Inhibition of IL-13 and IL-13Rα2 Expression by IL-32θ in Human Monocytic Cells Requires PKCδ and STAT3 Association

 , and
, and
Abstract
1. Introduction
2. Results
2.1. Microarray Identification of IL-13Rα2 and IL-13 as Down-Regulated Genes by IL-32θ
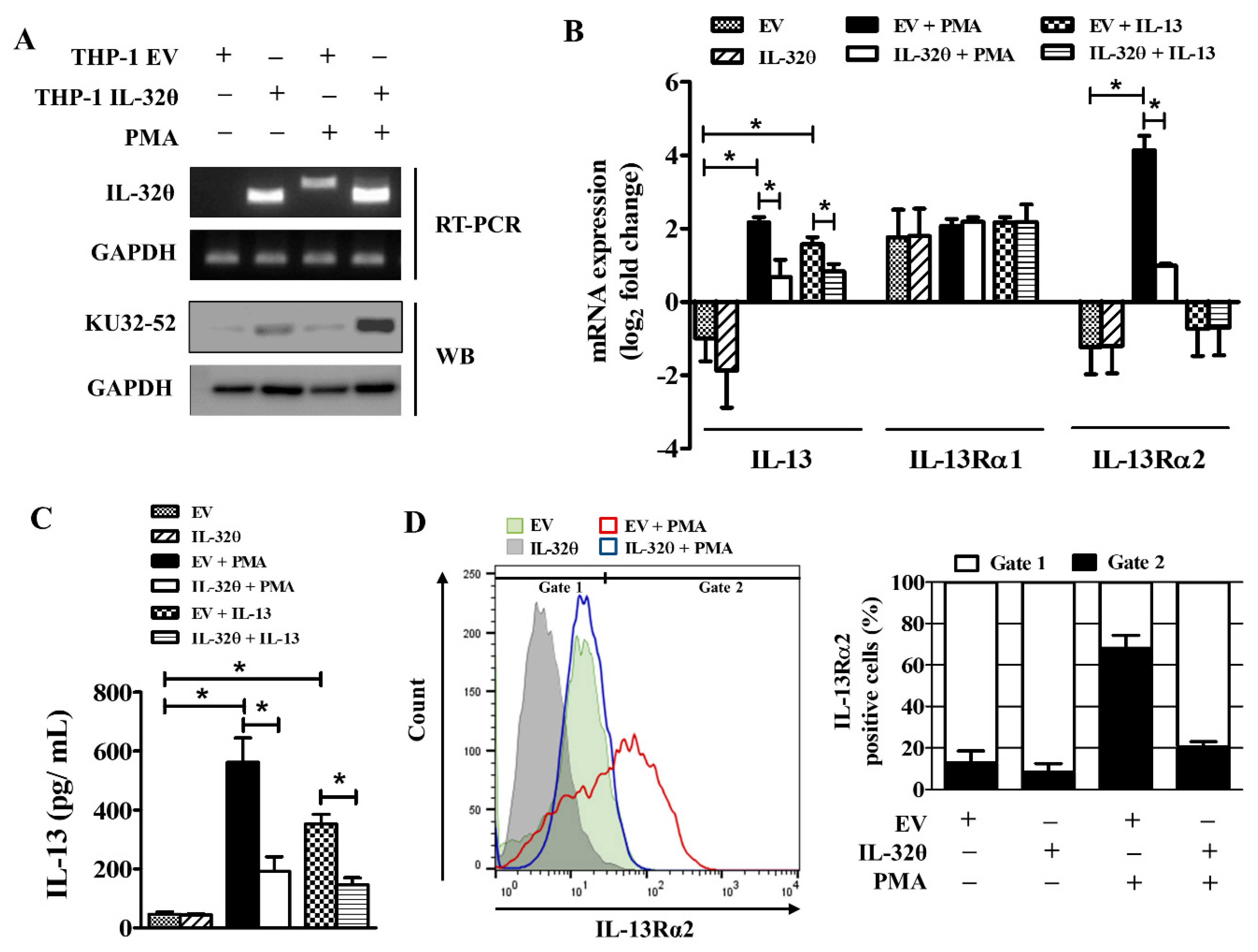
2.2. IL-32θ Negatively Regulates IL-13Rα2 and IL-13 in Both mRNA and Protein Expression
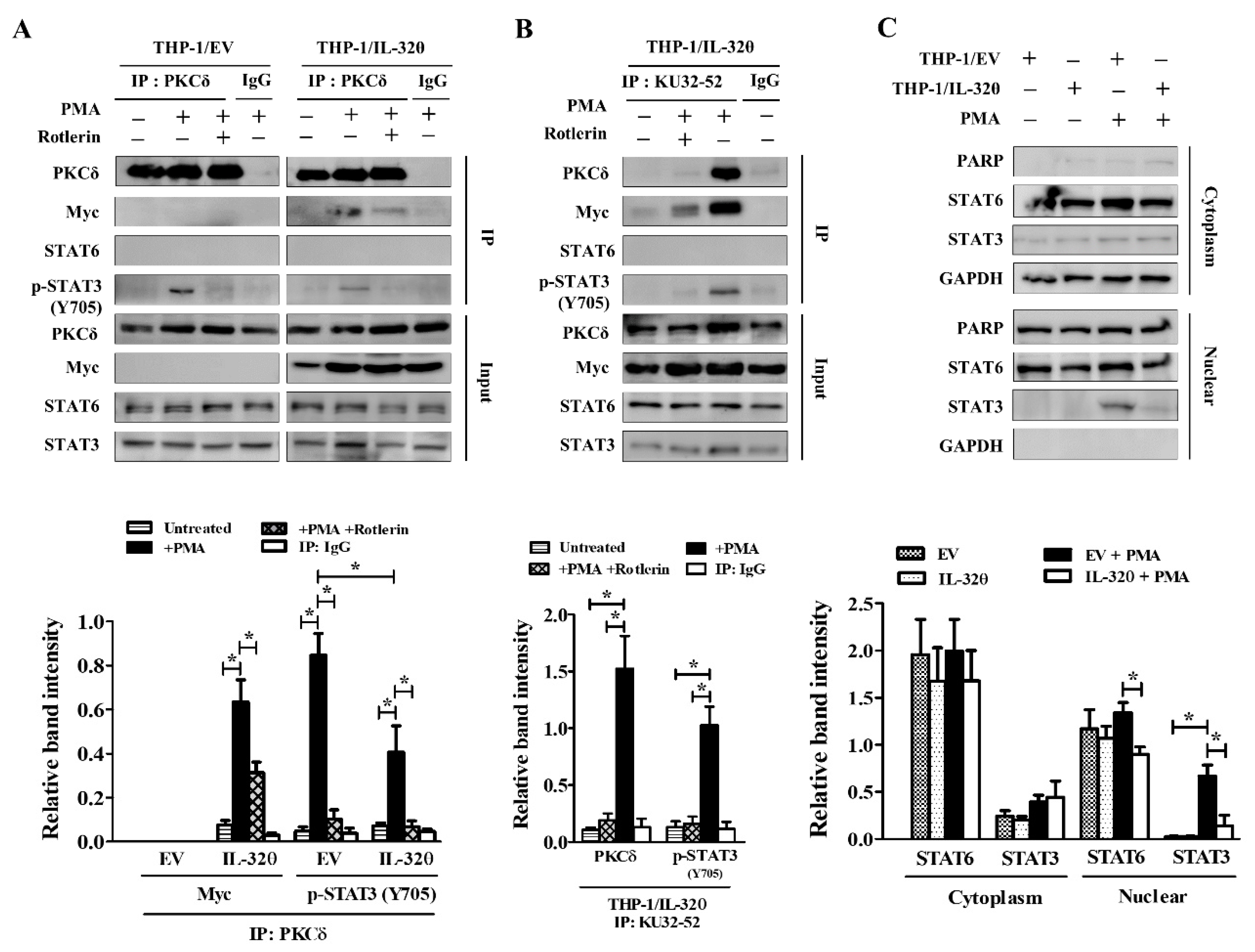
2.3. IL-32θ Directly Interacts with PKCδ, STAT3, but not STAT6
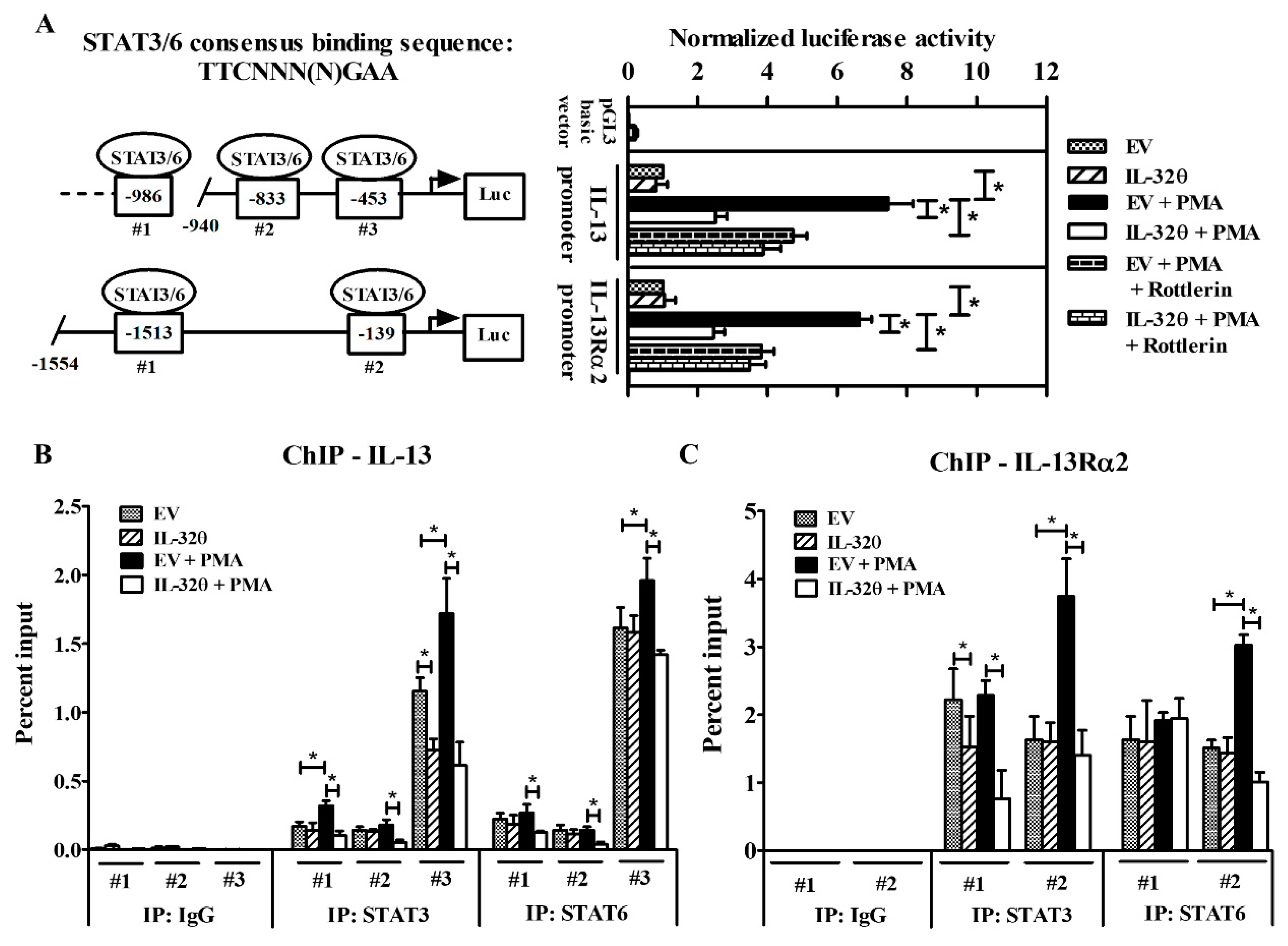
2.4. STAT3 Binding to IL-13Rα2 and IL-13 Promoters Was Suppressed by IL-32θ
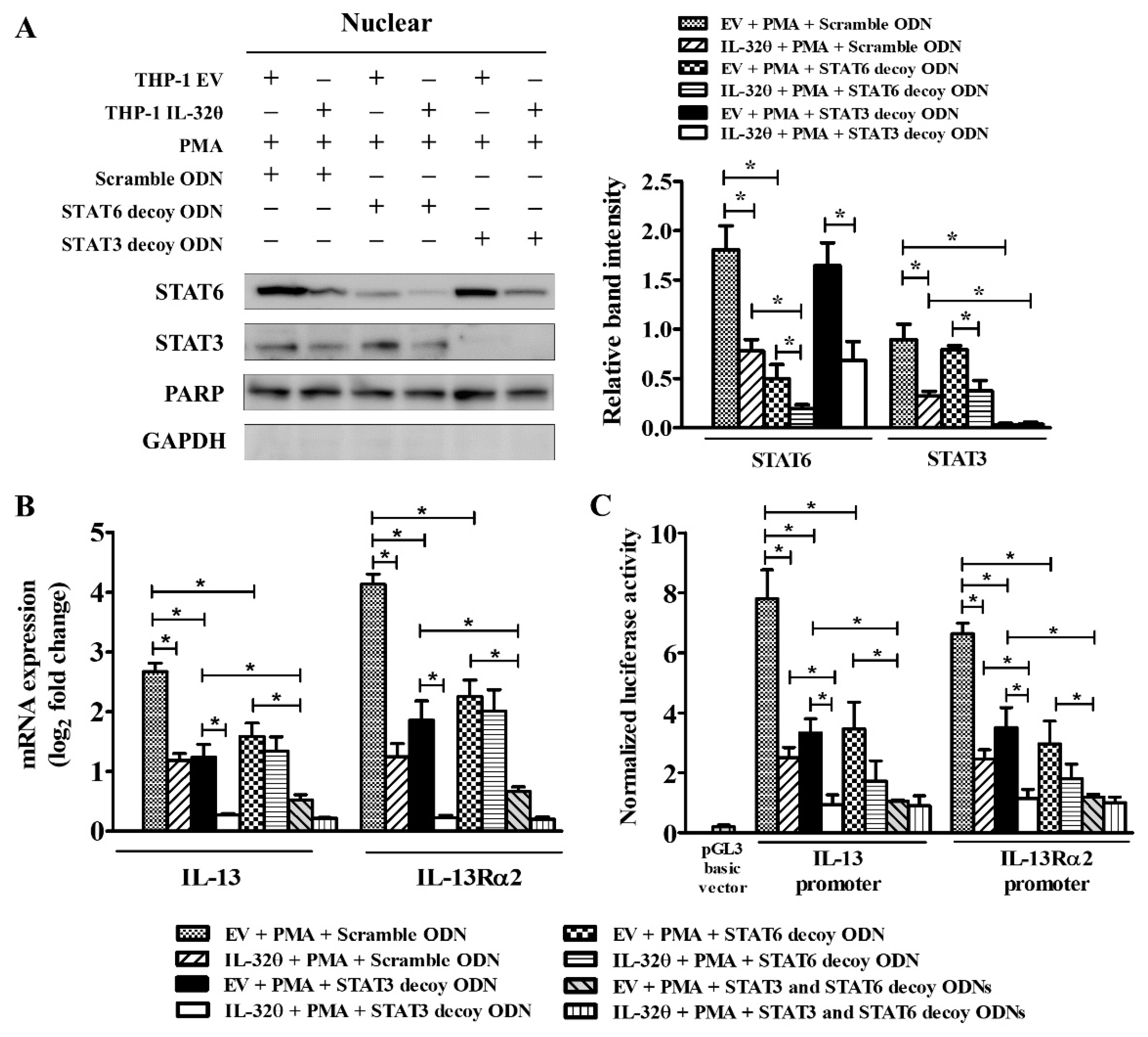
2.5. Blocking of STAT3 Binding Showed Additive Effect with IL-32θ
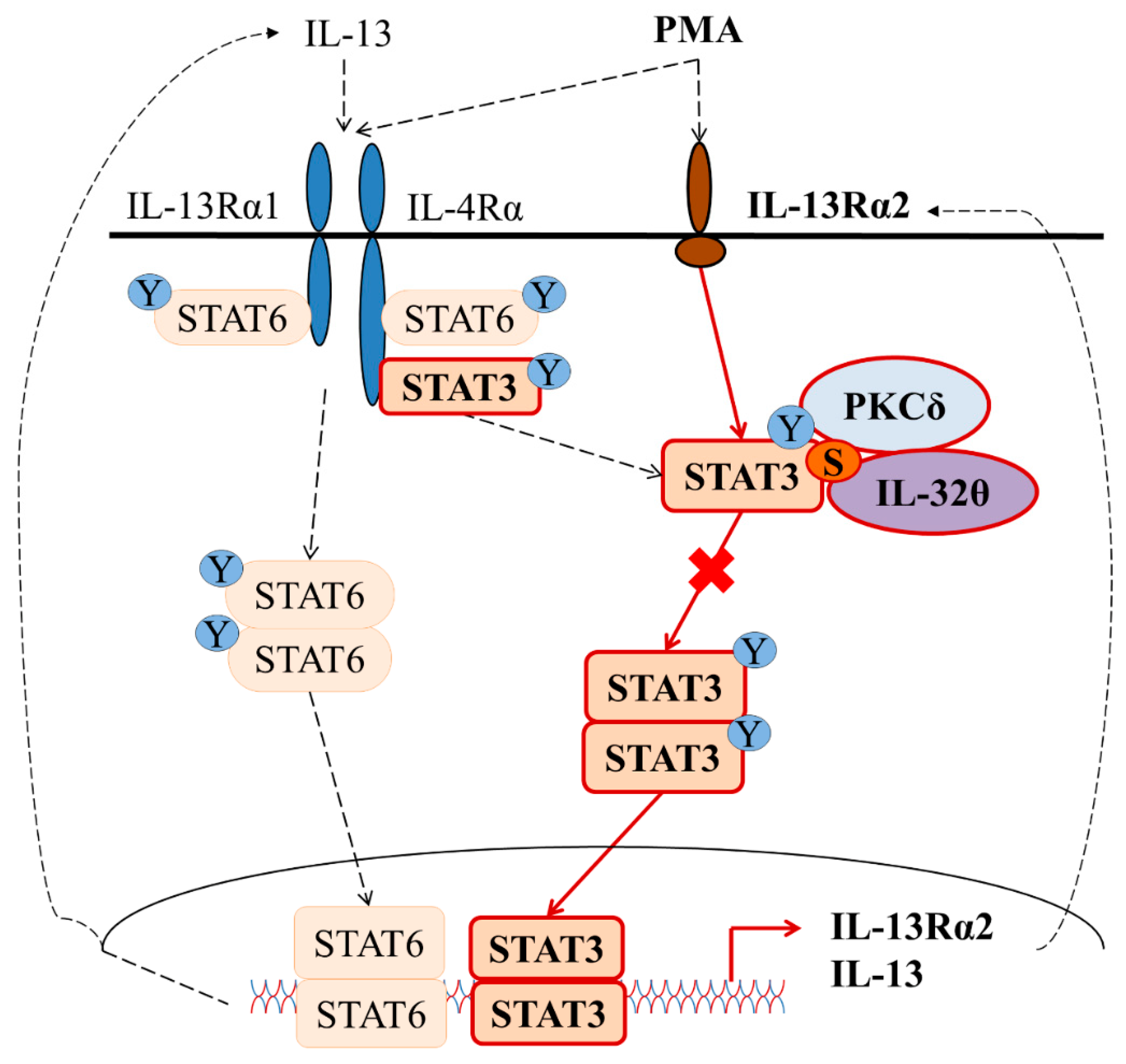
3. Discussion
4. Materials and Methods.
4.1. Cell Culture and Generation of IL-32θ-Stably Expressing Cell Line
4.2. Microarray
4.3. Reverse Transcription Quantitative PCR (RT-qPCR)
4.4. Nuclear and Cytoplasmic Fractionation
4.5. Immunoprecipitation and Western Blotting
4.6. Enzyme-Linked Immunosorbent Assay (ELISA)
4.7. Decoy Oligodeoxynucleotides (ODNs) and Transfection
4.8. Plasmid Constructs
4.9. Luciferase Assay
4.10. Chromatin Immunoprecipitation (ChIP) Assay
4.11. Flow Cytometry Analysis
4.12. Statistics
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CCL5 | Chemokine (C-C motif) ligand 5 |
| IL-4Rα | Interleukin-4 receptor subunit alpha |
| IL-13Rα1 | Interleukin-13 receptor subunit alpha 1 |
| JAK | Janus kinase |
| MAPK | Mitogen-activated protein kinase |
| NF-κB | Nuclear factor kappa light chain enhancer of activated B cells |
| PCR | Polymerase chain reaction |
| STAT | Signal transducer and activator of transcription |
| TNF-α | Tumor necrosis factor alpha |
| TYK | Tyrosine kinase |
References
- Hershey, G.K. IL-13 receptors and signaling pathways: An evolving web. J. Allergy Clin. Immunol. 2003, 111, 677–690. [Google Scholar] [CrossRef]
- Gour, N.; Wills-Karp, M. IL-4 and IL-13 signaling in allergic airway disease. Cytokine 2015, 75, 68–78. [Google Scholar] [CrossRef]
