, R.; Alkhouri, N.; De Vito, R.; Nobili, V.; Feldstein, A.E.; Alisi, A.; Yerian, L.; Lopez, R.; Feldstein, A.E.; et al. Development and validation of a new histological score for pediatric non-alcoholic fatty liver disease. J. Hepatol. 2012, 57, 1312–1318. [Google Scholar]
- Feldstein, A.E.; Charatcharoenwitthaya, P.; Treeprasertsuk, S.; Benson, J.T.; Enders, F.B.; Angulo, P. The natural history of non-alcoholic fatty liver disease in children: A follow-up study for up to 20 years. Gut 2009, 58, 1538–1544. [Google Scholar] [CrossRef] [PubMed]
- Hagström, H.; Stål, P.; Hultcrantz, R.; Hemmingsson, T.; Andreasson, A.; Hultcrantz, R.; Andreasson, A.; Stål, P.; Hagström, H.; Stål, P.; et al. Overweight in late adolescence predicts development of severe liver disease later in life: A 39years follow-up study. J. Hepatol. 2016, 65, 363–368. [Google Scholar] [CrossRef][Green Version]
- Kelsey, M.M.; Zeitler, P.S. Insulin Resistance of Puberty. Curr. Diab. Rep. 2016, 16, 64. [Google Scholar] [CrossRef] [PubMed]
- Temple, J.L.; Cordero, P.; Li, J.; Nguyen, V.; Oben, J.A. A Guide to Non-Alcoholic Fatty Liver Disease in Childhood and Adolescence. Int. J. Mol. Sci. 2016, 17, 947. [Google Scholar] [CrossRef] [PubMed]
- Soresi, M.; Noto, D.; Cefalù, A.B.; Martini, S.; Vigna, G.B.; Fonda, M.; Manzato, E.; Cattin, L.; Fellin, R.; Averna, M.R.; et al. Nonalcoholic fatty liver and metabolic syndrome in Italy: Results from a multicentric study of the Italian Arteriosclerosis society. Acta Diabetol. 2013, 50, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Katrina Loomis, A.; Kabadi, S.; Preiss, D.; Hyde, C.; Bonato, V.; Louis, M.S.; Desai, J.; Gill, J.M.R.; Welsh, P.; Waterworth, D.; et al. Body mass index and risk of nonalcoholic fatty liver disease: Two electronic health record prospective studies. J. Clin. Endocrinol. Metab. 2016, 101, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Calanna, S.; Scicali, R.; Di Pino, A.; Knop, F.K.; Piro, S.; Rabuazzo, A.M.; Purrello, F. Lipid and liver abnormalities in haemoglobin A1c-defined prediabetes and type 2 diabetes. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 670–676. [Google Scholar] [CrossRef]
- Loomba, R.; Abraham, M.; Unalp, A.; Wilson, L.; Lavine, J.; Doo, E.; Bass, N.M.; Nonalcoholic Steatohepatitis Clinical Research Network. Association between diabetes, family history of diabetes, and risk of nonalcoholic steatohepatitis and fibrosis. Hepatology 2012, 56, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389–397.e10. [Google Scholar] [CrossRef]
- Kim, D.; Kim, W.R. Nonobese Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2017, 15, 474–485. [Google Scholar] [CrossRef]
- Ruderman, N.; Chisholm, D.; Pi-Sunyer, X.; Schneider, S. The metabolically obese, normal-weight individual revisited. Diabetes 1998, 47, 699–713. [Google Scholar] [CrossRef]
- Sinn, D.H.; Gwak, G.-Y.; Park, H.N.; Kim, J.E.; Min, Y.W.; Kim, K.M.; Kim, Y.J.; Choi, M.S.; Lee, J.H.; Koh, K.C.; et al. Ultrasonographically Detected Non-Alcoholic Fatty Liver Disease Is an Independent Predictor for Identifying Patients With Insulin Resistance in Non-Obese, Non-Diabetic Middle-Aged Asian Adults. Am. J. Gastroenterol. 2012, 107, 561–567. [Google Scholar] [CrossRef]
- Calanna, S.; Piro, S.; Di Pino, A.; Maria Zagami, R.; Urbano, F.; Purrello, F.; Maria Rabuazzo, A. Beta and alpha cell function in metabolically healthy but obese subjects: Relationship with entero-insular axis. Obesity 2013, 21, 320–325. [Google Scholar] [CrossRef]
- Kumar, R.; Mohan, S. Non-alcoholic Fatty Liver Disease in Lean Subjects: Characteristics and Implications. J. Clin. Transl. Hepatol. 2017, 5, 216–223. [Google Scholar] [CrossRef]
- Wei, J.L.; Leung, J.C.-F.; Loong, T.C.-W.; Wong, G.L.-H.; Yeung, D.K.-W.; Chan, R.S.-M.; Chan, H.L.-Y.; Chim, A.M.-L.; Woo, J.; Chu, W.C.-W.; et al. Prevalence and Severity of Nonalcoholic Fatty Liver Disease in Non-Obese Patients: A Population Study Using Proton-Magnetic Resonance Spectroscopy. Am. J. Gastroenterol. 2015, 110, 1306–1314. [Google Scholar] [CrossRef]
- Enjoji, M.; Yasutake, K.; Kohjima, M.; Nakamuta, M. Nutrition and Nonalcoholic Fatty Liver Disease: The Significance of Cholesterol. Int. J. Hepatol. 2012, 2012, 1–6. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; De Michieli, F.; Cassader, M.; Rizzetto, M.; Durazzo, M.; Fagà, E.; Silli, B.; Pagano, G. Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology 2003, 37, 909–916. [Google Scholar] [CrossRef]
- Assy, N.; Nasser, G.; Kamayse, I.; Nseir, W.; Beniashvili, Z.; Djibre, A.; Grosovski, M. Soft drink consumption linked with fatty liver in the absence of traditional risk factors. Can. J. Gastroenterol. 2008, 22, 811–816. [Google Scholar] [CrossRef]
- Fracanzani, A.L.; Petta, S.; Lombardi, R.; Pisano, G.; Russello, M.; Consonni, D.; Di Marco, V.; Cammà, C.; Mensi, L.; Dongiovanni, P.; et al. Liver and Cardiovascular Damage in Patients With Lean Nonalcoholic Fatty Liver Disease, and Association With Visceral Obesity. Clin. Gastroenterol. Hepatol. 2017, 15, 1604–1611.e1. [Google Scholar] [CrossRef]
- Zhang, Q.-Q.; Lu, L.-G. Nonalcoholic Fatty Liver Disease: Dyslipidemia, Risk for Cardiovascular Complications, and Treatment Strategy. J. Clin. Transl. Hepatol. 2015, 3, 78–84. [Google Scholar]
- Kikkawa, K.; Nakajima, K.; Shimomura, Y.; Tokita, Y.; Machida, T.; Sumino, H.; Murakami, M. Small dense LDL cholesterol measured by homogeneous assay in Japanese healthy controls, metabolic syndrome and diabetes patients with or without a fatty liver. Clin. Chim. Acta 2015, 438, 70–79. [Google Scholar] [CrossRef]
- Imajo, K.; Hyogo, H.; Yoneda, M.; Honda, Y.; Kessoku, T.; Tomeno, W.; Ogawa, Y.; Taguri, M.; Mawatari, H.; Nozaki, Y.; et al. LDL-Migration Index (LDL-MI), an Indicator of Small Dense Low-Density Lipoprotein (sdLDL), Is Higher in Non-Alcoholic Steatohepatitis than in Non-Alcoholic Fatty Liver: A Multicenter Cross-Sectional Study. PLoS ONE 2014, 9, e115403. [Google Scholar] [CrossRef]
- Dias, C.B.; Moughan, P.J.; Wood, L.G.; Singh, H.; Garg, M.L. Postprandial lipemia: Factoring in lipemic response for ranking foods for their healthiness. Lipids Health Dis. 2017, 16, 178. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Durazzo, M.; Biroli, G.; Carello, M.; Fagà, E.; Pacini, G.; De Michieli, F.; Rabbione, L.; Premoli, A.; et al. Adipokines in NASH: Postprandial lipid metabolism as a link between adiponectin and liver disease. Hepatology 2005, 42, 1175–1183. [Google Scholar] [CrossRef]
- Katsiki, N.; Mikhailidis, D.P.; Mantzoros, C.S. Non-alcoholic fatty liver disease and dyslipidemia: An update. Metabolism 2016, 65, 1109–1123. [Google Scholar] [CrossRef]
- Nam, J.S.; Jo, S.; Kang, S.; Ahn, C.W.; Kim, K.R.; Park, J.S. Association between lipoprotein(a) and nonalcoholic fatty liver disease among Korean adults. Clin. Chim. Acta 2016, 461, 14–18. [Google Scholar] [CrossRef]
- Naik, A.; Košir, R.; Rozman, D. Genomic aspects of NAFLD pathogenesis. Genomics 2013, 102, 84–95. [Google Scholar] [CrossRef]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Bellentani, S.; Argo, C.K.; Ballestri, S.; Byrne, C.D.; Caldwell, S.H.; Cortez-Pinto, H.; Grieco, A.; Machado, M.V.; Miele, L.; et al. Epidemiological modifiers of non-alcoholic fatty liver disease: Focus on high-risk groups. Dig. Liver Dis. 2015, 47, 997–1006. [Google Scholar] [CrossRef]
