Interleukin-4 and Interleukin-13 Exacerbate Neurotoxicity of Prothrombin Kringle-2 in Cortex In Vivo via Oxidative Stress


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
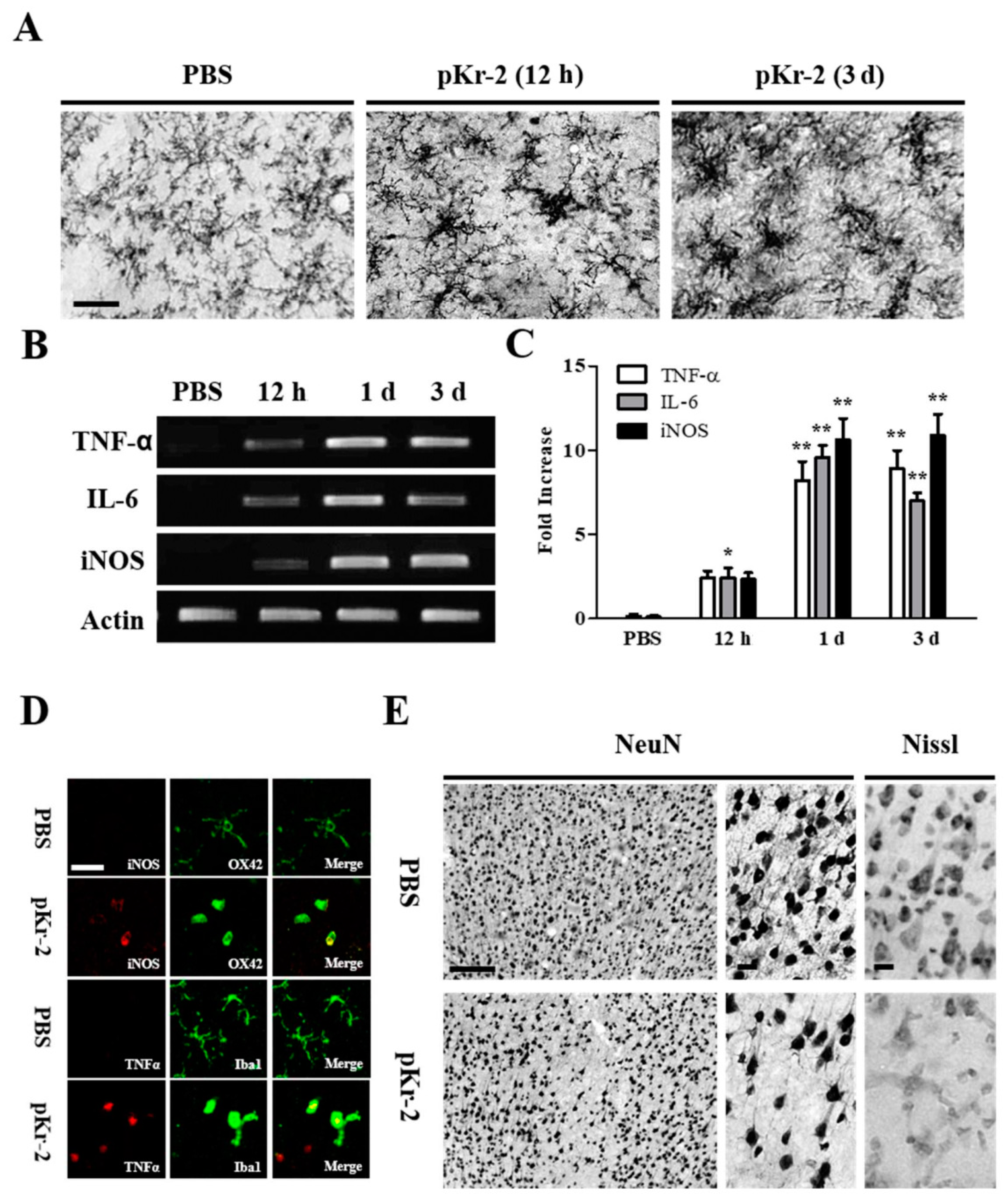
2.1. pKr-2 Induces Microglia/Macrophages Activation, Expression of Pro-Inflammatory Molecules, and Degeneration of Rat Cortical Neurons In Vivo
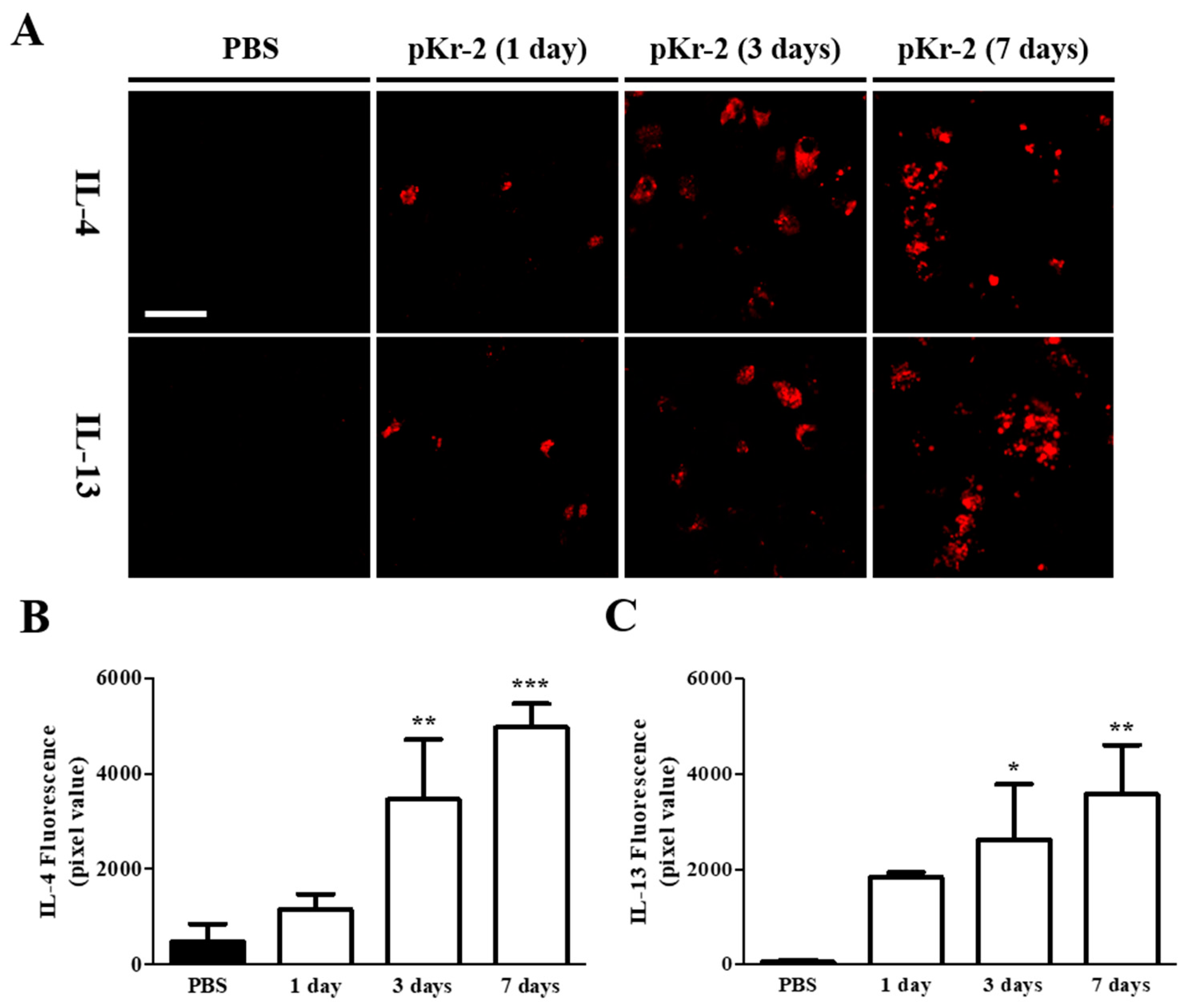
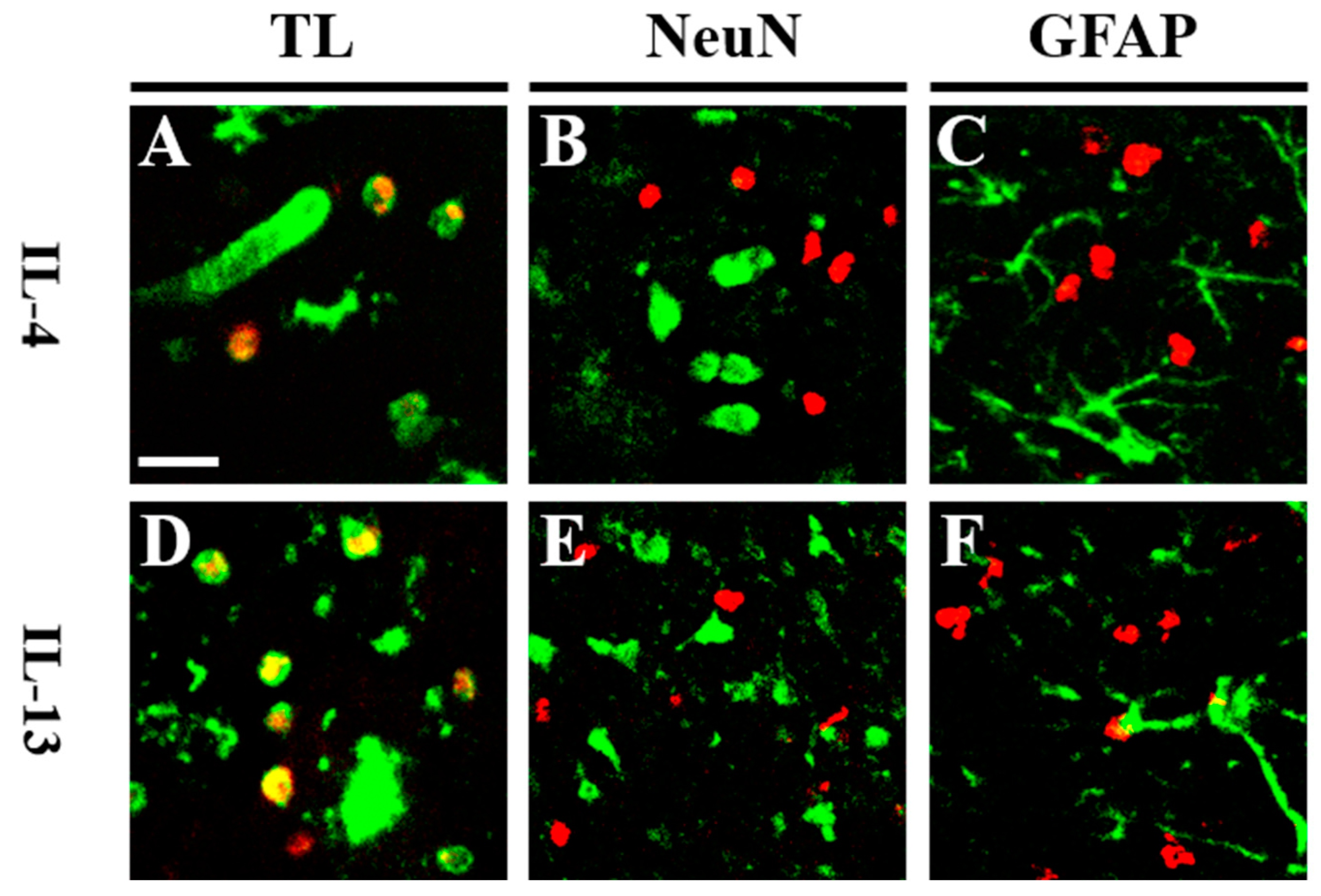
2.2. Levels of IL-4 and IL-13 Are Increased on TL+ Microglia/Macrophages in pKr-2-Injected Cerebral Cortex In Vivo
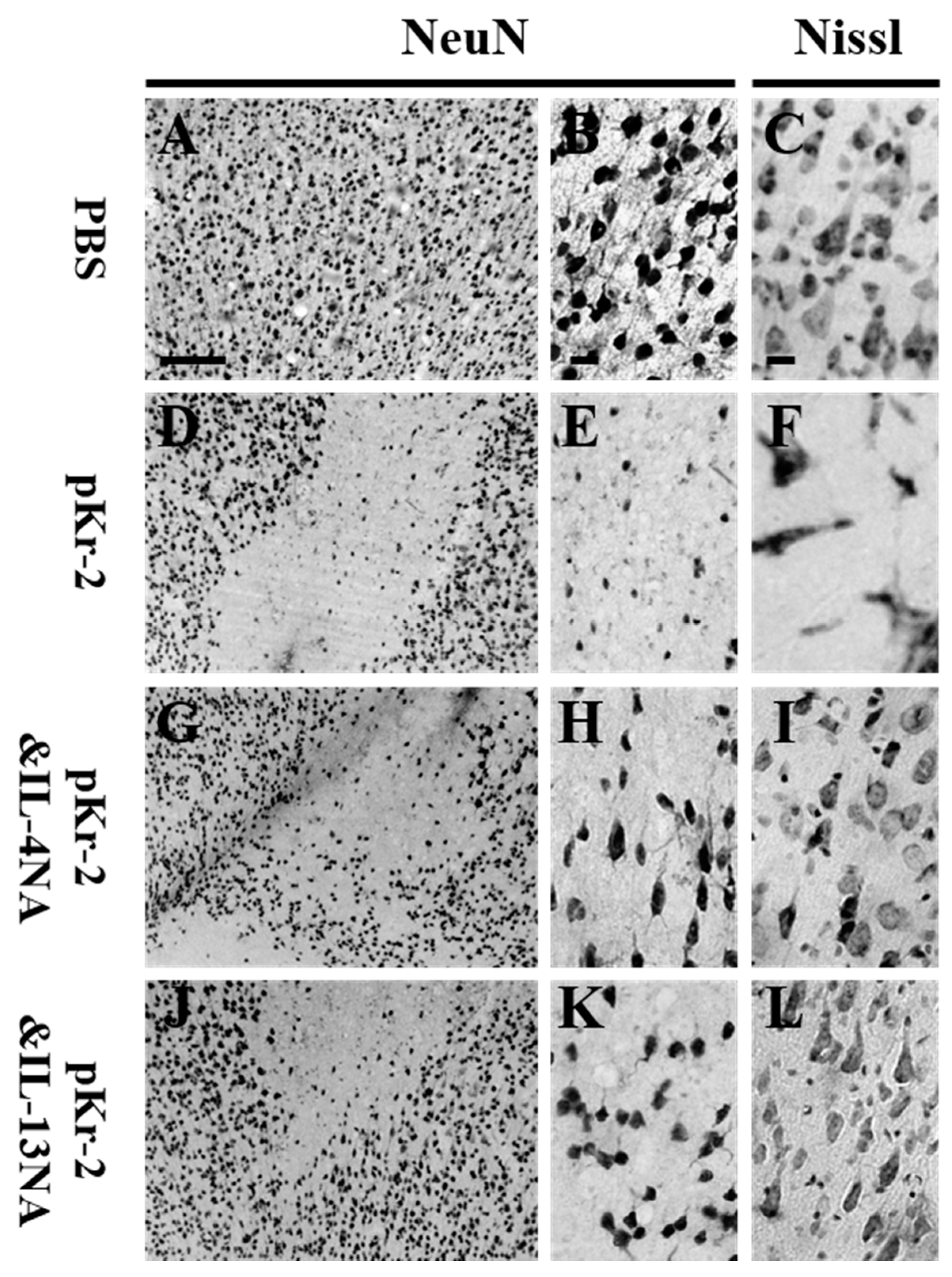
2.3. IL-4 and IL-13 Mediate Loss of Cortical Neurons in pKr-2-Tretaed Cortex In Vivo
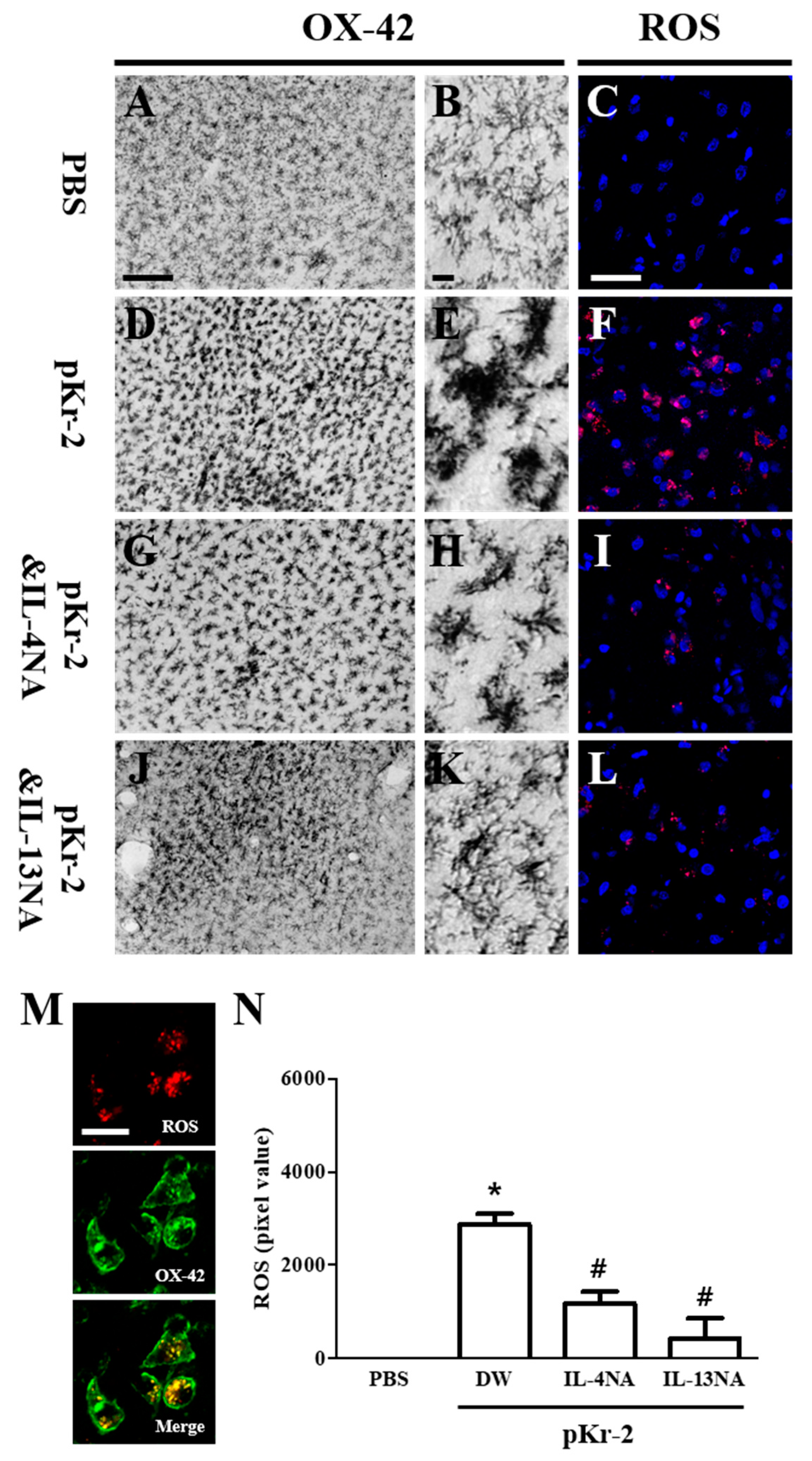
2.4. IL-4 and IL-13 Mediate Microglial Activation and ROS Production in pKr-2-Lesioned Cerebral Cortex In Vivo
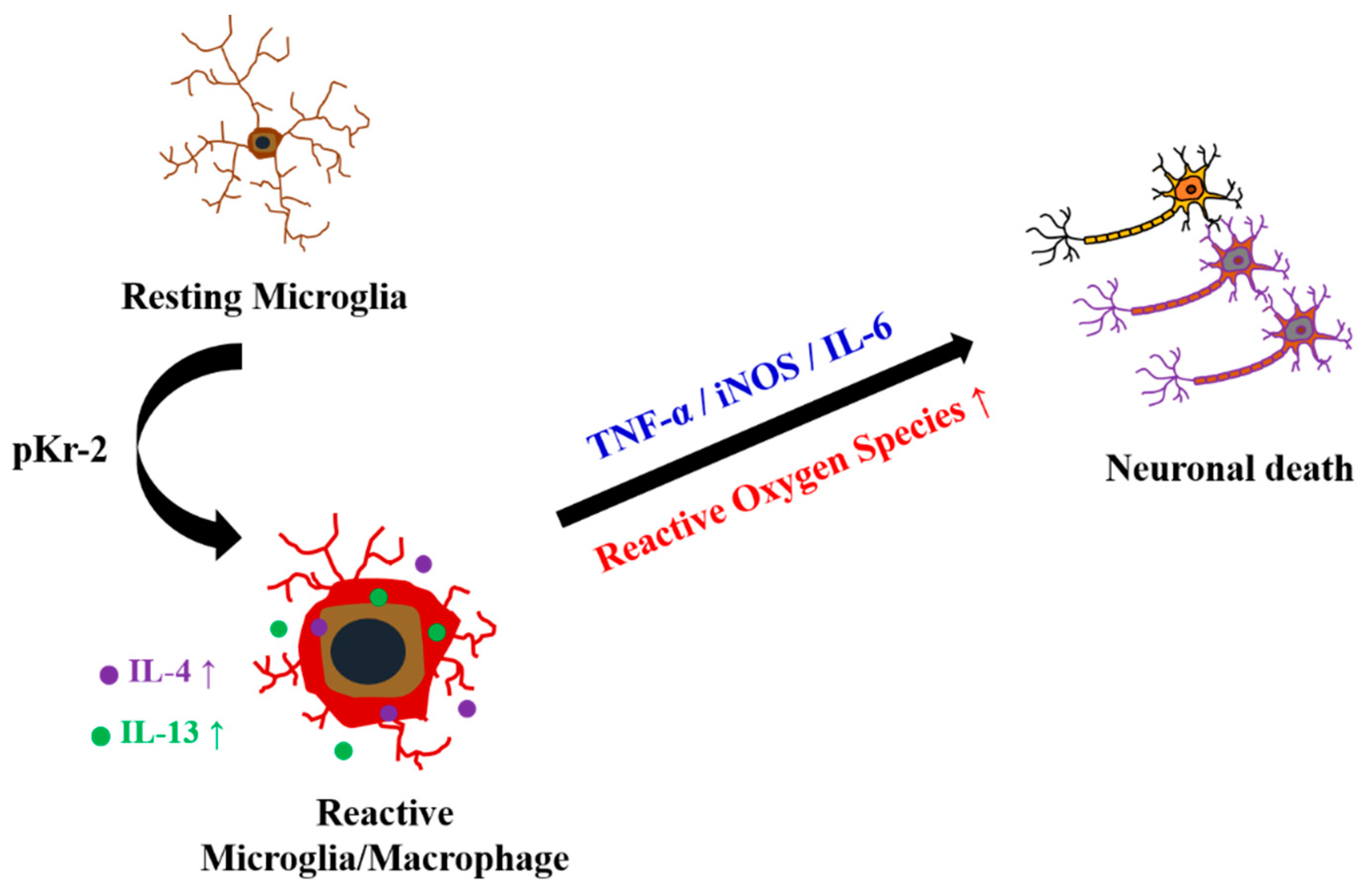
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Animals
4.3. Stereotaxic Injection of pKr-2 and IL-4 and IL-13 Neutralization
4.4. Immunohistochemstry and Immunofluorescence Staining
4.5. In Situ Detection of O2− and O2−-Derived Oxidants
4.6. Reverse-Transcription Polymerase Chain Reaction (RT-PCR) for Cytokines
4.7. Image J Analysis
4.8. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Libby, P.; Kobold, S. Inflammation: A Common Contributor to Cancer, Aging, and Cardiovascular Diseases. Cardiovasc. Res. 2019. [Google Scholar] [CrossRef]
- Kaur, D.; Sharma, V.; Deshmukh, R. Activation of microglia and astrocytes: A roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology 2019. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Stolp, H.B.; Liddelow, S.A.; Sa-Pereira, I.; Dziegielewska, K.M.; Saunders, N.R. Immune responses at brain barriers and implications for brain development and neurological function in later life. Front. Integr. Neurosci. 2013, 7, 61. [Google Scholar] [CrossRef] [PubMed]