- David, M.D.; Bertoglio, J.; Pierre, J. Functional characterization of IL-13 receptor alpha2 gene promoter: A critical role of the transcription factor STAT6 for regulated expression. Oncogene 2003, 22, 3386–3394. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Shukla, M.; Yakubenko, V.P.; Mulya, A.; Kundu, S.; Cathcart, M.K. IL-4 and IL-13 employ discrete signaling pathways for target gene expression in alternatively activated monocytes/macrophages. Free Radic. Biol. Med. 2013, 54, 1–16. [Google Scholar] [CrossRef]
- Kawakami, K.; Taguchi, J.; Murata, T.; Puri, R.K. The interleukin-13 receptor alpha2 chain: An essential component for binding and internalization but not for interleukin-13-induced signal transduction through the STAT6 pathway. Blood 2001, 97, 2673–2679. [Google Scholar] [CrossRef]
- Mandal, D.; Levine, A.D. Elevated IL-13Ralpha2 in intestinal epithelial cells from ulcerative colitis or colorectal cancer initiates MAPK pathway. Inflamm. Bowel Dis. 2010, 16, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Fichtner-Feigl, S.; Strober, W.; Kawakami, K.; Puri, R.K.; Kitani, A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat. Med. 2006, 12, 99–106. [Google Scholar] [CrossRef] [PubMed]
- He, C.H.; Lee, C.G.; Dela Cruz, C.S.; Lee, C.M.; Zhou, Y.; Ahangari, F.; Ma, B.; Herzog, E.L.; Rosenberg, S.A.; Li, Y.; et al. Chitinase 3-like 1 regulates cellular and tissue responses via IL-13 receptor alpha2. Cell Rep. 2013, 4, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, S.O.; Sharma, P.; Harbor, P.C.; Aman, M.J.; Vogelbaum, M.A.; Haque, S.J. IL-13R(alpha)2, a decoy receptor for IL-13 acts as an inhibitor of IL-4-dependent signal transduction in glioblastoma cells. Cancer Res. 2002, 62, 1103–1109. [Google Scholar] [PubMed]