- Lu, G.; Shimizu, I.; Cui, X.; Itonaga, M.; Tamaki, K.; Fukuno, H.; Inoue, H.; Honda, H.; Ito, S. Antioxidant and antiapoptotic activities of idoxifene and estradiol in hepatic fibrosis in rats. Life Sci. 2004, 74, 897–907. [Google Scholar] [CrossRef]
- Yasuda, M.; Shimizu, I.; Shiba, M.; Ito, S. Suppressive effects of estradiol on dimethylnitrosamine-induced fibrosis of the liver in rats. Hepatology 1999, 29, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Ballestri, S.; Nascimbeni, F.; Baldelli, E.; Marrazzo, A.; Romagnoli, D.; Lonardo, A. NAFLD as a Sexual Dimorphic Disease: Role of Gender and Reproductive Status in the Development and Progression of Nonalcoholic Fatty Liver Disease and Inherent Cardiovascular Risk. Adv. Ther. 2017, 34, 1291–1326. [Google Scholar] [CrossRef] [PubMed]
- Després, J.-P. Body Fat Distribution and Risk of Cardiovascular Disease. Circulation 2012, 126, 1301–1313. [Google Scholar] [CrossRef]
- Varlamov, O.; Bethea, C.L.; Roberts, C.T. Sex-Specific Differences in Lipid and Glucose Metabolism. Front. Endocrinol. (Lausanne) 2015, 5, 241. [Google Scholar] [CrossRef]
- Nielsen, S.; Guo, Z.; Johnson, C.M.; Hensrud, D.D.; Jensen, M.D. Splanchnic lipolysis in human obesity. J. Clin. Investig. 2004, 113, 1582–1588. [Google Scholar] [CrossRef] [PubMed]
- Turola, E.; Petta, S.; Vanni, E.; Milosa, F.; Valenti, L.; Critelli, R.; Miele, L.; Maccio, L.; Calvaruso, V.; Fracanzani, A.L.; et al. Ovarian senescence increases liver fibrosis in humans and zebrafish with steatosis. Dis. Model. Mech. 2015, 8, 1037–1046. [Google Scholar] [CrossRef]
- Lonardo, A.; Trande, P. Are there any sex differences in fatty liver? A study of glucose metabolism and body fat distribution. J. Gastroenterol. Hepatol. 2000, 15, 775–782. [Google Scholar] [CrossRef]
- Lundholm, L.; Zang, H.; Hirschberg, A.L.; Gustafsson, J.-Å.; Arner, P.; Dahlman-Wright, K. Key lipogenic gene expression can be decreased by estrogen in human adipose tissue. Fertil. Steril. 2008, 90, 44–48. [Google Scholar] [CrossRef]
- Klair, J.S.; Yang, J.D.; Abdelmalek, M.F.; Guy, C.D.; Gill, R.M.; Yates, K.; Unalp-Arida, A.; Lavine, J.E.; Clark, J.M.; Diehl, A.M.; et al. A longer duration of estrogen deficiency increases fibrosis risk among postmenopausal women with nonalcoholic fatty liver disease. Hepatology 2016, 64, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.-C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Unalp, A.; Behling, C.E.; Lavine, J.E.; Neuschwander-Tetri, B.A.; NASH Clinical Research NetworkA list of members of the Nonalcoholic Steatohepatitis Clinical Research Network can be found in the Appendix. Portal chronic inflammation in nonalcoholic fatty liver disease (NAFLD): A histologic marker of advanced NAFLD-Clinicopathologic correlations from the nonalcoholic steatohepatitis clinical research network. Hepatology 2009, 49, 809–820. [Google Scholar] [PubMed]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis Among a Largely Middle-Aged Population Utilizing Ultrasound and Liver Biopsy: A Prospective Study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Piscaglia, F.; Svegliati-Baroni, G.; Barchetti, A.; Pecorelli, A.; Marinelli, S.; Tiribelli, C.; Bellentani, S.; HCC-NAFLD Italian Study Group; Tiribelli, C.; Svegliati-Baroni, G.; et al. Clinical patterns of hepatocellular carcinoma in nonalcoholic fatty liver disease: A multicenter prospective study. Hepatology 2015, 63, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Paradis, V.; Zalinski, S.; Chelbi, E.; Guedj, N.; Degos, F.; Vilgrain, V.; Bedossa, P.; Belghiti, J. Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: A pathological analysis. Hepatology 2009, 49, 851–859. [Google Scholar] [CrossRef]
- Hester, D.; Golabi, P.; Paik, J.; Younossi, I.; Mishra, A.; Younossi, Z.M. Among Medicare Patients With Hepatocellular Carcinoma, Non–alcoholic Fatty Liver Disease is the Most Common Etiology and Cause of Mortality. J. Clin. Gastroenterol. 2019. [Google Scholar] [CrossRef]
- Hultcrantz, R.; Nasr, P.; Hagström, H.; Ekstedt, M.; Kechagias, S.; Stål, P.; Fredrikson, M.; Hagström, H.; Nasr, P.; Fredrikson, M.; et al. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2014, 61, 1547–1554. [Google Scholar]
- Pelusi, S.; Cespiati, A.; Rametta, R.; Pennisi, G.; Mannisto, V.; Rosso, C.; Baselli, G.; Dongiovanni, P.; Fracanzani, A.L.; Badiali, S.; et al. Prevalence and Risk Factors of Significant Fibrosis in Patients With Nonalcoholic Fatty Liver Without Steatohepatitis. Clin. Gastroenterol. Hepatol. 2019. [Google Scholar] [CrossRef]
- McPherson, S.; Hardy, T.; Henderson, E.; Burt, A.D.; Day, C.P.; Anstee, Q.M. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. J. Hepatol. 2015, 62, 1148–1155. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Maurantonio, M.; Marrazzo, A.; Rinaldi, L.; Adinolfi, L.E. Nonalcoholic fatty liver disease: Evolving paradigms. World J. Gastroenterol. 2017, 23, 6571–6592. [Google Scholar] [CrossRef]
- Arab, J.P.; Arrese, M.; Trauner, M. Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu. Rev. Pathol. Mech. Dis. 2018, 13, 321–350. [Google Scholar] [CrossRef]
- Donnelly, K.L. Sources of fatty acids stored in liver and secreted via lipoporteins in patients with NAFLD. J. Clin. Investig. 2012, 115, 1343–1351. [Google Scholar] [CrossRef]
- McGarry, J.D.; Brown, N.F. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur. J. Biochem. 1997, 244, 1–14. [Google Scholar] [CrossRef]
- Trauner, M.; Arrese, M.; Wagner, M. Fatty liver and lipotoxicity. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2010, 1801, 299–310. [Google Scholar] [CrossRef]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of Hepatic Mitochondrial Function in Humans with Non-Alcoholic Fatty Liver Is Lost in Steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef]
- Gallagher, E.J.; LeRoith, D. Minireview: IGF, Insulin, and Cancer. Endocrinology 2011, 152, 2546–2551. [Google Scholar] [CrossRef]
- Yamashita, T.; Honda, M.; Takatori, H.; Nishino, R.; Minato, H.; Takamura, H.; Ohta, T.; Kaneko, S. Activation of lipogenic pathway correlates with cell proliferation and poor prognosis in hepatocellular carcinoma. J. Hepatol. 2009, 50, 100–110. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Romeo, S.; Valenti, L. Hepatocellular carcinoma in nonalcoholic fatty liver: Role of environmental and genetic factors. World J. Gastroenterol. 2014, 20, 12945–12955. [Google Scholar] [CrossRef]
- Vinciguerra, M.; Carrozzino, F.; Peyrou, M.; Carlone, S.; Montesano, R.; Benelli, R.; Foti, M. Unsaturated fatty acids promote hepatoma proliferation and progression through downregulation of the tumor suppressor PTEN. J. Hepatol. 2009, 50, 1132–1141. [Google Scholar] [CrossRef]
- Marra, F.; Bertolani, C. Adipokines in liver diseases. Hepatology 2009, 50, 957–969. [Google Scholar] [CrossRef]