- Lucas, S.M.; Rothwell, N.J.; Gibson, R.M. The role of inflammation in CNS injury and disease. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S232–S240. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.C.; Ko, H.W.; Bok, E.; Park, E.S.; Huh, S.H.; Nam, J.H.; Jin, B.K. The role of neuroinflammation on the pathogenesis of Parkinson’s disease. BMB Rep. 2010, 43, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Eikelenboom, P.; van Exel, E.; Hoozemans, J.J.; Veerhuis, R.; Rozemuller, A.J.; van Gool, W.A. Neuroinflammation—An early event in both the history and pathogenesis of Alzheimer’s disease. Neurodegener. Dis. 2010, 7, 38–41. [Google Scholar] [CrossRef]
- Kim, S.R.; Chung, E.S.; Bok, E.; Baik, H.H.; Chung, Y.C.; Won, S.Y.; Joe, E.; Kim, T.H.; Kim, S.S.; Jin, M.Y.; et al. Prothrombin kringle-2 induces death of mesencephalic dopaminergic neurons in vivo and in vitro via microglial activation. J. Neurosci. Res. 2010, 88, 1537–1548. [Google Scholar] [CrossRef]
- Nam, J.H.; Park, K.W.; Park, E.S.; Lee, Y.B.; Lee, H.G.; Baik, H.H.; Kim, Y.S.; Maeng, S.; Park, J.; Jin, B.K. Interleukin-13/-4-induced oxidative stress contributes to death of hippocampal neurons in abeta1-42-treated hippocampus in vivo. Antioxid. Redox Signal. 2012, 16, 1369–1383. [Google Scholar] [CrossRef]
- Shin, W.H.; Lee, D.Y.; Park, K.W.; Kim, S.U.; Yang, M.S.; Joe, E.H.; Jin, B.K. Microglia expressing interleukin-13 undergo cell death and contribute to neuronal survival in vivo. Glia 2004, 46, 142–152. [Google Scholar] [CrossRef]
- Streit, W.J.; Mrak, R.E.; Griffin, W.S. Microglia and neuroinflammation: A pathological perspective. J. Neuroinflamm. 2004, 1, 14. [Google Scholar] [CrossRef][Green Version]
