Dating Whole Genome Duplication in Ceratopteris thalictroides and Potential Adaptive Values of Retained Gene Duplicates

 ,
,
Abstract
1. Introduction
2. Results
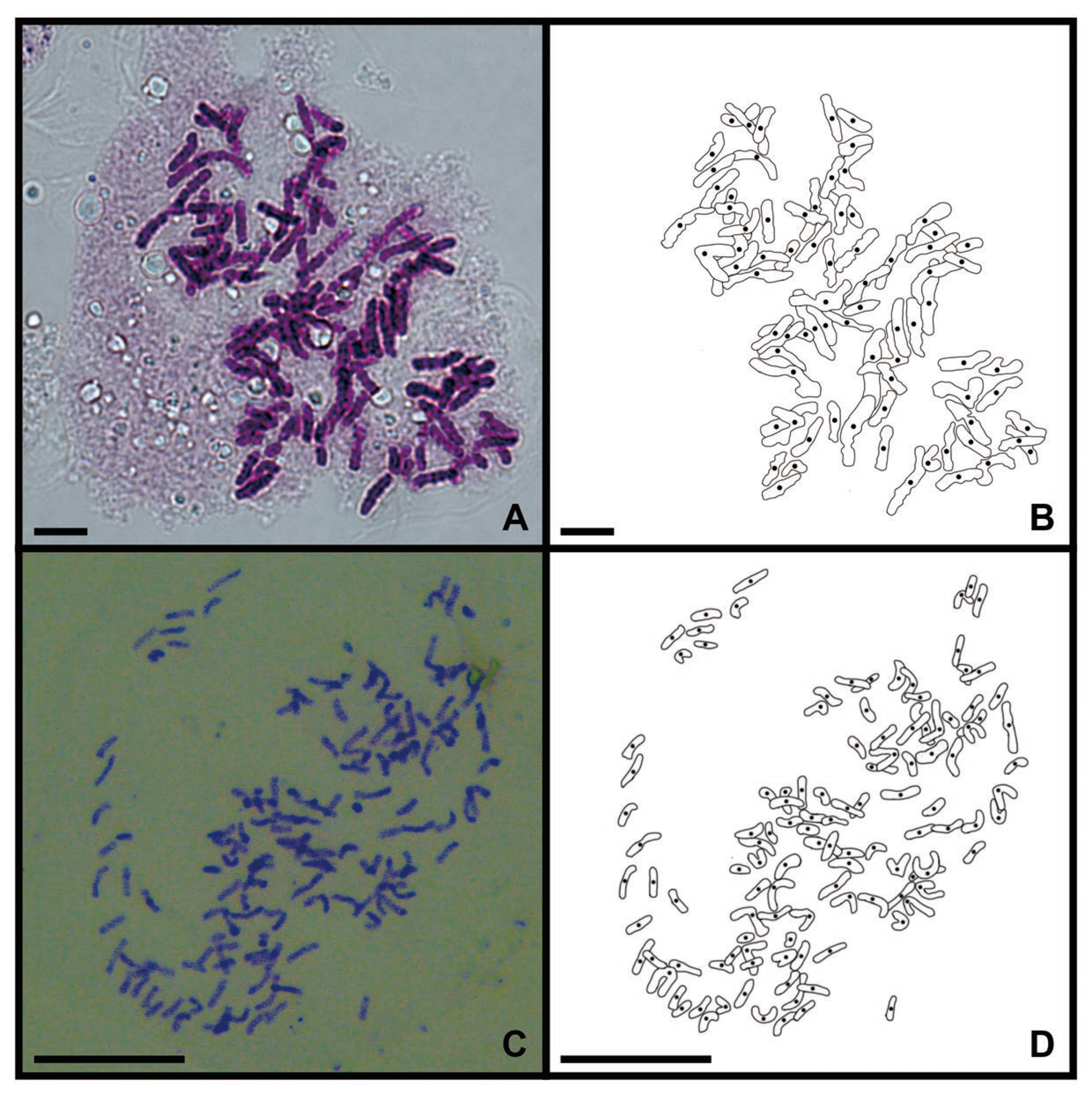
2.1. Chromosome Counting
2.2. Benchmarking Universal Single Copy Orthologs (BUSCO) Analysis
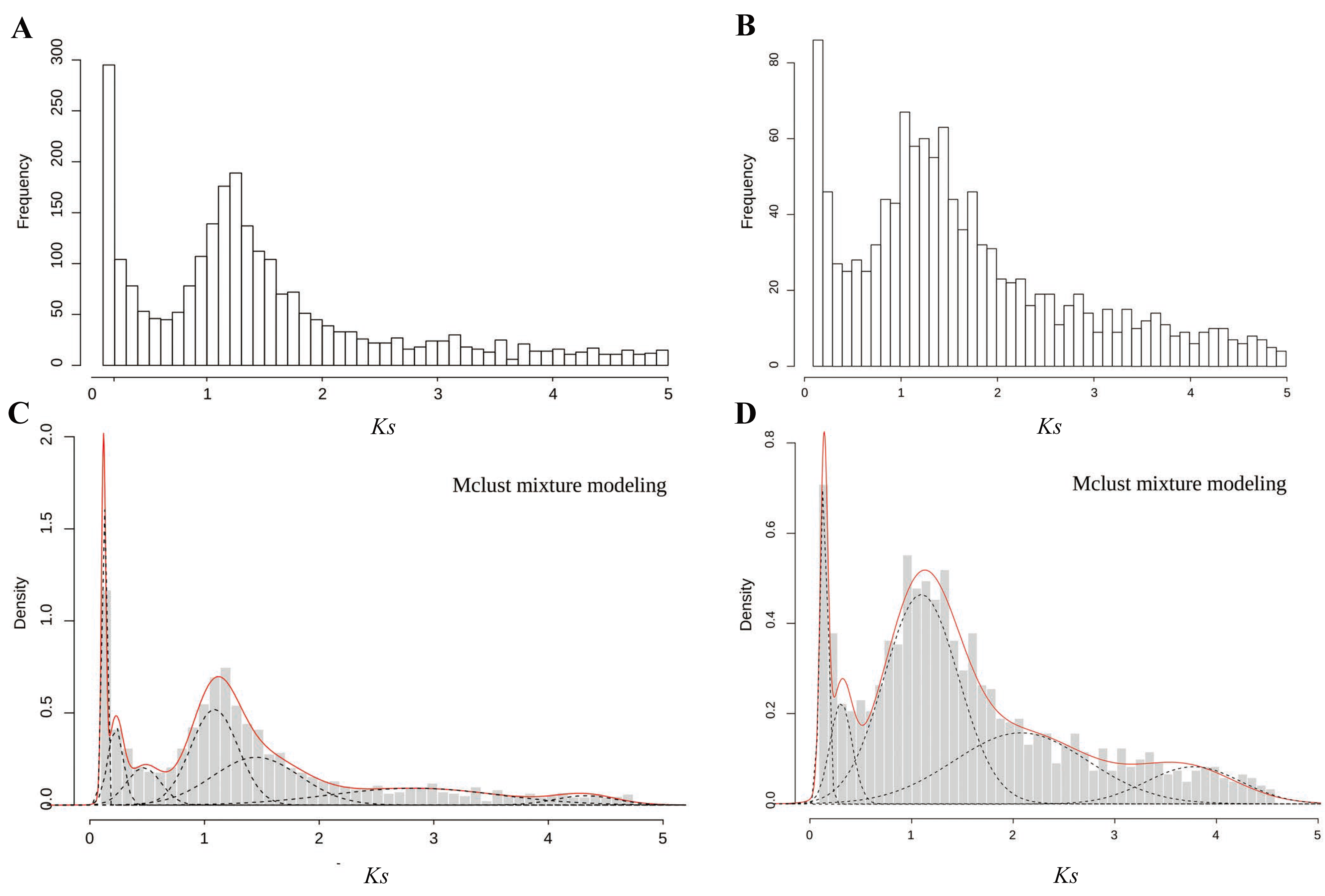
2.3. Identification of Pairs of Paralogous and WGD Events Estimated on the Basis of Ks Age Distributions
2.4. Functional Classifications of Retained Duplicates after WGD Events Revealed That Retained Genes Are Biased Rather Than Random
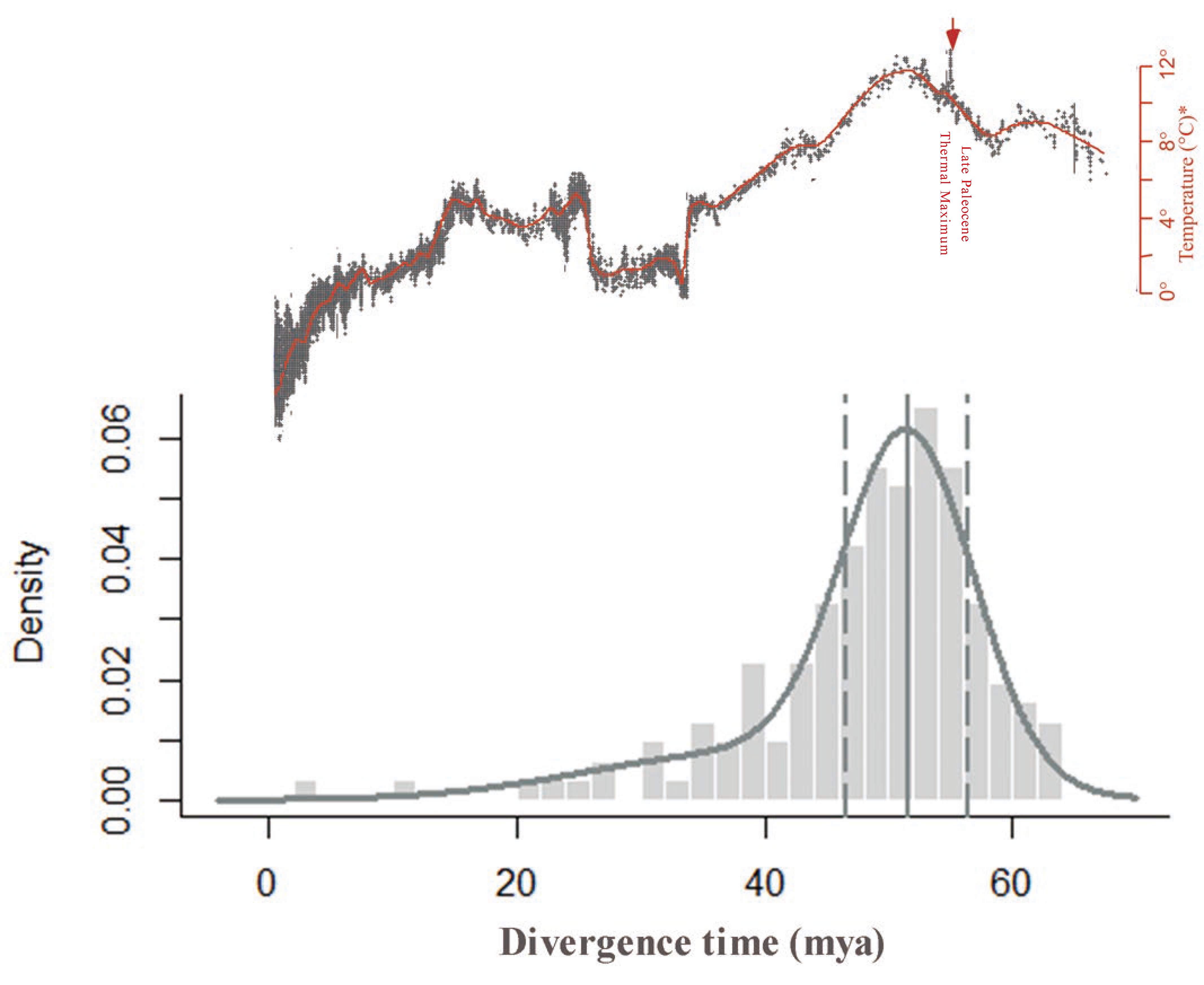
2.5. Dating the WGD Event in C. thalictroides Using Ks Distribution Peaks and Absolute Dating
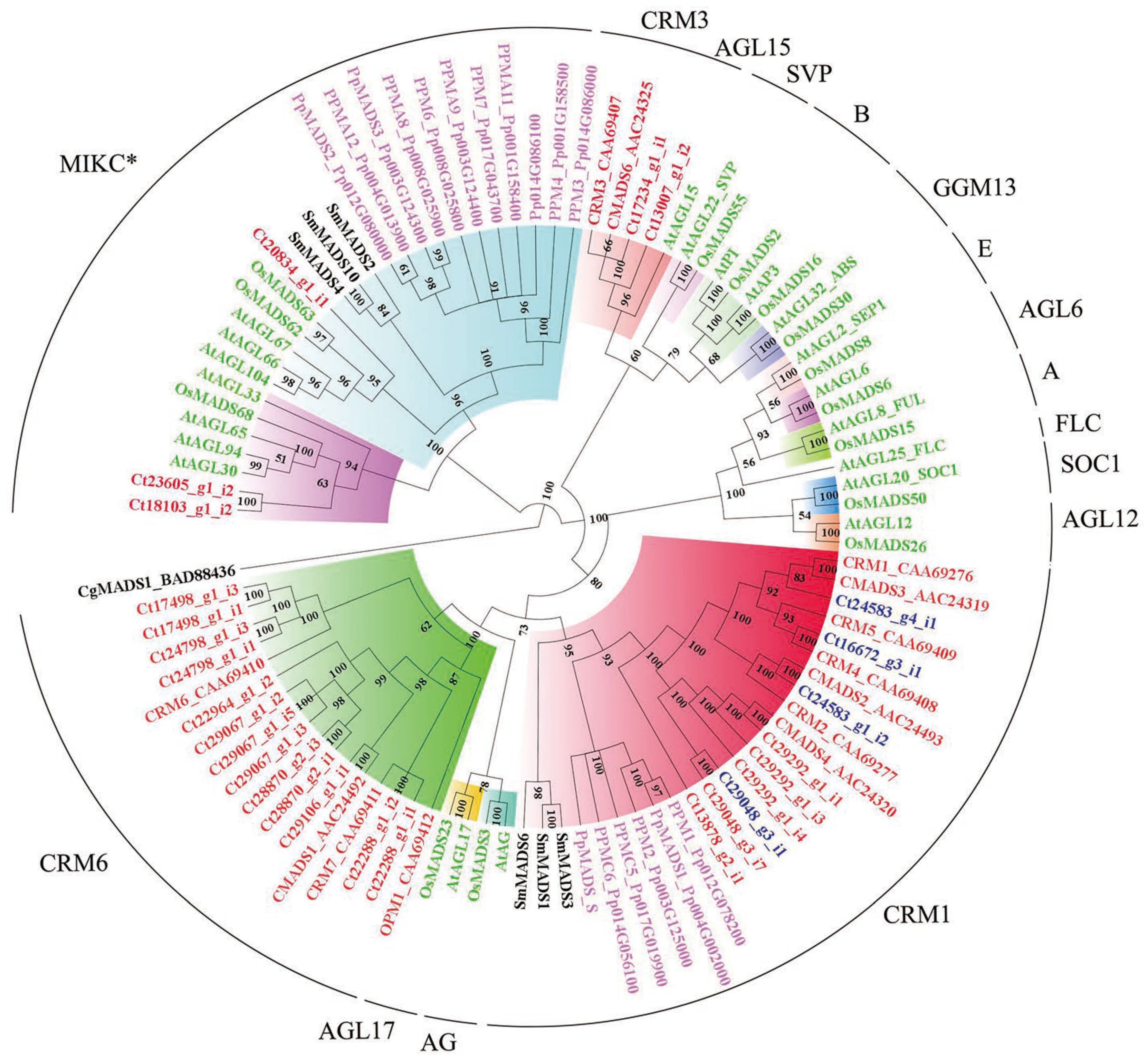
2.6. Phylogenomic Analysis of MADS-box Family Genes
3. Discussion
3.1. Inferring WGD from Ks Age Distribution
3.2. Absolute Dating of the WGD Event in C. thalictroides
3.3. WGDs Contribute to Evolutionary Success and Potential Adaptation of Ceratopteris
4. Materials and Methods
4.1. Chromosome Counting
4.2. Data Collection
4.3. BUSCO Analysis
4.4. Evaluation of WGD candidates on the Basis of Ks Age Distribution
4.5. Absolute Dating
4.6. Functional Enrichment
4.7. Identification of the MADS-box Gene Family in C. thalictroides
4.8. Phylogenetic Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| WGD | Whole-genome duplication |
| KT | Cretaceous–Tertiary |
| PETM | Paleocene-Eocene Thermal Maximum |
| Ks | synonymous substitutions per synonymous site |
| BUSCO | Benchmarking Universal Single Copy Orthologs |
| EST | expressed sequence tag |
References
- Soltis, D.E.; Albert, V.A.; Leebens-Mack, J.; Bell, C.D.; Paterson, A.H.; Zheng, C.; Sankoff, D.; Depamptilis, C.W.; Wall, P.K.; Soltis, P.S. Polyploidy and angiosperm diversification. Am. J. Bot. 2009, 96, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Bowers, J.E.; Wang, X.; Paterson, A.H. Angiosperm genome comparisons reveal early polyploidy in the monocot lineage. Proc. Natl. Acad. Sci. USA 2010, 107, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Wickett, N.J.; Ayyampalayam, S.; Chanderbali, A.S.; Landherr, L.; Ralph, P.E.; Tomsho, L.P.; Hu, Y.; Liang, H.; Soltis, P.S.; et al. Ancestral polyploidy in seed plants and angiosperms. Nature 2011, 473, 97–100. [Google Scholar] [CrossRef]