- Stender, S.; Kozlitina, J.; Nordestgaard, B.G.; Tybjærg-Hansen, A.; Hobbs, H.H.; Cohen, J.C. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat. Genet. 2017, 49, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C.J. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 2011, 53, 1883–1894. [Google Scholar] [CrossRef]
- Sookoian, S.; Castaño, G.O.; Burgueño, A.L.; Gianotti, T.F.; Rosselli, M.S.; Pirola, C.J. A nonsynonymous gene variant in the adiponutrin gene is associated with nonalcoholic fatty liver disease severity. J. Lipid Res. 2009, 50, 2111–2116. [Google Scholar] [CrossRef] [PubMed]
- Trépo, E.; Romeo, S.; Zucman-Rossi, J.; Nahon, P. PNPLA3 gene in liver diseases. J. Hepatol. 2016, 65, 399–412. [Google Scholar] [CrossRef]
- Salameh, H.; Raff, E.; Erwin, A.; Seth, D.; Nischalke, H.D.; Falleti, E.; Burza, M.A.; Leathert, J.; Romeo, S.; Molinaro, A.; et al. PNPLA3 Gene Polymorphism Is Associated With Predisposition to and Severity of Alcoholic Liver Disease. Am. J. Gastroenterol. 2015, 110, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Alisi, A.; Mancina, R.M.; Xing, C.; Petta, S.; Rametta, R.; Nobili, V.; del Menico, B.; Valenti, L.; Craxì, A.; Fracanzani, A.L.; et al. The rs2294918 E434K variant modulates patatin-like phospholipase domain-containing 3 expression and liver damage. Hepatology 2015, 63, 787–798. [Google Scholar]
- Bruschi, F.V.; Claudel, T.; Tardelli, M.; Caligiuri, A.; Stulnig, T.M.; Marra, F.; Trauner, M. The PNPLA3 I148M variant modulates the fibrogenic phenotype of human hepatic stellate cells. Hepatology 2017, 65, 1875–1890. [Google Scholar] [CrossRef] [PubMed]
- Mahdessian, H.; Taxiarchis, A.; Popov, S.; Silveira, A.; Franco-Cereceda, A.; Hamsten, A.; Eriksson, P.; van’t Hooft, F. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc. Natl. Acad. Sci. 2014, 111, 8913–8918. [Google Scholar] [CrossRef] [PubMed]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Reeves, H.L.; Burt, A.D.; Tiniakos, D.; McPherson, S.; Leathart, J.B.S.; Allison, M.E.D.; Alexander, G.J.; Piguet, A.-C.; Anty, R.; et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 4309. [Google Scholar] [CrossRef] [PubMed]
- Goffredo, M.; Caprio, S.; Feldstein, A.E.; D’Adamo, E.; Shaw, M.M.; Pierpont, B.; Savoye, M.; Zhao, H.; Bale, A.E.; Santoro, N. Role of TM6SF2 rs58542926 in the pathogenesis of nonalcoholic pediatric fatty liver disease: A multiethnic study. Hepatology 2016, 63, 117–125. [Google Scholar] [CrossRef]
- Pirola, C.J.; Sookoian, S. The dual and opposite role of the TM6SF2 -rs58542926 variant in protecting against cardiovascular disease and conferring risk for nonalcoholic fatty liver: A meta-analysis. Hepatology 2015, 62, 1742–1756. [Google Scholar] [CrossRef]
- Sliz, E.; Sebert, S.; Würtz, P.; Kangas, A.J.; Soininen, P.; Lehtimäki, T.; Kähönen, M.; Viikari, J.; Männikkö, M.; Ala-Korpela, M.; et al. NAFLD risk alleles in PNPLA3, TM6SF2, GCKR and LYPLAL1 show divergent metabolic effects. Hum. Mol. Genet. 2018, 27, 2214–2223. [Google Scholar] [CrossRef] [PubMed]
- Santoro, N.; Zhang, C.K.; Zhao, H.; Pakstis, A.J.; Kim, G.; Kursawe, R.; Dykas, D.J.; Bale, A.E.; Giannini, C.; Pierpont, B.; et al. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology 2012, 55, 781–789. [Google Scholar] [CrossRef]
- Beer, N.L.; Tribble, N.D.; McCulloch, L.J.; Roos, C.; Johnson, P.R.V.; Orho-Melander, M.; Gloyn, A.L. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum. Mol. Genet. 2009, 18, 4081–4088. [Google Scholar] [CrossRef]
- Wang, Y.; van der Tuin, S.; Tjeerdema, N.; van Dam, A.D.; Rensen, S.S.; Hendrikx, T.; Berbée, J.F.P.; Atanasovska, B.; Fu, J.; Hoekstra, M.; et al. Plasma cholesteryl ester transfer protein is predominantly derived from Kupffer cells. Hepatology 2015, 62, 1710–1722. [Google Scholar] [CrossRef]
- Aller, R.; Izaola, O.; Primo, D.; de Luis, D.; De Luis, D. Cholesteryl ester transfer protein variant (RS1800777) with liver histology in non-alcoholic fatty liver disease patients. Ann. Nutr. Metab. 2018, 73, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Bo, S.; Cassader, M.; De Michieli, F.; Gambino, R. Impact of sterol regulatory element-binding factor-1c polymorphism on incidence of nonalcoholic fatty liver disease and on the severity of liver disease and of glucose and lipid dysmetabolism. Am. J. Clin. Nutr. 2013, 98, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Borén, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016, 150, 1219–1230.e6. [Google Scholar] [CrossRef]
- Krawczyk, M.; Rau, M.; Schattenberg, J.M.; Bantel, H.; Pathil, A.; Demir, M.; Kluwe, J.; Boettler, T.; Lammert, F.; Geier, A.; et al. Combined effects of the PNPLA3 rs738409, TM6SF2 rs58542926, and MBOAT7 rs641738 variants on NAFLD severity: A multicenter biopsy-based study. J. Lipid Res. 2017, 58, 247–255. [Google Scholar] [CrossRef]
- Umano, G.R.; Caprio, S.; Di Sessa, A.; Chalasani, N.; Dykas, D.J.; Pierpont, B.; Bale, A.E.; Santoro, N. The rs626283 variant in the MBOAT7 gene is associated with insulin resistance and fatty liver in Caucasian obese youth. Am. J. Gastroenterol. 2018, 113, 376–383. [Google Scholar] [CrossRef]
- Hussain, M.M.; Rava, P.; Walsh, M.; Rana, M.; Iqbal, J. Multiple functions of microsomal triglyceride transfer protein. Nutr. Metab. (Lond.) 2012, 9, 14. [Google Scholar] [CrossRef]
- Gouda, W.; Ashour, E.; Shaker, Y.; Ezzat, W. MTP genetic variants associated with non-alcoholic fatty liver in metabolic syndrome patients. Genes Dis. 2017, 4, 222–228. [Google Scholar] [CrossRef]
- Marchesini, G.; Brizi, M.; Morselli-Labate, A.M.; Bianchi, G.; Bugianesi, E.; McCullough, A.J.; Forlani, G.; Melchionda, N. Association of nonalcoholic fatty liver disease with insulin resistance. Am. J. Med. 1999, 107, 450–455. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Mantovani, A.; Targher, G. Hypertension, diabetes, atherosclerosis and NASH: Cause or consequence? J. Hepatol. 2018, 68, 335–352. [Google Scholar] [CrossRef]
- Ballestri, S.; Zona, S.; Targher, G.; Romagnoli, D.; Baldelli, E.; Nascimbeni, F.; Roverato, A.; Guaraldi, G.; Lonardo, A. Nonalcoholic fatty liver disease is associated with an almost twofold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2016, 31, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Byrne, C.D.; Bonora, E.; Targher, G. Nonalcoholic Fatty Liver Disease and Risk of Incident Type 2 Diabetes: A Meta-analysis. Diabetes Care 2018, 41, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Falcon, A.; Storm, T.A.; Kazantzis, M.; Kay, M.A.; Doege, H.; Stahl, A.; Tsang, B.; Xu, H.; Ortegon, A.M. Silencing of Hepatic Fatty Acid Transporter Protein 5 in Vivo Reverses Diet-induced Non-alcoholic Fatty Liver Disease and Improves Hyperglycemia. J. Biol. Chem. 2008, 283, 22186–22192. [Google Scholar]
- Brown, M.S.; Goldstein, J.L. Selective versus Total Insulin Resistance: A Pathogenic Paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef] [PubMed]