- Janda, E.; Boi, L.; Carta, A.R. Microglial Phagocytosis and Its Regulation: A Therapeutic Target in Parkinson’s Disease? Front. Mol. Neurosci. 2018, 11, 144. [Google Scholar] [CrossRef] [PubMed]
- Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; Gordonov, S.; et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Shikamoto, Y.; Morita, T. Expression of factor X in both the rat brain and cells of the central nervous system. FEBS Lett. 1999, 463, 387–389. [Google Scholar] [CrossRef]
- Davie, E.W.; Fujikawa, K.; Kisiel, W. The coagulation cascade: Initiation, maintenance, and regulation. Biochemistry 1991, 30, 10363–10370. [Google Scholar] [CrossRef] [PubMed]
- Berzin, T.M.; Zipser, B.D.; Rafii, M.S.; Kuo-Leblanc, V.; Yancopoulos, G.D.; Glass, D.J.; Fallon, J.R.; Stopa, E.G. Agrin and microvascular damage in Alzheimer’s disease. Neurobiol. Aging 2000, 21, 349–355. [Google Scholar] [CrossRef]
- Shin, W.H.; Jeon, M.T.; Leem, E.; Won, S.Y.; Jeong, K.H.; Park, S.J.; McLean, C.; Lee, S.J.; Jin, B.K.; Jung, U.J.; et al. Induction of microglial toll-like receptor 4 by prothrombin kringle-2: A potential pathogenic mechanism in Parkinson’s disease. Sci. Rep. 2015, 5, 14764. [Google Scholar] [CrossRef]
- Sokolova, E.; Reiser, G. Prothrombin/thrombin and the thrombin receptors PAR-1 and PAR-4 in the brain: Localization, expression and participation in neurodegenerative diseases. Thromb. Haemost. 2008, 100, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Won, S.Y.; Choi, S.H.; Jin, B.K. Prothrombin kringle-2-induced oxidative stress contributes to the death of cortical neurons in vivo and in vitro: Role of microglial NADPH oxidase. J. Neuroimmunol. 2009, 214, 83–92. [Google Scholar] [CrossRef]