- Arima, K.; Sato, K.; Tanaka, G.; Kanaji, S.; Terada, T.; Honjo, E.; Kuroki, R.; Matsuo, Y.; Izuhara, K. Characterization of the interaction between interleukin-13 and interleukin-13 receptors. J. Biol. Chem. 2005, 280, 24915–24922. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, S.O.; Vogelbaum, M.A.; Haque, S.J. Aberrant Stat3 signaling by interleukin-4 in malignant glioma cells: Involvement of IL-13Ralpha2. Cancer Res. 2005, 65, 2956–2963. [Google Scholar] [CrossRef] [PubMed]
- Orchansky, P.L.; Kwan, R.; Lee, F.; Schrader, J.W. Characterization of the cytoplasmic domain of interleukin-13 receptor-alpha. J. Biol. Chem. 1999, 274, 20818–20825. [Google Scholar] [CrossRef]
- Umeshita-Suyama, R.; Sugimoto, R.; Akaiwa, M.; Arima, K.; Yu, B.; Wada, M.; Kuwano, M.; Nakajima, K.; Hamasaki, N.; Izuhara, K. Characterization of IL-4 and IL-13 signals dependent on the human IL-13 receptor alpha chain 1: Redundancy of requirement of tyrosine residue for STAT3 activation. Int. Immunol. 2000, 12, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Bhattacharjee, A.; Roy, B.; Feldman, G.M.; Cathcart, M.K. Role of protein kinase C isoforms in the regulation of interleukin-13-induced 15-lipoxygenase gene expression in human monocytes. J. Biol. Chem. 2004, 279, 15954–15960. [Google Scholar] [CrossRef] [PubMed]
- Akira, S. Roles of STAT3 defined by tissue-specific gene targeting. Oncogene 2000, 19, 2607–2611. [Google Scholar] [CrossRef]
- Zhong, Z.; Wen, Z.; Darnell, J.E., Jr. Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef]
- Shen, Y.; Schlessinger, K.; Zhu, X.; Meffre, E.; Quimby, F.; Levy, D.E.; Darnell, J.E., Jr. Essential role of STAT3 in postnatal survival and growth revealed by mice lacking STAT3 serine 727 phosphorylation. Mol. Cell. Biol. 2004, 24, 407–419. [Google Scholar] [CrossRef]
- Qin, H.R.; Kim, H.J.; Kim, J.Y.; Hurt, E.M.; Klarmann, G.J.; Kawasaki, B.T.; Duhagon Serrat, M.A.; Farrar, W.L. Activation of signal transducer and activator of transcription 3 through a phosphomimetic serine 727 promotes prostate tumorigenesis independent of tyrosine 705 phosphorylation. Cancer Res. 2008, 68, 7736–7741. [Google Scholar] [CrossRef]
- Decker, T.; Kovarik, P. Serine phosphorylation of STATs. Oncogene 2000, 19, 2628–2637. [Google Scholar] [CrossRef]