- Biddinger, S.B.; Hernandez-Ono, A.; Rask-Madsen, C.; Haas, J.T.; Alemán, J.O.; Suzuki, R.; Scapa, E.F.; Agarwal, C.; Carey, M.C.; Stephanopoulos, G.; et al. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab. 2015, 27, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Hijmans, B.S.; Grefhorst, A.; Oosterveer, M.H.; Groen, A.K. Zonation of glucose and fatty acid metabolism in the liver: Mechanism and metabolic consequences. Biochimie 2014, 96, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Wilson, L.; Kleiner, D.E.; Cummings, O.W.; Brunt, E.M.; Ünalp, A.; NASH Clinical Research Network. Relationship of steatosis grade and zonal location to histological features of steatohepatitis in adult patients with non-alcoholic fatty liver disease. J. Hepatol. 2008, 48, 829–834. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Makhlouf, H.R. Histology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis in Adults and Children. Clin. Liver Dis. 2016, 20, 293–312. [Google Scholar] [CrossRef]
- Sanders, F.W.B.; Griffin, J.L. De novo lipogenesis in the liver in health and disease: More than just a shunting yard for glucose. Biol. Rev. 2016, 91, 452–468. [Google Scholar] [CrossRef]
- Perseghin, G.; Lattuada, G.; De Cobelli, F.; Esposito, A.; Costantino, F.; Canu, T.; Scifo, P.; De Taddeo, F.; Maffi, P.; Secchi, A.; et al. Reduced intrahepatic fat content is associated with increased whole-body lipid oxidation in patients with type 1 diabetes. Diabetologia 2005, 48, 2615–2621. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Rinehart, J.; Shulman, G.I.; Petersen, M.C.; Madiraju, A.K.; Gassaway, B.M.; Marcel, M.; Nasiri, A.R.; Butrico, G.; Marcucci, M.J.; et al. Insulin receptor Thr 1160 phosphorylation mediates lipid-induced hepatic insulin resistance Find the latest version: Insulin receptor Thr 1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J. Clin. Investig. 2016, 126, 4361–4371. [Google Scholar] [CrossRef]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Galadari, A.; Thayyullathil, F. Role of ceramide in diabetes mellitus: Evidence and mechanisms: MedSök Region Skåne. Lipids Health Dis. 2013, 12, 98. [Google Scholar] [CrossRef] [PubMed]
- Bi, P.; Kuang, S. Notch signaling as a novel regulator of metabolism. Trends Endocrinol. Metab. 2015, 26, 248–255. [Google Scholar] [CrossRef]
- Kopan, R.; Ilagan, M.X.G. The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef]
- Guruharsha, K.G.; Kankel, M.W.; Artavanis-Tsakonas, S. The Notch signalling system: Recent insights into the complexity of a conserved pathway. Nat. Rev. Genet. 2012, 13, 654–666. [Google Scholar] [CrossRef]
- Geisler, F.; Strazzabosco, M. Emerging roles of Notch signaling in liver disease. Hepatology 2015, 61, 382–392. [Google Scholar] [CrossRef]
- Zhu, C.; Kim, K.; Wang, X.; Bartolome, A.; Salomao, M.; Dongiovanni, P.; Meroni, M.; Graham, M.J.; Yates, K.P.; Diehl, A.M.; et al. Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci. Transl. Med. 2018, 10, eaat0344. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S. Notch and Nonalcoholic Fatty Liver and Fibrosis. N. Engl. J. Med. 2019, 380, 681–683. [Google Scholar] [CrossRef]
- Bi, P.; Shan, T.; Liu, W.; Yue, F.; Yang, X.; Liang, X.-R.; Wang, J.; Li, J.; Carlesso, N.; Liu, X.; et al. Inhibition of Notch signaling promotes browning of white adipose tissue and ameliorates obesity. Nat. Med. 2014, 20, 911–918. [Google Scholar] [CrossRef]
- Pajvani, U.B.; Shawber, C.J.; Samuel, V.T.; Birkenfeld, A.L.; Shulman, G.I.; Kitajewski, J.; Accili, D. Inhibition of Notch signaling ameliorates insulin resistance in a FoxO1-dependent manner. Nat. Med. 2011, 17, 961–967. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Jacot, E.; Jequier, E.; Maeder, E.; Wahren, J.; Felber, J.P. The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes 1981, 30, 1000–1007. [Google Scholar] [CrossRef]
- Yu, R.; Shi, Q.; Liu, L.; Chen, L. Relationship of sarcopenia with steatohepatitis and advanced liver fibrosis in non-alcoholic fatty liver disease: A meta-analysis. BMC Gastroenterol. 2018, 18, 51. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Ciminnisi, S.; Di Marco, V.; Cabibi, D.; Cammà, C.; Licata, A.; Marchesini, G.; Craxì, A. Sarcopenia is associated with severe liver fibrosis in patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2017, 45, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Febbraio, M.A. Muscles, exercise and obesity: Skeletal muscle as a secretory organ. Nat. Rev. Endocrinol. 2012, 8, 457–465. [Google Scholar] [CrossRef]
- Pappachan, J.M.; Babu, S.; Krishnan, B.; Ravindran, N.C. Non-alcoholic Fatty Liver Disease: A Clinical Update. J. Clin. Transl. Hepatol. 2017, 5, 384–393. [Google Scholar] [CrossRef]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar] [CrossRef]
- Basarnoglu, M.; Basarnoglu, G.; Bugianesi, E. Carbohydrate intake and nonalcoholic fatty liver disease: Fructose as a weapon of mass destruction. HepatoBiliary Surg Nutr 2015, 4, 109–116. [Google Scholar]
- Adeli, K.; Su, Q.; Rutledge, A.C.; Dekker, M.J.; Baker, C. Fructose: A highly lipogenic nutrient implicated in insulin resistance, hepatic steatosis, and the metabolic syndrome. Am. J. Physiol. Metab. 2010, 299, E685–E694. [Google Scholar]
- Basaranoglu, M.; Basaranoglu, G.; Sabuncu, T.; Sentürk, H. Fructose as a key player in the development of fatty liver disease. World J. Gastroenterol. 2013, 19, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.B.; Lavine, J.E. Dietary fructose in nonalcoholic fatty liver disease. Hepatology 2013, 57, 2525–2531. [Google Scholar] [CrossRef]
- Barrera, F.; George, J. The role of diet and nutritional intervention for the management of patients with NAFLD. Clin. Liver Dis. 2014, 18, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Lambertz, J.; Weiskirchen, S.; Landert, S.; Weiskirchen, R. Fructose: A dietary sugar in crosstalk with microbiota contributing to the development and progression of non-alcoholic liver disease. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Tarantino, G.; Savastano, S.; Colao, A. Hepatic steatosis, low-grade chronic inflammation and hormone/growth factor/adipokine imbalance. World J. Gastroenterol. 2010, 16, 4773–4783. [Google Scholar] [CrossRef]
- Finelli, C.; Tarantino, G. What is the role of adiponectin in obesity related non-alcoholic fatty liver disease? World J. Gastroenterol. 2013, 19, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Biroli, G.; Carello, M.; Faga, E.; Pacini, G.; De Michieli, F.; Cassader, M.; Durazzo, M.; Rizzetto, M.; et al. Hypoadiponectinemia Predicts the Severity of Hepatic Fibrosis and Pancreatic Beta-Cell Dysfunction in Nondiabetic Nonobese Patients with Nonalcoholic Steatohepatitis. Am. J. Gastroenterol. 2005, 100, 2438–2446. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Toulis, K.A.; Goulis, D.G.; Zavos, C.; Kountouras, J. Serum total adiponectin in nonalcoholic fatty liver disease: A systematic review and meta-analysis. Metabolism 2011, 60, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Minokoshi, Y.; Kim, Y.-B.; Peroni, O.D.; Fryer, L.G.D.; Müller, C.; Carling, D.; Kahn, B.B. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002, 415, 339–343. [Google Scholar] [CrossRef]
- Abenavoli, L.; Peta, V. Role of adipokines and cytokines in non-alcoholic fatty liver disease. Rev. Recent Clin. Trials 2014, 9, 134–140. [Google Scholar] [CrossRef]