- Won, S.Y.; Kim, S.R.; Maeng, S.; Jin, B.K. Interleukin-13/Interleukin-4-induced oxidative stress contributes to death of prothrombinkringle-2 (pKr-2)-activated microglia. J. Neuroimmunol. 2013, 265, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Chittezhath, M.; Shalova, I.N.; Lim, J.Y. Macrophage polarization and plasticity in health and disease. Immunol. Res. 2012, 53, 11–24. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Young, D.A.; Lowe, L.D.; Booth, S.S.; Whitters, M.J.; Nicholson, L.; Kuchroo, V.K.; Collins, M. IL-4, IL-10, IL-13, and TGF-beta from an altered peptide ligand-specific Th2 cell clone down-regulate adoptive transfer of experimental autoimmune encephalomyelitis. J. Immunol. 2000, 164, 3563–3572. [Google Scholar] [CrossRef]
- Kawahara, K.; Suenobu, M.; Yoshida, A.; Koga, K.; Hyodo, A.; Ohtsuka, H.; Kuniyasu, A.; Tamamaki, N.; Sugimoto, Y.; Nakayama, H. Intracerebral microinjection of interleukin-4/interleukin-13 reduces beta-amyloid accumulation in the ipsilateral side and improves cognitive deficits in young amyloid precursor protein 23 mice. Neuroscience 2012, 207, 243–260. [Google Scholar] [CrossRef]
- Park, K.W.; Baik, H.H.; Jin, B.K. Interleukin-4-induced oxidative stress via microglial NADPH oxidase contributes to the death of hippocampal neurons in vivo. Curr. Aging Sci. 2008, 1, 192–201. [Google Scholar] [CrossRef]
- Park, K.W.; Baik, H.H.; Jin, B.K. IL-13-induced oxidative stress via microglial NADPH oxidase contributes to death of hippocampal neurons in vivo. J. Immunol. 2009, 183, 4666–4674. [Google Scholar] [CrossRef]
- Shimizu, E.; Kawahara, K.; Kajizono, M.; Sawada, M.; Nakayama, H. IL-4-induced selective clearance of oligomeric beta-amyloid peptide(1-42) by rat primary type 2 microglia. J. Immunol. 2008, 181, 6503–6513. [Google Scholar] [CrossRef]