- Badouin, H.; Gouzy, J.; Grassa, C.J.; Murat, F.; Staton, S.E.; Cottret, L.; Lelandais-Briere, C.; Owens, G.L.; Carrere, S.; Mayjonade, B.; et al. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 2017, 546, 148–152. [Google Scholar] [CrossRef]
- Van de Peer, Y.; Mizrachi, E.; Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 2017, 18, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Blanc, G.; Wolfe, K.H. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 2004, 16, 1667–1678. [Google Scholar] [CrossRef] [PubMed]
- Mun, J.H.; Kwon, S.J.; Yang, T.J.; Seol, Y.J.; Jin, M.; Kim, J.A.; Lim, M.H.; Kim, J.S.; Baek, S.; Choi, B.S.; et al. Genome-wide comparative analysis of the Brassica rapa gene space reveals genome shrinkage and differential loss of duplicated genes after whole genome triplication. Genome Biol. 2009, 10, R111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, G.; Guo, X.; Guo, X.; Fan, L. Two ancient rounds of polyploidy in rice genome. J. Zhejiang Univ. Sci. 2005, 6B, 87–90. [Google Scholar] [CrossRef]
- Vanneste, K.; Sterck, L.; Myburg, A.A.; Van de Peer, Y.; Mizrachi, E. Horsetails are ancient polyploids: Evidence from Equisetum giganteum. Plant Cell 2015, 27, 1567–1578. [Google Scholar] [CrossRef]
- Fawcett, J.A.; Maere, S.; Van de Peer, Y. Plants with double genomes might have had a better chance to survive the Cretaceous–Tertiary extinction event. Proc. Natl. Acad. Sci. USA 2009, 106, 5737–5742. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Xi, Z.; Amorim, A.M.; Sugumaran, M.; Rest, J.S.; Liu, L.; Davis, C.C. Widespread ancient whole genome duplications in Malpighiales coincide with Eocene global climatic upheaval. New Phytol. 2019, 221, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.; Wang, H.; Guo, C.; Zhang, N.; Zeng, L.; Chen, Y.; Ma, H.; Qi, J. Wide-spread whole genome duplications contribute to genome complexity and species diversity in angiosperms. Mol. Plant 2018, 11, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Van de Peer, Y.; Maere, S.; Meyer, A. The evolutionary significance of ancient genome duplications. Nat. Rev. Genet. 