- Adolph, T.E.; Grander, C.; Grabherr, F.; Tilg, H. Adipokines and Non-Alcoholic Fatty Liver Disease: Multiple Interactions. Int. J. Mol. Sci. 2017, 18, 1649. [Google Scholar] [CrossRef] [PubMed]
- Crespo, J.; Cayón, A.; Fernández-Gil, P.; Hernández-Guerra, M.; Mayorga, M.; Domínguez-Díez, A.; Fernández-Escalante, J.C.; Pons-Romero, F. Gene expression of tumor necrosis factor [alpha ] and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology 2001, 34, 1158–1163. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen Recognition by the Innate Immune System. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Brenner, D.A. Toll-like receptors and adaptor molecules in liver disease: Update. Hepatology 2008, 48, 322–335. [Google Scholar] [CrossRef]
- Lawrence, T. The Nuclear Factor NF- B Pathway in Inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef]
- Arrese, M.; Cabrera, D.; Kalergis, A.M.; Feldstein, A.E. Innate Immunity and Inflammation in NAFLD/NASH. Dig. Dis. Sci. 2016, 61, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, G.I.; Langley, K.G.; Berglund, N.A.; Kammoun, H.L.; Reibe, S.; Estevez, E.; Weir, J.; Mellett, N.A.; Pernes, G.; Conway, J.R.W.; et al. Evidence that TLR4 Is Not a Receptor for Saturated Fatty Acids but Mediates Lipid-Induced Inflammation by Reprogramming Macrophage Metabolism. Cell Metab. 2018, 27, 1096–1110.e5. [Google Scholar] [CrossRef]
- Britton, L.; Bridle, K.; Reiling, J.; Santrampurwala, N.; Wockner, L.; Ching, H.; Stuart, K.; Subramaniam, V.N.; Jeffrey, G.; St. Pierre, T.; et al. Hepatic iron concentration correlates with insulin sensitivity in nonalcoholic fatty liver disease. Hepatol. Commun. 2018, 2, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Eaton, J.W.; Qian, M. Molecular bases of cellular iron toxicity. Free Radic. Biol. Med. 2002, 32, 833–840. [Google Scholar] [CrossRef]
- Puntarulo, S. Iron, oxidative stress and human health. Mol. Aspects Med. 2005, 26, 299–312. [Google Scholar] [CrossRef]
- MacDonald, G.A.; Bridle, K.R.; Ward, P.J.; Walker, N.I.; Houglum, K.; George, D.K.; Smith, J.L.; Powell, L.W.; Crawford, D.H.; Ramm, G.A. Lipid peroxidation in hepatic steatosis in humans is associated with hepatic fibrosis and occurs predominately in acinar zone 3. J. Gastroenterol. Hepatol. 2001, 16, 599–606. [Google Scholar] [CrossRef]
- Maliken, B.D.; Nelson, J.E.; Klintworth, H.M.; Beauchamp, M.; Yeh, M.M.; Kowdley, K.V. Hepatic reticuloendothelial system cell iron deposition is associated with increased apoptosis in nonalcoholic fatty liver disease. Hepatology 2013, 57, 1806–1813. [Google Scholar] [CrossRef]
- Nelson, J.E.; Wilson, L.; Brunt, E.M.; Yeh, M.M.; Kleiner, D.E.; Unalp-Arida, A.; Kowdley, K.V.; Nonalcoholic Steatohepatitis Clinical Research Network. Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology 2011, 53, 448–457. [Google Scholar] [CrossRef]
- Handa, P.; Morgan-Stevenson, V.; Maliken, B.D.; Nelson, J.E.; Washington, S.; Westerman, M.; Yeh, M.M.; Kowdley, K.V. Iron overload results in hepatic oxidative stress, immune cell activation, and hepatocellular ballooning injury, leading to nonalcoholic steatohepatitis in genetically obese mice. Am. J. Physiol. Liver Physiol. 2016, 310, G117–G127. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Belt, P.; Wilson, L.A.; Yeh, M.M.; Neuschwander-Tetri, B.A.; Chalasani, N.; Sanyal, A.J.; Nelson, J.E.; NASH Clinical Research Network. Serum ferritin is an independent predictor of histologic severity and advanced fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2012, 55, 77–85. [Google Scholar] [CrossRef]
- Licata, A.; Nebbia, M.E.; Cabibbo, G.; Iacono, G.L.; Barbaria, F.; Brucato, V.; Alessi, N.; Porrovecchio, S.; Di Marco, V.; Craxì, A.; et al. Hyperferritinemia is a risk factor for steatosis in chronic liver disease. World J. Gastroenterol. 2009, 15, 2132–2138. [Google Scholar] [CrossRef]
- Buzzetti, E.; Petta, S.; Manuguerra, R.; Luong, T.V.; Cabibi, D.; Corradini, E.; Craxì, A.; Pinzani, M.; Tsochatzis, E.; Pietrangelo, A. Evaluating the association of serum ferritin and hepatic iron with disease severity in non-alcoholic fatty liver disease. Liver Int. 2019. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Crawford, D.H.; Stuart, K.; House, M.J.; St. Pierre, T.G.; Webb, M.; Ching, H.L.I.; Kava, J.; Bynevelt, M.; MacQuillan, G.C.; et al. The impact of phlebotomy in nonalcoholic fatty liver disease: A prospective, randomized, controlled trial. Hepatology 2015, 61, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Fracanzani, A.L.; Dongiovanni, P.; Rovida, S.; Rametta, R.; Fatta, E.; Pulixi, E.A.; Maggioni, M.; Fargion, S. A randomized trial of iron depletion in patients with nonalcoholic fatty liver disease and hyperferritinemia. World J. Gastroenterol. 2014, 20, 3002. [Google Scholar] [CrossRef]
- Schaap, F.G.; Trauner, M.; Jansen, P.L.M. Bile acid receptors as targets for drug development. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Arab, J.P.; Karpen, S.J.; Dawson, P.A.; Arrese, M.; Trauner, M.; Arab, J.P.; Dawson, P.A.; Arrese, M. Bile acids and nonalcoholic fatty liver disease: Molecular insights and therapeutic perspectives. Hepatology 2016, 65, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Perino, A.; Schoonjans, K. TGR5 and Immunometabolism: Insights from Physiology and Pharmacology. Trends Pharmacol. Sci. 2015, 36, 847–857. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332–338. [Google Scholar] [CrossRef]
- Inagaki, T.; Moschetta, A.; Lee, Y.-K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Kohli, R. Bile acid metabolism and signaling: Potential therapeutic target for nonalcoholic fatty liver disease. Clin. Transl. Gastroenterol. 2018, 9, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Mazzoccoli, G.; De Cosmo, S.; Mazza, T. The biological clock: A pivotal hub in non-alcoholic fatty liver disease pathogenesis. Front. Physiol. 2018, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.Y. The Suprachiasmatic Nucleus and the Circadian Timing System. Prog. Mol. Biol. Transl. Sci. 2013, 119, 1–28. [Google Scholar]
- Reinke, H.; Asher, G. Circadian Clock Control of Liver Metabolic Functions. Gastroenterology 2016, 150, 574–580. [Google Scholar] [CrossRef]
- Kettner, N.M.; Voicu, H.; Finegold, M.J.; Coarfa, C.; Sreekumar, A.; Putluri, N.; Katchy, C.A.; Lee, C.; Moore, D.D.; Fu, L. Circadian Homeostasis of Liver Metabolism Suppresses Hepatocarcinogenesis. Cancer Cell 2016, 30, 909–924. [Google Scholar] [CrossRef]
- Tahara, Y.; Shibata, S. Circadian rhythms of liver physiology and disease: Experimental and clinical evidence. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 217–226. [Google Scholar] [CrossRef]
- Zagami, R.M.; Di Pino, A.; Urbano, F.; Piro, S.; Purrello, F.; Rabuazzo, A.M. Low circulating vitamin D levels are associated with increased arterial stiffness in prediabetic subjects identified according to HbA1c. Atherosclerosis 2015, 243, 395–401. [Google Scholar] [CrossRef]
- Sung, K.-C.; Ryu, S.; Lee, J.-Y.; Kim, J.-Y.; Wild, S.H.; Byrne, C.D. Effect of exercise on the development of new fatty liver and the resolution of existing fatty liver. J. Hepatol. 2016, 65, 791–797. [Google Scholar] [CrossRef]