- Hazan-Halevy, I.; Harris, D.; Liu, Z.; Liu, J.; Li, P.; Chen, X.; Shanker, S.; Ferrajoli, A.; Keating, M.J.; Estrov, Z. STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood 2010, 115, 2852–2863. [Google Scholar] [CrossRef]
- Mellor, H.; Parker, P.J. The extended protein kinase C superfamily. Biochem. J. 1998, 332, 281–292. [Google Scholar] [CrossRef]
- Novotny-Diermayr, V.; Zhang, T.; Gu, L.; Cao, X. Protein kinase C delta associates with the interleukin-6 receptor subunit glycoprotein (gp) 130 via Stat3 and enhances Stat3-gp130 interaction. J. Biol. Chem. 2002, 277, 49134–49142. [Google Scholar] [CrossRef]
- Gartsbein, M.; Alt, A.; Hashimoto, K.; Nakajima, K.; Kuroki, T.; Tennenbaum, T. The role of protein kinase C delta activation and STAT3 Ser727 phosphorylation in insulin-induced keratinocyte proliferation. J. Cell Sci. 2006, 119, 470–481. [Google Scholar] [CrossRef][Green Version]
- Ren, J.; Wang, Q.; Morgan, S.; Si, Y.; Ravichander, A.; Dou, C.; Kent, K.C.; Liu, B. Protein kinase C-delta (PKCdelta) regulates proinflammatory chemokine expression through cytosolic interaction with the NF-kappaB subunit p65 in vascular smooth muscle cells. J. Biol. Chem. 2014, 289, 9013–9026. [Google Scholar] [CrossRef] [PubMed]
- Kikkawa, U.; Matsuzaki, H.; Yamamoto, T. Protein kinase C delta (PKC delta): Activation mechanisms and functions. J. Biochem. 2002, 132, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Zhang, T.; Kee, W.H.; Li, W.; Cao, X. Protein kinase C delta associates with and phosphorylates Stat3 in an interleukin-6-dependent manner. J. Biol. Chem. 1999, 274, 24392–24400. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, A.; Xu, B.; Frank, D.A.; Feldman, G.M.; Cathcart, M.K. Monocyte 15-lipoxygenase expression is regulated by a novel cytosolic signaling complex with protein kinase C delta and tyrosine-phosphorylated Stat3. J. Immunol. 2006, 177, 3771–3781. [Google Scholar] [CrossRef]