2009, 10, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Kassahn, K.S.; Dang, V.T.; Wilkins, S.J.; Perkins, A.C.; Ragan, M.A. Evolution of gene function and regulatory control after whole-genome duplication: Comparative analyses in vertebrates. Genome Res. 2009, 19, 1404–1418. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Defoort, J.; Tasdighian, S.; Maere, S.; Van De Peer, Y.; De Smet, R. Gene duplicability of core genes is highly consistent across all angiosperms. Plant Cell 2016, 28, 326–344. [Google Scholar] [CrossRef]
- Lee, H.L.; Irish, V.F. Gene duplication and loss in a MADS box gene transcription factor circuit. Mol. Biol. Evol. 2011, 28, 3367–3380. [Google Scholar] [CrossRef] [PubMed]
- Wood, T.E.; Takebayashi, N.; Barker, M.S.; Mayrose, I.; Greenspoon, P.B.; Rieseberg, L.H. The frequency of polyploid speciation in vascular plants. Proc. Natl. Acad. Sci. USA 2009, 106, 13875–13879. [Google Scholar] [CrossRef]
- Soltis, P.S.; Marchant, D.B.; Van de Peer, Y.; Soltis, D.E. Polyploidy and genome evolution in plants. Curr. Opin. Genet. Dev. 2015, 35, 119–125. [Google Scholar] [CrossRef]
- Wagner, W.H.; Wagner, F.S. Polyploidy: Biological Relevance; Lewis, W.H., Ed.; Plenum: New York, NY, USA, 1980; pp. 199–214. [Google Scholar]
- Klekowski, E.J.; Baker, H.G. Evolutionary significance of polyploidy in the Pteridophyta. Science 1966, 153, 305–307. [Google Scholar] [CrossRef]
- Clark, J.; Hidalgo, O.; Pellicer, J.; Liu, H.; Marquardt, J.; Robert, Y.; Christenhusz, M.; Zhang, S.; Gibby, M.; Leitch, I.J.; et al. Genome evolution of ferns: Evidence for relative stasis of genome size across the fern phylogeny. New Phytol. 2016, 210, 1072–1082. [Google Scholar] [CrossRef]
- Schneider, H.; Liu, H.M.; Chang, Y.F.; Ohlsen, D.; Perrie, L.R.; Shepherd, L.; Kessler, M.; Karger, D.N.; Hennequin, S.; Marquardt, J.; et al. Neo- and Paleopolyploidy contribute to the species diversity of Asplenium—The most species rich genus of ferns. J. Syst. Evol. 2017, 55, 353–364. [Google Scholar] [CrossRef]
- Li, F.W.; Brouwer, P.; Carretero-Paulet, L.; Cheng, S.; de Vires, J.; Delaux, P.M.; Eily, A.; Koppers, N.; Kuo, L.Y.; Li, Z.; et al. Fern genomes elucidate land plant evolution and cyanobacterial symbioses. Nat. Plants 2018, 4, 460–472. [Google Scholar] [CrossRef]