- Yang, M.S.; Park, E.J.; Sohn, S.; Kwon, H.J.; Shin, W.H.; Pyo, H.K.; Jin, B.; Choi, K.S.; Jou, I.; Joe, E.H. Interleukin-13 and -4 induce death of activated microglia. Glia 2002, 38, 273–280. [Google Scholar] [CrossRef]
- Bok, E.; Cho, E.J.; Chung, E.S.; Shin, W.H.; Jin, B.K. Interleukin-4 Contributes to Degeneration of Dopamine Neurons in the Lipopolysaccharide-treated Substantia Nigra in vivo. Exp. Neurobiol. 2018, 27, 309–319. [Google Scholar] [CrossRef]
- Choi, S.H.; Lee, D.Y.; Kim, S.U.; Jin, B.K. Thrombin-induced oxidative stress contributes to the death of hippocampal neurons in vivo: Role of microglial NADPH oxidase. J. Neurosci. 2005, 25, 4082–4090. [Google Scholar] [CrossRef]
- Mann, K.G. Prothrombin. Methods Enzymol. 1976, 45, 123–156. [Google Scholar]
- Taneda, H.; Andoh, K.; Nishioka, J.; Takeya, H.; Suzuki, K. Blood coagulation factor Xa interacts with a linear sequence of the kringle 2 domain of prothrombin. J. Biochem. 1994, 116, 589–597. [Google Scholar] [CrossRef]
- Soifer, S.J.; Peters, K.G.; O’Keefe, J.; Coughlin, S.R. Disparate temporal expression of the prothrombin and thrombin receptor genes during mouse development. Am. J. Pathol. 1994, 144, 60–69. [Google Scholar]
- Weinstein, J.R.; Gold, S.J.; Cunningham, D.D.; Gall, C.M. Cellular localization of thrombin receptor mRNA in rat brain: Expression by mesencephalic dopaminergic neurons and codistribution with prothrombin mRNA. J. Neurosci. 1995, 15, 2906–2919. [Google Scholar] [CrossRef]
- Fenton, J.W., 2nd. Thrombin. Ann. N. Y. Acad. Sci. 1986, 485, 5–15. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Kisler, K.; Montagne, A.; Toga, A.W.; Zlokovic, B.V. The role of brain vasculature in neurodegenerative disorders. Nat. Neurosci. 2018, 21, 1318–1331. [Google Scholar] [CrossRef]