- Kang, J.W.; Park, Y.S.; Lee, D.H.; Kim, M.S.; Bak, Y.; Ham, S.Y.; Park, S.H.; Kim, H.; Ahn, J.H.; Hong, J.T.; et al. Interaction network mapping among IL-32 isoforms. Biochimie 2014, 101, 248–251. [Google Scholar] [CrossRef]
- Bak, Y.; Kang, J.W.; Kim, M.S.; Park, Y.S.; Kwon, T.; Kim, S.; Hong, J.; Yoon, D.Y. IL-32theta downregulates CCL5 expression through its interaction with PKCdelta and STAT3. Cell. Signal. 2014, 26, 3007–3015. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Kang, J.W.; Lee, D.H.; Bak, Y.; Park, Y.S.; Song, Y.S.; Ham, S.Y.; Oh, D.K.; Hong, J.; Yoon, D.Y. IL-32theta negatively regulates IL-1beta production through its interaction with PKCdelta and the inhibition of PU.1 phosphorylation. FEBS Lett. 2014, 588, 2822–2829. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Kang, J.W.; Jeon, J.S.; Kim, J.K.; Kim, J.W.; Hong, J.; Yoon, D.Y. IL-32theta gene expression in acute myeloid leukemia suppresses TNF-alpha production. Oncotarget 2015, 6, 40747–40761. [Google Scholar] [CrossRef][Green Version]
- Daines, M.O.; Hershey, G.K. A novel mechanism by which interferon-gamma can regulate interleukin (IL)-13 responses. Evidence for intracellular stores of IL-13 receptor alpha -2 and their rapid mobilization by interferon-gamma. J. Biol. Chem. 2002, 277, 10387–10393. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Bhattacharjee, A.; Xu, B.; Ford, D.; Maizel, A.L.; Cathcart, M.K. IL-13 signal transduction in human monocytes: Phosphorylation of receptor components, association with Jaks, and phosphorylation/activation of Stats. J. Leukoc. Biol. 2002, 72, 580–589. [Google Scholar] [PubMed]
- Kontny, E.; Kurowska, M.; Szczepanska, K.; Maslinski, W. Rottlerin, a PKC isozyme-selective inhibitor, affects signaling events and cytokine production in human monocytes. J. Leukoc. Biol. 2000, 67, 249–258. [Google Scholar] [CrossRef]
- Kraus, J.; Borner, C.; Hollt, V. Distinct palindromic extensions of the 5′-TTC...GAA-3′ motif allow STAT6 binding in vivo. FASEB J. 2003, 17, 304–306. [Google Scholar] [CrossRef]