- Barker, M.S.; Wolf, P.G. Unfurling fern biology in the genomics age. Bioscience 2010, 60, 177–185. [Google Scholar] [CrossRef]
- Lynch, M.; Conery, J.S. The evolutionary fate and consequences of duplicate genes. Science 2000, 290, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Leroux, O.; Eeckhout, S.; Viane, R.L.; Popper, Z.A. Ceratopteris richardii (C-Fern): A model for investigating adaptive modification of vascular plant cell walls. Front. Plant Sci. 2013, 4, 367. [Google Scholar] [CrossRef] [PubMed]
- Sessa, E.B.; Banks, J.A.; Barker, M.S.; Der, J.P.; Duffy, A.M.; Graham, S.W.; Hasebe, M.; Langdale, J.; Li, F.W.; Marchant, D.B.; et al. Between two fern genomes. GigaScience 2014, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; He, Z.; Xu, S.; Li, X.; Guo, W.; Yang, Y.; Zhou, R.; Shi, S. Transcriptome analyses provide insights into the phylogeny and adaptive evolution of the mangrove fern genus Acrostichum. Sci. Rep. 2016, 6, 35634. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Jin, D.; Shu, J.P.; Zhou, X.L.; Lei, M.; Wei, R.; Shang, H.; Wei, H.J.; Zhang, R.; Liu, L.; et al. Large scale phylogenomic analysis resolves a backbone phylogeny in ferns. GigaScience 2018, 7, 1–11. [Google Scholar] [CrossRef]
- Zachos, J.; Pagani, M.; Sloan, L.; Thomas, E.; Billups, K. Trends, rhythms, and aberrations in global climate 65 Ma to present. Science 2001, 292, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Gramzow, L.; Theissen, G. A hitchhiker’s guide to the MADS world of plants. Genome Boil. 2010, 11, 214. [Google Scholar] [CrossRef]
- Nakazato, T.; Jung, M.K.; Housworth, E.A.; Rieseberg, L.H.; Gastony, G.J. Genetic map-based analysis of genome structure in the homosporous fern Ceratopteris richardii. Genetics 2006, 173, 1585–1597. [Google Scholar] [CrossRef]
- Barker, M.S. Evolutionary genomic analyses of ferns reveal that high chromosome numbers are a product of high retention and fewer rounds of polyploidy relative to angiosperms. Am. Fern J. 2009, 99, 136–141. [Google Scholar]
- Vanneste, K.; Van de Peer, Y.; Maere, S. Inference of genome duplications from age distributions revisited. Mol. Biol. Evol. 2013, 30, 177–190. [Google Scholar] [CrossRef]