- Festoff, B.W.; Sajja, R.K.; van Dreden, P.; Cucullo, L. HMGB1 and thrombin mediate the blood-brain barrier dysfunction acting as biomarkers of neuroinflammation and progression to neurodegeneration in Alzheimer’s disease. J. Neuroinflamm. 2016, 13, 194. [Google Scholar] [CrossRef]
- Chung, Y.C.; Kruyer, A.; Yao, Y.; Feierman, E.; Richards, A.; Strickland, S.; Norris, E.H. Hyperhomocysteinemia exacerbates Alzheimer’s disease pathology by way of the beta-amyloid fibrinogen interaction. J. Thromb. Haemost. 2016, 14, 1442–1452. [Google Scholar] [CrossRef]
- Bachiller, S.; Jimenez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef]
- Park, K.W.; Lee, H.G.; Jin, B.K.; Lee, Y.B. Interleukin-10 endogenously expressed in microglia prevents lipopolysaccharide-induced neurodegeneration in the rat cerebral cortex in vivo. Exp. Mol. Med. 2007, 39, 812–819. [Google Scholar] [CrossRef]
- Cash, E.; Minty, A.; Ferrara, P.; Caput, D.; Fradelizi, D.; Rott, O. Macrophage-inactivating IL-13 suppresses experimental autoimmune encephalomyelitis in rats. J. Immunol. 1994, 153, 4258–4267. [Google Scholar] [PubMed]
- Szczepanik, A.M.; Funes, S.; Petko, W.; Ringheim, G.E. IL-4, IL-10 and IL-13 modulate A beta(1–42)-induced cytokine and chemokine production in primary murine microglia and a human monocyte cell line. J. Neuroimmunol. 2001, 113, 49–62. [Google Scholar] [CrossRef]
- Casella, G.; Garzetti, L.; Gatta, A.T.; Finardi, A.; Maiorino, C.; Ruffini, F.; Martino, G.; Muzio, L.; Furlan, R. IL4 induces IL6-producing M2 macrophages associated to inhibition of neuroinflammation in vitro and in vivo. J. Neuroinflamm. 2016, 13, 139. [Google Scholar] [CrossRef]