- Prenner, S.; Rinella, M.E. Moderate Exercise for Nonalcoholic Fatty Liver Disease. JAMA Intern. Med. 2016, 176, 1083. [Google Scholar] [CrossRef]
- Marchesini, G.; Petta, S.; Dalle Grave, R. Diet, weight loss, and liver health in nonalcoholic fatty liver disease: Pathophysiology, evidence, and practice. Hepatology 2016, 63, 2032–2043. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 367–378.e5. [Google Scholar] [CrossRef]
- Harrison, S.A.; Fecht, W.; Brunt, E.M.; Neuschwander-Tetri, B.A. Orlistat for overweight subjects with nonalcoholic steatohepatitis: A randomized, prospective trial. Hepatology 2009, 49, 80–86. [Google Scholar] [CrossRef]
- Haufe, S.; Engeli, S.; Kast, P.; Böhnke, J.; Utz, W.; Haas, V.; Hermsdorf, M.; Mähler, A.; Wiesner, S.; Birkenfeld, A.L.; et al. Randomized comparison of reduced fat and reduced carbohydrate hypocaloric diets on intrahepatic fat in overweight and obese human subjects. Hepatology 2011, 53, 1504–1514. [Google Scholar] [CrossRef]
- Boden, G. High- or low-carbohydrate diets: Which is better for weight loss, insulin resistance, and fatty livers? Gastroenterology 2009, 136, 1490–1492. [Google Scholar] [CrossRef]
- Bower, G.; Toma, T.; Harling, L.; Jiao, L.R.; Efthimiou, E.; Darzi, A.; Athanasiou, T.; Ashrafian, H. Bariatric Surgery and Non-Alcoholic Fatty Liver Disease: A Systematic Review of Liver Biochemistry and Histology. Obes. Surg. 2015, 25, 2280–2289. [Google Scholar] [CrossRef]
- Zelber-Sagi, S.; Salomone, F.; Mlynarsky, L. The Mediterranean dietary pattern as the diet of choice for non-alcoholic fatty liver disease: Evidence and plausible mechanisms. Liver Int. 2017, 37, 936–949. [Google Scholar] [CrossRef]
- Targher, G.; Byrne, C.D. Ad Libitum Mediterranean or Low-Fat Diets as Treatments for Nonalcoholic Fatty Liver Disease? Hepatology 2018, 68, 1668–1671. [Google Scholar] [CrossRef] [PubMed]
- Properzi, C.; O’Sullivan, T.A.; Sherriff, J.L.; Ching, H.L.; Jeffrey, G.P.; Buckley, R.F.; Tibballs, J.; MacQuillan, G.C.; Garas, G.; Adams, L.A. Ad Libitum Mediterranean and Low-Fat Diets Both Significantly Reduce Hepatic Steatosis: A Randomized Controlled Trial. Hepatology 2018, 68, 1741–1754. [Google Scholar] [CrossRef] [PubMed]
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.-T.; Corrà, U.; Cosyns, B.; Deaton, C.; et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice. Eur. Heart J. 2016, 37, 2315–2381. [Google Scholar] [CrossRef] [PubMed]
- Perumpail, B.J.; Cholankeril, R.; Yoo, E.R.; Kim, D.; Ahmed, A. An Overview of Dietary Interventions and Strategies to Optimize the Management of Non-Alcoholic Fatty Liver Disease. Diseases 2017, 5, 23. [Google Scholar] [CrossRef]
- Saab, S.; Mallam, D.; Cox, G.A.; Tong, M.J. Impact of coffee on liver diseases: A systematic review. Liver Int. 2014, 34, 495–504. [Google Scholar] [CrossRef]
- Mantovani, A. Time to revise the definition of NAFLD: A purist vision. Dig. Liver Dis. 2019, 51, 457–458. [Google Scholar] [CrossRef]
- Griswold, M.G.; Fullman, N.; Hawley, C.; Arian, N.; Zimsen, S.R.M.; Tymeson, H.D.; Venkateswaran, V.; Tapp, A.D.; Forouzanfar, M.H.; Salama, J.S.; et al. Alcohol use and burden for 195 countries and territories, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2018, 392, 1015–1035. [Google Scholar] [CrossRef]
- Aithal, G.P.; Thomas, J.A.; Kaye, P.V.; Lawson, A.; Ryder, S.D.; Spendlove, I.; Austin, A.S.; Freeman, J.G.; Morgan, L.; Webber, J. Randomized, Placebo-Controlled Trial of Pioglitazone in Nondiabetic Subjects With Nonalcoholic Steatohepatitis. Gastroenterology 2008, 135, 1176–1184. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, Vitamin E, or Placebo for Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef]
- Shah, P.; Mudaliar, S. Pioglitazone: Side effect and safety profile. Expert Opin. Drug Saf. 2010, 9, 347–354. [Google Scholar] [CrossRef]
- Bugianesi, E.; Gentilcore, E.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; David, E.; Rizzetto, M.; Marchesini, G. A Randomized Controlled Trial of Metformin versus Vitamin E or Prescriptive Diet in Nonalcoholic Fatty Liver Disease. Am. J. Gastroenterol. 2005, 100, 1082–1090. [Google Scholar] [CrossRef]
- Haukeland, J.W.; Konopski, Z.; Eggesbø, H.B.; von Volkmann, H.L.; Raschpichler, G.; Bjøro, K.; Haaland, T.; Løberg, E.M.; Birkeland, K. Metformin in patients with non-alcoholic fatty liver disease: A randomized, controlled trial. Scand. J. Gastroenterol. 2009, 44, 853–860. [Google Scholar] [CrossRef]
- Shields, W.W.; Thompson, K.E.; Grice, G.A.; Harrison, S.A.; Coyle, W.J. The Effect of Metformin and Standard Therapy versus Standard Therapy alone in Nondiabetic Patients with Insulin Resistance and Nonalcoholic Steatohepatitis (NASH): A Pilot Trial. Therap. Adv. Gastroenterol. 2009, 2, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.-H. Metformin and Risk of Hepatocellular Carcinoma in Taiwanese Patients with Type 2 Diabetes. Diabetes 2018, 38, 2018–2027. [Google Scholar] [CrossRef]
- Fujita, K.; Iwama, H.; Miyoshi, H.; Tani, J.; Oura, K.; Tadokoro, T.; Sakamoto, T.; Nomura, T.; Morishita, A.; Yoneyama, H.; et al. Diabetes mellitus and metformin in hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 6100–6113. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Abouda, G.; Aldersley, M.A.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.E.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Marchesini, G. Time for Glucagon like peptide-1 receptor agonists treatment for patients with NAFLD? J. Hepatol. 2016, 64, 262–264. [Google Scholar] [CrossRef] [PubMed]
- Seghieri, M.; Christensen, A.S.; Andersen, A.; Solini, A.; Knop, F.K.; Vilsbøll, T. Future Perspectives on GLP-1 Receptor Agonists and GLP-1/glucagon Receptor Co-agonists in the Treatment of NAFLD. Front. Endocrinol. (Lausanne) 2018, 9. [Google Scholar] [CrossRef]
- Cui, J.; Philo, L.; Nguyen, P.; Hofflich, H.; Hernandez, C.; Bettencourt, R.; Richards, L.; Salotti, J.; Bhatt, A.; Hooker, J.; et al. Sitagliptin vs. placebo for non-alcoholic fatty liver disease: A randomized controlled trial. J. Hepatol. 2016, 65, 369–376. [Google Scholar] [CrossRef]
- Scheen, A.J. Cardiovascular Effects of New Oral Glucose-Lowering Agents. Circ. Res. 2018, 122, 1439–1459. [Google Scholar] [CrossRef] [PubMed]
- Kuchay, M.S.; Krishan, S.; Mishra, S.K.; Farooqui, K.J.; Singh, M.K.; Wasir, J.S.; Bansal, B.; Kaur, P.; Jevalikar, G.; Gill, H.K.; et al. Effect of Empagliflozin on Liver Fat in Patients With Type 2 Diabetes and Nonalcoholic Fatty Liver Disease: A Randomized Controlled Trial (E-LIFT Trial). Diabetes Care 2018, 41, dc180165. [Google Scholar] [CrossRef] [PubMed]
- Takase, T.; Nakamura, A.; Miyoshi, H.; Yamamoto, C.; Atsumi, T. Amelioration of fatty liver index in patients with type 2 diabetes on ipragliflozin: An association with glucose-lowering effects. Endocr. J. 2017, 64, 363–367. [Google Scholar] [CrossRef]
- Shibuya, T.; Fushimi, N.; Kawai, M.; Yoshida, Y.; Hachiya, H.; Ito, S.; Kawai, H.; Ohashi, N.; Mori, A. Luseogliflozin improves liver fat deposition compared to metformin in type 2 diabetes patients with non-alcoholic fatty liver disease: A prospective randomized controlled pilot study. Diabetes, Obes. Metab. 2018, 20, 438–442. [Google Scholar] [CrossRef]