- Van de Peer, Y. Computational approaches to unveiling ancient genome duplications. Nat. Rev. Genet. 2004, 5, 752. [Google Scholar] [CrossRef] [PubMed]
- Vanneste, K.; Baele, G.; Maere, S.; Van de Peer, Y. Analysis of 41 plant genomes supports a wave of successful genome duplications in association with the Cretaceous-Paleogene boundary. Genome Res. 2014, 24, 1334–1347. [Google Scholar] [CrossRef]
- Tang, H.; Wang, X.; Bowers, J.E.; Ming, R.; Alam, M.; Paterson, A.H. Unraveling ancient hexaploidy through multiply-aligned angiosperm gene maps. Genome Res. 2008, 18, 1944–1954. [Google Scholar] [CrossRef] [PubMed]
- Hickok, L.G.; Warne, T.R.; Fribourg, R.S. The biology of the fern Ceratopteris and its use as a model system. Int. J. Plant Sci. 1955, 156, 332–345. [Google Scholar] [CrossRef]
- Tewksbury, J.J.; Huey, R.B.; Deutsch, C.A. Putting the heat on tropical animals. Science 2008, 320, 1296–1297. [Google Scholar] [CrossRef]
- Zachos, J.C.; Dickens, G.R.; Zeebe, R.E. An early Cenozoic perspective on greenhouse warming and carbon-cycle dynamics. Nature 2008, 451, 279–283. [Google Scholar] [CrossRef]
- Wendel, J.F. Genome evolution in polyploids. Plant Mol. Evol. 2000, 42, 225–249. [Google Scholar]
- Ramsey, J. Polyploidy and ecological adaptation in wild yarrow. Proc. Natl. Acad. Sci. USA 2011, 108, 7096–7101. [Google Scholar] [CrossRef]
- Chao, D.Y.; Dilkes, B.P.; Luo, H.; Douglas, A.; Yakubova, E.; Lahner, B.; Salt, D.E. Polyploids exhibit higher potassium uptake and salinity tolerance in Arabidopsis. Science 2013, 341, 658–659. [Google Scholar] [CrossRef]
- Diallo, A.M.; Nielsen, L.R.; Kjaer, E.D.; Patersen, K.K.; Raebild, A. Polyploidy can confer superiority to West African Acacia senegal (L.) Willd. trees. Front. Plant Sci. 2016, 7, 821. [Google Scholar] [CrossRef]
- Warne, T.R.; Lloyd, R.M. The role of spore germination and gametophyte development in habitat selection: Temperaturere sponses in certain temperate and tropical ferns. Bull. Torrey Bot. Club 1980, 107, 57–64. [Google Scholar] [CrossRef]