- Payne, N.L.; Dantanarayana, A.; Sun, G.; Moussa, L.; Caine, S.; McDonald, C.; Herszfeld, D.; Bernard, C.C.; Siatskas, C. Early intervention with gene-modified mesenchymal stem cells overexpressing interleukin-4 enhances anti-inflammatory responses and functional recovery in experimental autoimmune demyelination. Cell Adh. Migr. 2012, 6, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Ochoa-Reparaz, J.; Rynda, A.; Ascon, M.A.; Yang, X.; Kochetkova, I.; Riccardi, C.; Callis, G.; Trunkle, T.; Pascual, D.W. IL-13 production by regulatory T cells protects against experimental autoimmune encephalomyelitis independently of autoantigen. J. Immunol. 2008, 181, 954–968. [Google Scholar] [CrossRef]
- Lee, S.I.; Jeong, S.R.; Kang, Y.M.; Han, D.H.; Jin, B.K.; Namgung, U.; Kim, B.G. Endogenous expression of interleukin-4 regulates macrophage activation and confines cavity formation after traumatic spinal cord injury. J. Neurosci. Res. 2010, 88, 2409–2419. [Google Scholar] [CrossRef] [PubMed]
- Lukens, J.R.; Gurung, P.; Shaw, P.J.; Barr, M.J.; Zaki, M.H.; Brown, S.A.; Vogel, P.; Chi, H.; Kanneganti, T.D. The NLRP12 Sensor Negatively Regulates Autoinflammatory Disease by Modulating Interleukin-4 Production in T Cells. Immunity 2015, 42, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Kaler, L.J.; Proctor, T.M.; Teuscher, C.; Vandenbark, A.A.; Offner, H. IL-13-mediated gender difference in susceptibility to autoimmune encephalomyelitis. J. Immunol. 2008, 180, 2679–2685. [Google Scholar] [CrossRef]
- Morrison, B.E.; Marcondes, M.C.; Nomura, D.K.; Sanchez-Alavez, M.; Sanchez-Gonzalez, A.; Saar, I.; Kim, K.S.; Bartfai, T.; Maher, P.; Sugama, S.; et al. Cutting edge: IL-13Ralpha1 expression in dopaminergic neurons contributes to their oxidative stress-mediated loss following chronic peripheral treatment with lipopolysaccharide. J. Immunol. 2012, 189, 5498–5502. [Google Scholar] [CrossRef]