- Ehret, G.B.; Reichenbach, P.; Schindler, U.; Horvath, C.M.; Fritz, S.; Nabholz, M.; Bucher, P. DNA binding specificity of different STAT proteins. Comparison of in vitro specificity with natural target sites. J. Biol. Chem. 2001, 276, 6675–6688. [Google Scholar] [CrossRef]
- Holden, N.S.; Squires, P.E.; Kaur, M.; Bland, R.; Jones, C.E.; Newton, R. Phorbol ester-stimulated NF-kappaB-dependent transcription: Roles for isoforms of novel protein kinase C. Cell. Signal. 2008, 20, 1338–1348. [Google Scholar] [CrossRef]
- Vasamsetti, S.B.; Karnewar, S.; Kanugula, A.K.; Thatipalli, A.R.; Kumar, J.M.; Kotamraju, S. Metformin inhibits monocyte-to-macrophage differentiation via AMPK-mediated inhibition of STAT3 activation: Potential role in atherosclerosis. Diabetes 2015, 64, 2028–2041. [Google Scholar] [CrossRef]
- Fan, H.; Rothstein, T.L. Lymphokine dependence of STAT3 activation produced by surface immunoglobulin cross-linking and by phorbol ester plus calcium ionophore treatment in B cells. Eur. J. Immunol. 2001, 31, 665–671. [Google Scholar] [CrossRef]
- Keen, J.C.; Cianferoni, A.; Florio, G.; Guo, J.; Chen, R.; Roman, J.; Wills-Karp, M.; Casolaro, V.; Georas, S.N. Characterization of a novel PMA-inducible pathway of interleukin-13 gene expression in T cells. Immunology 2006, 117, 29–37. [Google Scholar] [CrossRef]
- Nolan, T.; Hands, R.E.; Bustin, S.A. Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 2006, 1, 1559–1582. [Google Scholar] [CrossRef]
- Kim, K.H.; Shim, J.H.; Seo, E.H.; Cho, M.C.; Kang, J.W.; Kim, S.H.; Yu, D.Y.; Song, E.Y.; Lee, H.G.; Sohn, J.H.; et al. Interleukin-32 monoclonal antibodies for immunohistochemistry, Western blotting, and ELISA. J. Immunol. Methods 2008, 333, 38–50. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Sen, M.; Thomas, S.M.; Kim, S.; Yeh, J.I.; Ferris, R.L.; Johnson, J.T.; Duvvuri, U.; Lee, J.; Sahu, N.; Joyce, S.; et al. First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: Implications for cancer therapy. Cancer Discov. 2012, 2, 694–705. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Accession | Gene Description | Gene Symbol | Ratio Value (log2) | GO Biological Process Term |