- Muenster, T.; Pahnke, J.; Di Rosa, A.; Kim, J.T.; Martin, W.; Saedler, H.; Theißen, G. Floral homeotic genes were recruited from homologous MADS-box genes preexisting in the common ancestor of ferns and seed plants. Proc. Natl. Acad. Sci. USA 1997, 94, 2415–2420. [Google Scholar] [CrossRef]
- Hasebe, M.; Wen, C.-K.; Kato, M.; Banks, J.A. Characterization of MADS homeotic genes in the fern Ceratopteris richardii. Proc. Natl. Acad. Sci. USA 1998, 95, 6222–6227. [Google Scholar] [CrossRef]
- Theissen, G.; Becker, A.; Di Rosa, A.; Kanno, A.; Kim, J.T.; Münster, T.; Winter, K.U.; Saedler, H. A short history of MADS-box genes in plants. Plant Mol. Evol. 2000, 42, 115–149. [Google Scholar]
- Veron, A.S.; Kaufmann, K. Bornberg-Bauer, E. Evidence of interaction network evolution by wholegenome duplications: A case study in MADS-box proteins. Mol. Biol. Evol. 2007, 24, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Airoldi, C.A.; Davies, B. Gene duplication and the evolution of plant MADS-box transcription factors. J. Genet. Genomics 2012, 39, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, G.; Nayar, S. A survey of MIKC type MADS-box genes in non-seed plants: Algae, Bryophytes, Lycophytes and Ferns. Front. Plant Sci. 2018, 9, 510. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Simao, F.A.; Waterhouse, R.M.; Ioannidi, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Sollars, E.S.A.; Harper, A.L.; Kelly, L.J.; Sambles, C.M.; Ramirez-Gonzalez, R.H.; Swarbreck, D.; Kaithakottil, G.; Cooper, E.D.; Uauy, C.; Havlickova, L.; et al. Genome sequence and genetic diversity of European ash trees. Nature 2017, 541, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Fraley, C.; Raftery, A.E. MCLUST Version 3: An R Package for Normal Mixture Modeling and Model-based Clustering; Department of Statistic, University of Washington: Seattle, WA, USA, 2006. [Google Scholar]
- Zar, J.H. Biostatistical Analysis, 4th ed.; Prentice Hall International: Englewood Cliffs, NJ, USA, 1999. [Google Scholar]
- Moreno-Hagelsieb, G.; Latimer, K. Choosing BLAST options for better detection of orthologs as reciprocal best hits. Bioinformatics 2007, 24, 319–324. [Google Scholar] [CrossRef]