- Park, K.W.; Lee, D.Y.; Joe, E.H.; Kim, S.U.; Jin, B.K. Neuroprotective role of microglia expressing interleukin-4. J. Neurosci. Res. 2005, 81, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Ji, K.A.; Yang, M.S.; Jeong, H.K.; Min, K.J.; Kang, S.H.; Jou, I.; Joe, E.H. Resident microglia die and infiltrated neutrophils and monocytes become major inflammatory cells in lipopolysaccharide-injected brain. Glia 2007, 55, 1577–1588. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.Y.; Jeong, J.Y.; Kim, K.I.; Won, S.Y.; Chung, Y.C.; Nam, J.H.; Cho, E.J.; Ahn, T.B.; Bok, E.; Shin, W.H.; et al. Inhibition of Microglia-Derived Oxidative Stress by Ciliary Neurotrophic Factor Protects Dopamine Neurons In Vivo from MPP(+) Neurotoxicity. Int. J. Mol. Sci. 2018, 19, 3543. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, J.Y.; Chung, Y.C.; Jin, B.K. Interleukin-4 and Interleukin-13 Exacerbate Neurotoxicity of Prothrombin Kringle-2 in Cortex In Vivo via Oxidative Stress. Int. J. Mol. Sci. 2019, 20, 1927. https://doi.org/10.3390/ijms20081927
Jeong JY, Chung YC, Jin BK. Interleukin-4 and Interleukin-13 Exacerbate Neurotoxicity of Prothrombin Kringle-2 in Cortex In Vivo via Oxidative Stress. International Journal of Molecular Sciences. 2019; 20(8):1927. https://doi.org/10.3390/ijms20081927
Chicago/Turabian StyleJeong, Jae Yeong, Young Cheul Chung, and Byung Kwan Jin. 2019. "Interleukin-4 and Interleukin-13 Exacerbate Neurotoxicity of Prothrombin Kringle-2 in Cortex In Vivo via Oxidative Stress" International Journal of Molecular Sciences 20, no. 8: 1927. https://doi.org/10.3390/ijms20081927
APA StyleJeong, J. Y., Chung, Y. C., & Jin, B. K. (2019). Interleukin-4 and Interleukin-13 Exacerbate Neurotoxicity of Prothrombin Kringle-2 in Cortex In Vivo via Oxidative Stress. International Journal of Molecular Sciences, 20(8), 1927. https://doi.org/10.3390/ijms20081927