|---|---|---|---|---|
| Down-regulation | ||||
| NM_005988 | Small proline-rich protein 2A | SPRR2A | −3.866 | Epidermis development // keratinocyte differentiation |
| NM_002985 | Chemokine (C-C motif) ligand 5 | CCL5 | −2.876 | Chemotaxis // inflammatory response // immune response |
| NM_001712 | Carcinoembryonic antigen-related cell adhesion molecule 1 | CEACAM1 | −2.612 | Angiogenesis // integrin-mediated signaling pathway // cell migration |
| NM_000640 | Interleukin 13 receptor, alpha 2 | IL13RA2 | −2.399 | --- |
| NM_002160 | Tenascin C | TNC | −2.314 | Cell adhesion // signal transduction // response to wounding |
| NM_005546 | Interleukin 2-inducible T-cell kinase | ITK | −3.250 | Cellular defense response // signal transduction // intracellular signaling pathway |
| NR_029635 | MicroRNA 221 | MIR221 | −2.242 | --- |
| NM_001558 | Interleukin 10 receptor, alpha | IL10RA | −1.526 | Blood coagulation // response to lipopolysaccharide |
| NM_000201 | Intercellular adhesion molecule 1 | ICAM1 | −1.231 | Regulation of cell adhesion |
| NM_000878 | Interleukin 2 receptor, beta | IL2RB | −1.158 | Cytokine-mediated signaling pathway // positive regulation of survival gene product expression |
| NM_006850 | Interleukin 24 | IL24 | −1.106 | Apoptosis |
| NM_001562 | Interleukin 18 (Interferon-gamma-inducing factor) | IL18 | −1.045 | Angiogenesis // immune response // positive regulation of interferon-gamma production |
| NM_000576 | Interleukin 1, beta | IL1B | −1.024 | Positive regulation of protein phosphorylation // inflammatory response // signal transduction |
| NM_002188 | Interleukin 13 | IL13 | −0.550 | Inflammatory response // cell-cell signaling // positive regulation of protein secretion |
| Primer Names | Primer Sequences (5′ to 3′) | Products Size (bp) | |
|---|---|---|---|
| GAPDH mRNA | Forward | TGGTATCGTGGAAGGACTCATGAC | 189 |
| Reverse | ATGCCAGTGAGCTTCCCGTTCAGC | ||
| IL-13 mRNA | Forward | CATGGCGCTTTTGTTGACCA | 181 |