- Eriksson, J.W.; Lundkvist, P.; Jansson, P.-A.; Johansson, L.; Kvarnström, M.; Moris, L.; Miliotis, T.; Forsberg, G.-B.; Risérus, U.; Lind, L.; et al. Effects of dapagliflozin and n-3 carboxylic acids on non-alcoholic fatty liver disease in people with type 2 diabetes: A double-blind randomised placebo-controlled study. Diabetologia 2018, 61, 1923–1934. [Google Scholar] [CrossRef]
- Scheen, A.J. Effect of sodium-glucose cotransporter type 2 inhibitors on liver fat in patients with type 2 diabetes: Hepatic beyond cardiovascular and renal protection? Ann. Transl. Med. 2018, 6, S68. [Google Scholar] [CrossRef]
- Ferrannini, E.; Muscelli, E.; Frascerra, S.; Baldi, S.; Mari, A.; Heise, T.; Broedl, U.C.; Woerle, H.-J. Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J. Clin. Investig. 2014, 124, 499–508. [Google Scholar] [CrossRef]
- Bonnet, F.; Scheen, A.J. Effects of SGLT2 inhibitors on systemic and tissue low-grade inflammation: The potential contribution to diabetes complications and cardiovascular disease. Diabetes Metab. 2018, 44, 457–464. [Google Scholar] [CrossRef]
- Lonardo, A.; Bellentani, S.; Ratziu, V.; Loria, P. Insulin resistance in nonalcoholic steatohepatitis: Necessary but not sufficient – death of a dogma from analysis of therapeutic studies? Expert Rev. Gastroenterol. Hepatol. 2011, 5, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Perumpail, B.; Li, A.; John, N.; Sallam, S.; Shah, N.; Kwong, W.; Cholankeril, G.; Kim, D.; Ahmed, A. The Role of Vitamin E in the Treatment of NAFLD. Diseases 2018, 6, 86. [Google Scholar] [CrossRef]
- Sato, K.; Gosho, M.; Yamamoto, T.; Kobayashi, Y.; Ishii, N.; Ohashi, T.; Nakade, Y.; Ito, K.; Fukuzawa, Y.; Yoneda, M. Vitamin E has a beneficial effect on nonalcoholic fatty liver disease: A meta-analysis of randomized controlled trials. Nutrition 2015, 31, 923–930. [Google Scholar] [CrossRef]
- Wei, J.; Lei, G.-H.; Fu, L.; Zeng, C.; Yang, T.; Peng, S.-F. Association between Dietary Vitamin C Intake and Non-Alcoholic Fatty Liver Disease: A Cross-Sectional Study among Middle-Aged and Older Adults. PLoS ONE 2016, 11, e0147985. [Google Scholar] [CrossRef]
- Shidfar, F.; Faghihi, A.; Amiri, H.L.; Mousavi, S.N. Regression of Nonalcoholic Fatty Liver Disease with Zinc and Selenium Co-supplementation after Disease Progression in Rats. Iran. J. Med. Sci. 2018, 43, 26–31. [Google Scholar]
- Couet, C.; Delarue, J.; Ritz, P.; Antoine, J.M.; Lamisse, F. Effect of dietary fish oil on body fat mass and basal fat oxidation in healthy adults. Int. J. Obes. Relat. Metab. Disord. 1997, 21, 637–643. [Google Scholar] [CrossRef] [PubMed]
- de Castro, G.S.; Calder, P.C. Non-alcoholic fatty liver disease and its treatment with n-3 polyunsaturated fatty acids. Clin. Nutr. 2018, 37, 37–55. [Google Scholar] [CrossRef]
- Scorletti, E.; Byrne, C.D. Omega-3 fatty acids and non-alcoholic fatty liver disease: Evidence of efficacy and mechanism of action. Mol. Aspects Med. 2018, 64, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Spadaro, L.; Magliocco, O.; Spampinato, D.; Piro, S.; Oliveri, C.; Alagona, C.; Papa, G.; Rabuazzo, A.M.; Purrello, F. Effects of n-3 polyunsaturated fatty acids in subjects with nonalcoholic fatty liver disease. Dig. Liver Dis. 2008, 40, 194–199. [Google Scholar] [CrossRef]
- Hodson, L.; Bhatia, L.; Scorletti, E.; Smith, D.E.; Jackson, N.C.; Shojaee-Moradie, F.; Umpleby, M.; Calder, P.C.; Byrne, C.D. Docosahexaenoic acid enrichment in NAFLD is associated with improvements in hepatic metabolism and hepatic insulin sensitivity: A pilot study. Eur. J. Clin. Nutr. 2017, 71, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Zein, C.O.; Yerian, L.M.; Gogate, P.; Lopez, R.; Kirwan, J.P.; Feldstein, A.E.; McCullough, A.J. Pentoxifylline improves nonalcoholic steatohepatitis: A randomized placebo-controlled trial. Hepatology 2011, 54, 1610–1619. [Google Scholar] [CrossRef]
- Floch, M.H. Probiotics and Prebiotics. Gastroenterol. Hepatol. (N. Y.) 2014, 10, 680–681. [Google Scholar]
- Loman, B.R.; Hernández-Saavedra, D.; An, R.; Rector, R.S. Prebiotic and probiotic treatment of nonalcoholic fatty liver disease: A systematic review and meta-analysis. Nutr. Rev. 2018, 76, 822–839. [Google Scholar] [CrossRef]
- Kobyliak, N.; Abenavoli, L.; Mykhalchyshyn, G.; Kononenko, L.; Boccuto, L.; Kyriienko, D.; Dynnyk, O. A Multi-strain Probiotic Reduces the Fatty Liver Index, Cytokines and Aminotransferase levels in NAFLD Patients: Evidence from a Randomized Clinical Trial. J. Gastrointestin. Liver Dis. 2018, 27, 41–49. [Google Scholar] [PubMed]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef]
- Abenavoli, L.; Falalyeyeva, T.; Boccuto, L.; Tsyryuk, O.; Kobyliak, N. Obeticholic Acid: A New Era in the Treatment of Nonalcoholic Fatty Liver Disease. Pharmaceuticals 2018, 11, 104. [Google Scholar] [CrossRef]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and Safety of the Farnesoid X Receptor Agonist Obeticholic Acid in Patients With Type 2 Diabetes and Nonalcoholic Fatty Liver Disease. Gastroenterology 2013, 145, 574–582.e1. [Google Scholar] [CrossRef] [PubMed]
- Lindor, K.D.; Kowdley, K.V.; Heathcote, E.J.; Harrison, M.E.; Jorgensen, R.; Angulo, P.; Lymp, J.F.; Burgart, L.; Colin, P. Ursodeoxycholic acid for treatment of nonalcoholic steatohepatitis: Results of a randomized trial. Hepatology 2004, 39, 770–778. [Google Scholar] [CrossRef]
- Dufour, J.; Oneta, C.M.; Gonvers, J.; Bihl, F.; Cerny, A.; Cereda, J.; Zala, J.; Helbling, B.; Steuerwald, M.; Zimmermann, A. Randomized Placebo-Controlled Trial of Ursodeoxycholic Acid With Vitamin E in Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. 2006, 4, 1537–1543. [Google Scholar] [CrossRef]
- Athyros, V.G.; Tziomalos, K.; Gossios, T.D.; Griva, T.; Anagnostis, P.; Kargiotis, K.; Pagourelias, E.D.; Theocharidou, E.; Karagiannis, A.; Mikhailidis, D.P.; et al. Safety and efficacy of long-term statin treatment for cardiovascular events in patients with coronary heart disease and abnormal liver tests in the Greek Atorvastatin and Coronary Heart Disease Evaluation (GREACE) Study: A post-hoc analysis. Lancet 2010, 376, 1916–1922. [Google Scholar] [CrossRef]
- Tikkanen, M.J.; Fayyad, R.; Faergeman, O.; Olsson, A.G.; Wun, C.-C.; Laskey, R.; Kastelein, J.J.; Holme, I.; Pedersen, T.R.; IDEAL Investigators. IDEAL Investigators Effect of intensive lipid lowering with atorvastatin on cardiovascular outcomes in coronary heart disease patients with mild-to-moderate baseline elevations in alanine aminotransferase levels. Int. J. Cardiol. 2013, 168, 3846–3852. [Google Scholar] [CrossRef] [PubMed]
- Athyros, V.G.; Boutari, C.; Stavropoulos, K.; Anagnostis, P.; Imprialos, K.P.; Doumas, M.; Karagiannis, A. Statins: An Under-Appreciated Asset for the Prevention and the Treatment of NAFLD or NASH and the Related Cardiovascular Risk. Curr. Vasc. Pharmacol. 2018, 16, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Scicali, R.; Di Pino, A.; Ferrara, V.; Urbano, F.; Piro, S.; Rabuazzo, A.M.; Purrello, F. New treatment options for lipid-lowering therapy in subjects with type 2 diabetes. Acta Diabetol. 2018, 55, 209–218. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Petta, S.; Mannisto, V.; Mancina, R.M.; Pipitone, R.; Karja, V.; Maggioni, M.; Kakela, P.; Wiklund, O.; Mozzi, E.; et al. Statin use and non-alcoholic steatohepatitis in at risk individuals. J. Hepatol. 2015, 63, 705–712. [Google Scholar] [CrossRef]