- Schuettpelz, E.; Schneider, H.; Smith, A.R.; Hovenkamp, P.; Prado, J.; Rouhan, G.; Salino, A.; Sundue, M.; Almeida, T.E.; Parris, B.; et al. A community-derived classification for extant lycophytes and ferns. J. Syst. Evol. 2016, 54, 563–603. [Google Scholar]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef]
- Krassilov, V.; Bacchia, F. Cenomanian florule of Nammoura, Lebanon. Cretac. Res. 2000, 21, 785–799. [Google Scholar] [CrossRef]
- Collinson, M.E. Cainozoic ferns and their distribution. Brittonia 2001, 53, 172–235. [Google Scholar] [CrossRef]
- Bonde, S.D.; Kumaran, K.P.N. The oldest macrofossil record of the mangrove fern Acrostichum L. from the Late Cretaceous Deccan Intertrappean beds of India. Cretac. Res. 2002, 23, 149–152. [Google Scholar] [CrossRef]
- De Smet, R.; Van de Peer, Y. Redundancy and rewiring of genetic networks following genome-wide duplication events. Curr. Opin. Plant Biol. 2012, 15, 168–176. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Paterson, A.H. Genome and gene duplications and gene expression divergence: A view from plants. Ann. N. Y. Acad. Sci. 2012, 1256, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2. 0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree v1.4.2, a Graphical Viewer of Phylogenetic Trees; University of Edinburg: Edinburgh, UK, 2014; Available online: http://tree.bio.ed.ac.uk/software/figtree (accessed on 8 October 2018).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Total Reads (Clean) | Number of Contigs | Total Number of Unigenes | N50 (bp) | Mean Length (bp) |
|---|---|---|---|---|---|
| Triploid a | 35,528,634 | 69,929 | 60,823 | 787 | 576.00 |
| Diploid b | 31,741,082 | 74,728 | 83,202 | 1610 | 912.26 |
| Goniophlebium niponicumb | 38,786,214 | 54,152 | 58,494 | 1663 | 951.92 |
| Woodwardia proliferab | 40,967,322 | 69,931 | 74,564 | 1557 | 859.72 |
| Dennstaedtia pilosellab | 45,618,446 | 84,813 | 89,185 | 1582 | 831.56 |
| Cheilanthes chusanab | 51,851,066 | 49,449 | 52,782 | 1727 | 1012.63 |
| Acrostichum aureumb | 43,422,574 | 46,189 | 50,594 | 1729 | 1043.2 |
| Osmolindsaea odoratab | 46,808,646 | 113,778 | 130,549 | 1521 | 845.96 |
| Alsophila podophyllab | 48,768,608 | 66,254 | 72,404 | 1580 | 904.62 |
| Species | BUSCO Notation Assessment Results |
|---|---|