| Reverse | AGCTGTCAGGTTGATGCTCC | ||
| IL-13Rα1 mRNA | Forward | CATGAAGAGGATGCTGTGAA | 172 |
| Reverse | TAGGGATCACAACCACCACA | ||
| IL-13Rα2 mRNA | Forward | ATATTTACTCTGTTCTTGGA | 132 |
| Reverse | ATATTTTGTCCATCAGCCTT | ||
| IL-13 (ChIP: STAT3/6) #1 | Forward | AAACGAGGGAAGAGCAGGAA | 143 |
| Reverse | CCCTTTGCTCACCAGTCTCT | ||
| IL-13 (ChIP: STAT3/6) #2 | Forward | TGGCACTAGGAACACTTACA | 136 |
| Reverse | AGAGTGGCTGGAAGTAGTGT | ||
| IL-13 (ChIP: STAT3/6) #3 | Forward | TTT ACT CTT CTA TTG CCT GT | 104 |
| Reverse | TAT GGG AAT TTG GGG AGT TT | ||
| IL-13Rα2 (ChIP: STAT3/6) #1 | Forward | TGGGTTAGTGGCATTGAAAG | 109 |
| Reverse | AAAAAAAACCTCCAGGAAGT | ||
| IL-13Rα2 (ChIP: STAT3/6) #2 | Forward | GAAAACACTGGCATACTAAG | 132 |
| Reverse | TGCATTCAAAATAGGTCAGC | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pham, T.-H.; Bak, Y.; Oh, J.-W.; Hong, J.; Lee, S.; Hong, J.T.; Yoon, D.-Y. Inhibition of IL-13 and IL-13Rα2 Expression by IL-32θ in Human Monocytic Cells Requires PKCδ and STAT3 Association. Int. J. Mol. Sci. 2019, 20, 1949. https://doi.org/10.3390/ijms20081949
Pham T-H, Bak Y, Oh J-W, Hong J, Lee S, Hong JT, Yoon D-Y. Inhibition of IL-13 and IL-13Rα2 Expression by IL-32θ in Human Monocytic Cells Requires PKCδ and STAT3 Association. International Journal of Molecular Sciences. 2019; 20(8):1949. https://doi.org/10.3390/ijms20081949
Chicago/Turabian StylePham, Thu-Huyen, Yesol Bak, Jae-Wook Oh, Jingi Hong, Seungyeoun Lee, Jin Tae Hong, and Do-Young Yoon. 2019. "Inhibition of IL-13 and IL-13Rα2 Expression by IL-32θ in Human Monocytic Cells Requires PKCδ and STAT3 Association" International Journal of Molecular Sciences 20, no. 8: 1949. https://doi.org/10.3390/ijms20081949
APA StylePham, T.-H., Bak, Y., Oh, J.-W., Hong, J., Lee, S., Hong, J. T., & Yoon, D.-Y. (2019). Inhibition of IL-13 and IL-13Rα2 Expression by IL-32θ in Human Monocytic Cells Requires PKCδ and STAT3 Association. International Journal of Molecular Sciences, 20(8), 1949. https://doi.org/10.3390/ijms20081949