- McGlynn, K.A.; Divine, G.W.; Sahasrabuddhe, V.V.; Engel, L.S.; VanSlooten, A.; Wells, K.; Yood, M.U.; Alford, S.H. Statin use and risk of hepatocellular carcinoma in a U.S. population. Cancer Epidemiol. 2014, 38, 523–527. [Google Scholar] [CrossRef]
- Del Ben, M.; Baratta, F.; Polimeni, L.; Pastori, D.; Loffredo, L.; Averna, M.; Violi, F.; Angelico, F. Under-prescription of statins in patients with non-alcoholic fatty liver disease. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 161–167. [Google Scholar] [CrossRef]
- Averna, M. The effect of ezetimibe on NAFLD. Atheroscler. Suppl. 2015, 17, 27–34. [Google Scholar] [CrossRef]
- Nakade, Y.; Murotani, K.; Inoue, T.; Kobayashi, Y.; Yamamoto, T.; Ishii, N.; Ohashi, T.; Ito, K.; Fukuzawa, Y.; Yoneda, M. Ezetimibe for the treatment of non-alcoholic fatty liver disease: A meta-analysis. Hepatol. Res. 2017, 47, 1417–1428. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Kimura, T.; Fujimori, N.; Nagaya, T.; Komatsu, M.; Tanaka, E. Current status, problems, and perspectives of non-alcoholic fatty liver disease research. World J. Gastroenterol. 2019, 25, 163–177. [Google Scholar] [CrossRef]
- Loomba, R.; Lawitz, E.; Mantry, P.S.; Jayakumar, S.; Caldwell, S.H.; Arnold, H.; Diehl, A.M.; Djedjos, C.S.; Han, L.; Myers, R.P.; et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology 2017, 67, 549. [Google Scholar] [CrossRef]
- Friedman, S.L.; Ratziu, V.; Harrison, S.A.; Abdelmalek, M.F.; Aithal, G.P.; Caballeria, J.; Francque, S.; Farrell, G.; Kowdley, K.V.; Craxi, A.; et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018, 67, 1754–1767. [Google Scholar] [CrossRef] [PubMed]
- Gawrieh, S.; Chalasani, N. Emerging Treatments for Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Clin. Liver Dis. 2018, 22, 189–199. [Google Scholar] [CrossRef]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-α and -δ, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159.e5. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C.J. Elafibranor for the treatment of NAFLD: One pill, two molecular targets and multiple effects in a complex phenotype. Ann. Hepatol. 2016, 15, 604–609. [Google Scholar] [PubMed]
- Safadi, R.; Konikoff, F.M.; Mahamid, M.; Zelber-Sagi, S.; Halpern, M.; Gilat, T.; Oren, R.; Group, F. The Fatty Acid-Bile Acid Conjugate Aramchol Reduces Liver Fat Content in Patients With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Ladron-De-Guevara, L.; Safadi, R.; Poordad, F.; Fuster, F.; Flores-Figueroa, J.; Harrison, S.A.; Arrese, M.; Fargion, S.; Ben-Bashat, D.; et al. One-year results of the global phase 2b randomized placebo-controlled arrest trial of aramchol, a stearoyl CoA desaturase inhibitor, in patients with NASH. In Proceedings of the AASLD Liver Meeting, San Francisco, CA, USA, 9–13 November 2018. [Google Scholar]
- Shiffman, M.; Freilich, B.; Vuppalanchi, R.; Watt, K.; Chan, J.L.; Spada, A.; Hagerty, D.T.; Schiff, E. Randomised clinical trial: Emricasan versus placebo significantly decreases ALT and caspase 3/7 activation in subjects with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2019, 49, 64–73. [Google Scholar] [CrossRef]
- Sanyal, A.; Abdelmalek, M.F.; Diehl, A.M.; Caldwell, S.; Shiffman, M.L.; Ghalib, R.; Lawitz, E.; Rockey, D.C.; Schall, R.A.; Jia, C.; et al. Efficacy and safety of simtuzumab for the treatment of nonalcoholic steatohepatitis with bridging fibrosis or cirrhosis: Results of two phase 2b, dose-ranging, randomized, placebo-controlled trials. J. Hepatol. 2017, 66, S54. [Google Scholar] [CrossRef]
- Meex, R.C.; Hoy, A.J.; Morris, A.; Brown, R.D.; Lo, J.C.Y.; Burke, M.; Goode, R.J.A.; Kingwell, B.A.; Kraakman, M.J.; Febbraio, M.A.; et al. Fetuin B Is a Secreted Hepatocyte Factor Linking Steatosis to Impaired Glucose Metabolism. Cell Metab. 2015, 22, 1078–1089. [Google Scholar] [CrossRef]


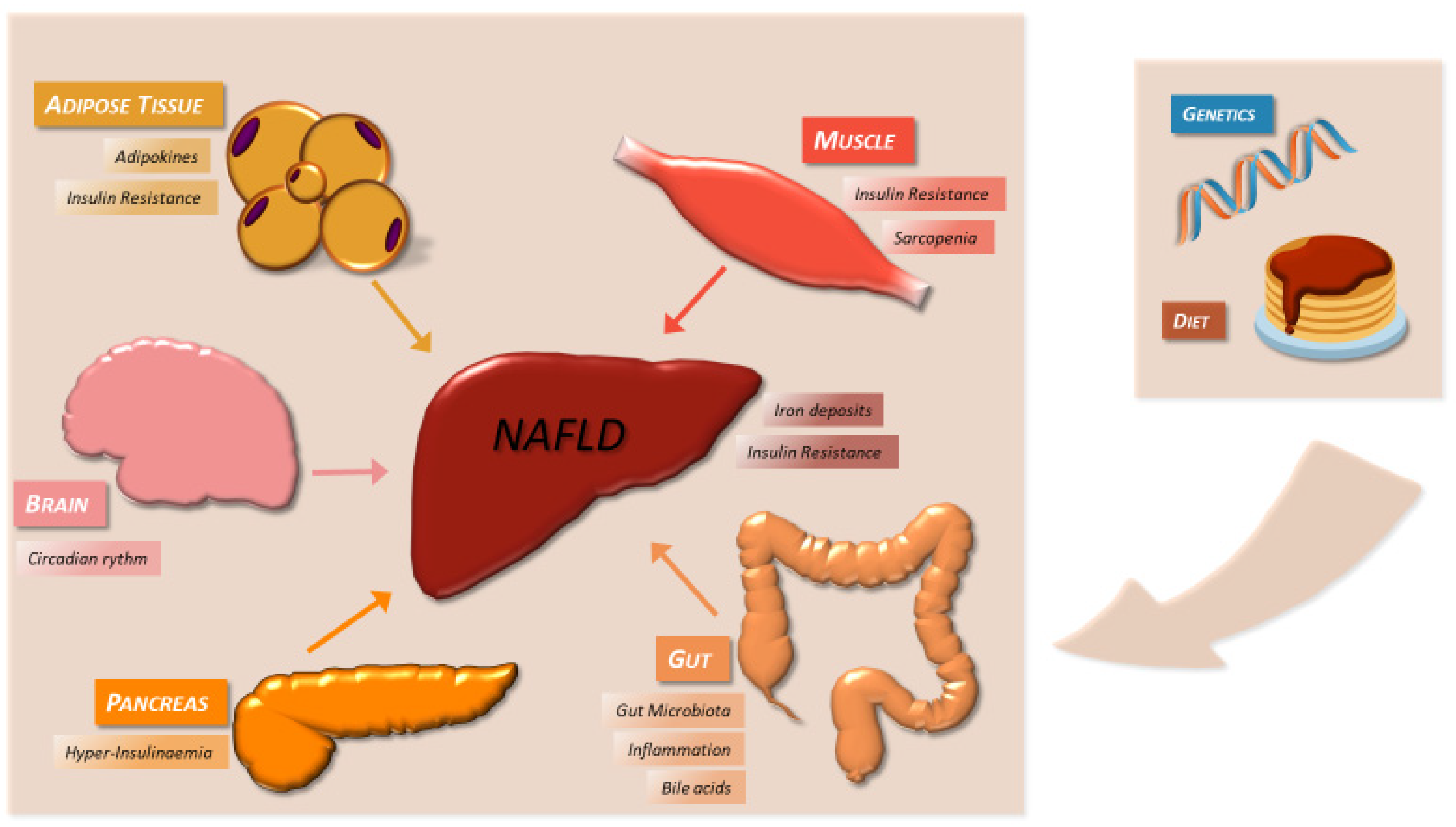{kind=link}
| Conditions Associated with Liver Steatosis | Mechanism of Action | References |
|---|---|---|
| Alcohol (>20 g/day (women) or >30 g/day (men)) | Redox state shift: fatty acid oxidation inhibition, induction of lipogenesis Altered VLDL secretion in the liver | [22] |
| HCV | Altered VLDL secretion in the liver Insulin resistance Mitochondrial dysfunction and oxidative stress | [23] |
| Medications (e.g., methotrexate, corticosteroids, valproate) | Fatty acid oxidation inhibition, induction of lipogenesis Mitochondrial dysfunction Impaired hepatic lipid secretion Insulin resistance | [24] |
| Lipid metabolism disorders: a/hypo-betalipoproteinaemia, Wolman’s disease | Impaired hepatic lipid secretion Impaired hydrolysis of cholesteryl esters and triglycerides | [25,26] |
| Metal storage disorders: Wilson’s disease | Copper-induced mitochondrial dysfunction | [27] |
| Autoimmune hepatitis | Drug-mediated effects | [28] |
| Coeliac disease | Weight gain on gluten-free diet Impaired hepatic lipid mobilization Intestinal malabsorption | [29] |
| Endocrine disorders: hypothyroidism, hypopituitarism, polycystic ovary syndrome | Reduced hepatic lipid utilization Insulin resistance Impaired insulin secretion | [30,31,32,33] |
| Starvation, parenteral nutrition | Impaired hepatic lipid secretion Reduced mitochondrial beta-oxidation | [34] |
| Lipodystrophy | Insulin resistance and ectopic fat accumulation | [35] |
| NAFLD | NASH | |
|---|---|---|
| Obesity | 51% | 82% |
| Diabetes mellitus | 23% | 47% |
| Metabolic syndrome | 41% | 71% |
| Hyperlipidemia/dyslipidemia | 69% | 72% |
| Hypertension | 39.34% | 67.97% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchisello, S.; Di Pino, A.; Scicali, R.; Urbano, F.; Piro, S.; Purrello, F.; Rabuazzo, A.M. Pathophysiological, Molecular and Therapeutic Issues of Nonalcoholic Fatty Liver Disease: An Overview. Int. J. Mol. Sci. 2019, 20, 1948. https://doi.org/10.3390/ijms20081948
Marchisello S, Di Pino A, Scicali R, Urbano F, Piro S, Purrello F, Rabuazzo AM. Pathophysiological, Molecular and Therapeutic Issues of Nonalcoholic Fatty Liver Disease: An Overview. International Journal of Molecular Sciences. 2019; 20(8):1948. https://doi.org/10.3390/ijms20081948
Chicago/Turabian StyleMarchisello, Simona, Antonino Di Pino, Roberto Scicali, Francesca Urbano, Salvatore Piro, Francesco Purrello, and Agata Maria Rabuazzo. 2019. "Pathophysiological, Molecular and Therapeutic Issues of Nonalcoholic Fatty Liver Disease: An Overview" International Journal of Molecular Sciences 20, no. 8: 1948. https://doi.org/10.3390/ijms20081948
APA StyleMarchisello, S., Di Pino, A., Scicali, R., Urbano, F., Piro, S., Purrello, F., & Rabuazzo, A. M. (2019). Pathophysiological, Molecular and Therapeutic Issues of Nonalcoholic Fatty Liver Disease: An Overview. International Journal of Molecular Sciences, 20(8), 1948. https://doi.org/10.3390/ijms20081948