| Diploid | C: 63.7% [S:39.7%, D:24%], F:5.9%, M: 30.4%, n: 1440 |
| Triploid | C: 34.3% [S:30.5%, D:3.8%], F:12.2%, M: 53.5%, n: 1440 |
| No. of Duplicates | No. of Components | Bayesian Information Criterion | Mixture Means (Ks) | Variance (Ks) | Proportion |
|---|---|---|---|---|---|
| Diploid | |||||
| 8364 | 7 | 5998.011 | 0.128 | 0.0004 | 0.091 |
| 8364 | 7 | 5998.011 | 0.238 | 0.0048 | 0.074 |
| 8364 | 7 | 5998.011 | 0.499 | 0.0248 | 0.079 |
| 8364 | 7 | 5998.011 | 1.148 | 0.0461 | 0.278 |
| 8364 | 7 | 5998.011 | 1.526 | 0.1619 | 0.265 |
| 8364 | 7 | 5998.011 | 2.989 | 0.6015 | 0.176 |
| 8364 | 7 | 5998.011 | 4.561 | 0.0790 | 0.036 |
| Triploid | |||||
| 3088 | 5 | 3380.834 | 0.154 | 0.0016 | 0.075 |
| 3088 | 5 | 3380.834 | 0.338 | 0.0135 | 0.066 |
| 3088 | 5 | 3380.834 | 1.199 | 0.1591 | 0.464 |
| 3088 | 5 | 3380.834 | 2.268 | 0.5133 | 0.282 |
| 3088 | 5 | 3380.834 | 4.019 | 0.2962 | 0.112 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, R.; Wang, F.-G.; Zhang, J.; Shang, H.; Liu, L.; Wang, H.; Zhao, G.-H.; Shen, H.; Yan, Y.-H. Dating Whole Genome Duplication in Ceratopteris thalictroides and Potential Adaptive Values of Retained Gene Duplicates. Int. J. Mol. Sci. 2019, 20, 1926. https://doi.org/10.3390/ijms20081926
Zhang R, Wang F-G, Zhang J, Shang H, Liu L, Wang H, Zhao G-H, Shen H, Yan Y-H. Dating Whole Genome Duplication in Ceratopteris thalictroides and Potential Adaptive Values of Retained Gene Duplicates. International Journal of Molecular Sciences. 2019; 20(8):1926. https://doi.org/10.3390/ijms20081926
Chicago/Turabian StyleZhang, Rui, Fa-Guo Wang, Jiao Zhang, Hui Shang, Li Liu, Hao Wang, Guo-Hua Zhao, Hui Shen, and Yue-Hong Yan. 2019. "Dating Whole Genome Duplication in Ceratopteris thalictroides and Potential Adaptive Values of Retained Gene Duplicates" International Journal of Molecular Sciences 20, no. 8: 1926. https://doi.org/10.3390/ijms20081926
APA StyleZhang, R., Wang, F.-G., Zhang, J., Shang, H., Liu, L., Wang, H., Zhao, G.-H., Shen, H., & Yan, Y.-H. (2019). Dating Whole Genome Duplication in Ceratopteris thalictroides and Potential Adaptive Values of Retained Gene Duplicates. International Journal of Molecular Sciences, 20(8), 1926. https://doi.org/10.3390/ijms20081926