Curcumin and Intestinal Inflammatory Diseases: Molecular Mechanisms of Protection


{kind=link}
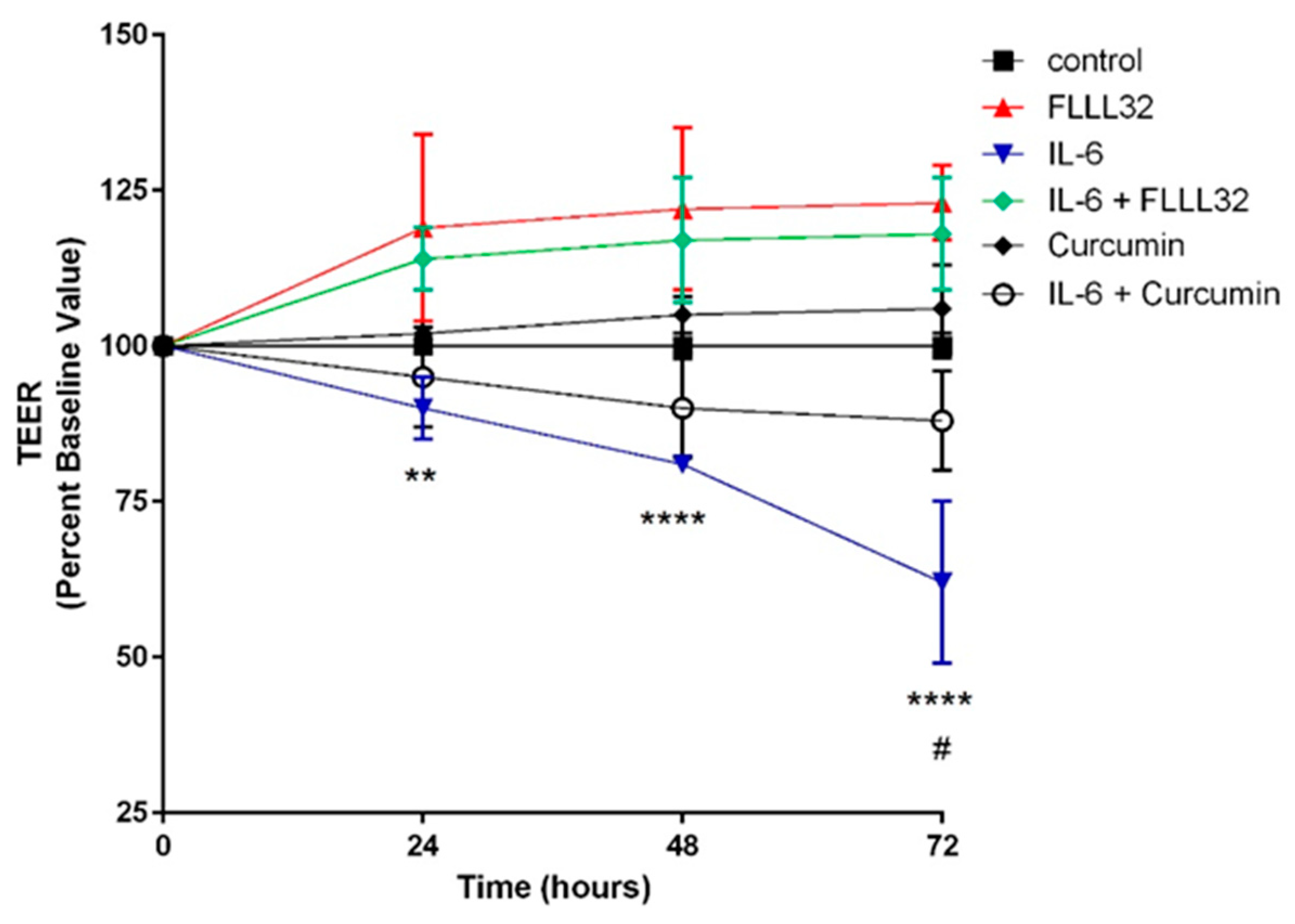{kind=link}
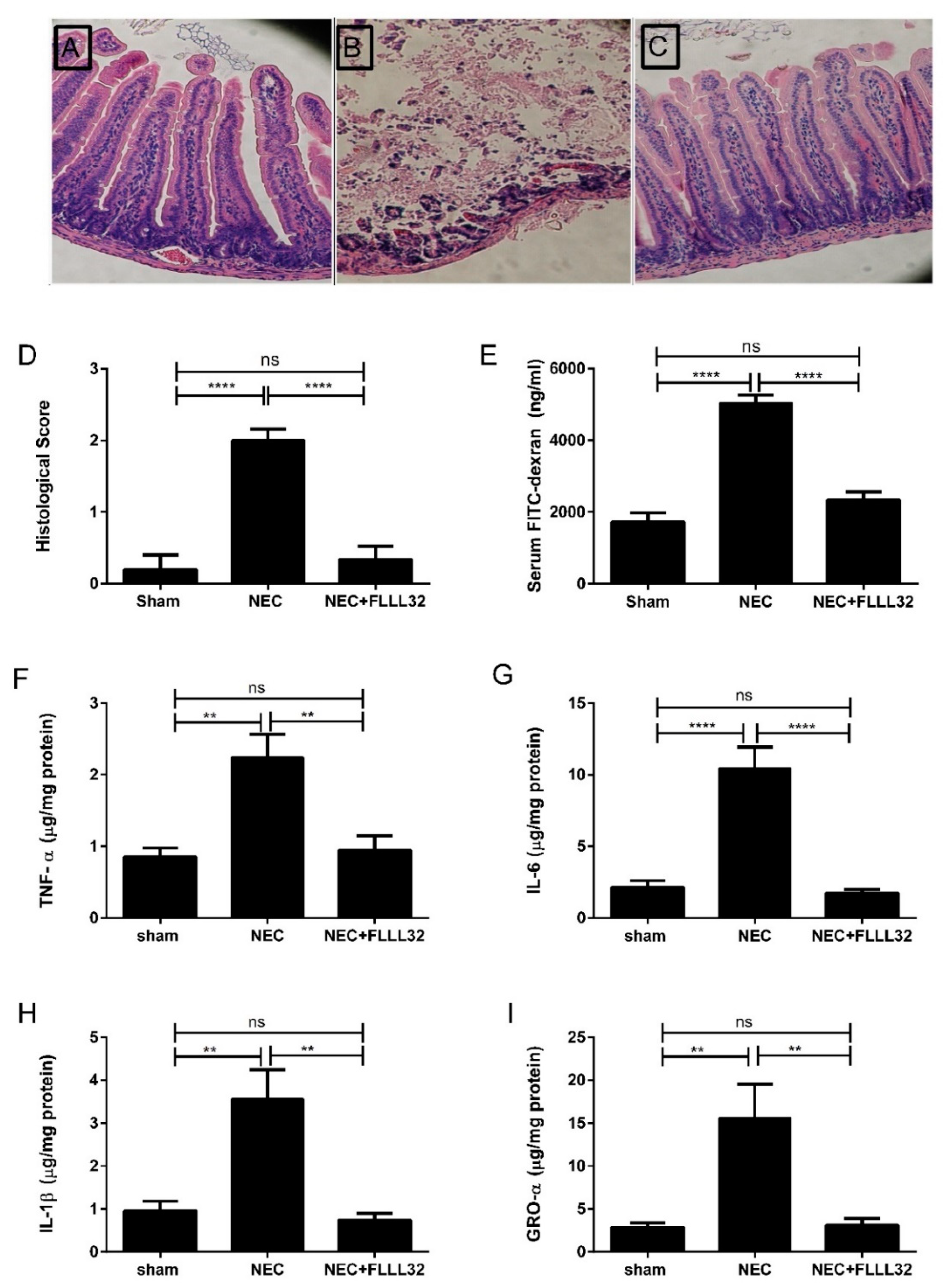{kind=link}
Abstract
1. Introduction
2. Intestinal Microbiome in Intestinal Inflammatory Diseases
2.1. Microbiome in NEC
2.2. Microbiome in IBD
3. Signal Transduction in Intestinal Inflammatory Diseases
3.1. NF-κB Signaling
3.2. AP-1 Signaling
3.3. TLR4 Induction
4. Molecular Mechanisms of Injury in Intestinal Inflammatory Diseases
4.1. Pathogenesis of NEC
4.2. Pathogenesis of IBD
5. The Effects of Curcumin on Intestinal Inflammatory Diseases
5.1. Antibacterial and Microbiome Effects
5.2. Effects on Signal Transduction
5.3. Effects on Inflammation and Immunomodulation
5.4. Antioxidant Effects
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AIEC | adherent invasive E. coli |
| AMP | antimicrobial protein |
| APC | antigen-presenting cell |
| AP-1 | activator protein 1 |
| ATF-2 | activating transcription factor 2 |
| CD | Crohn’s disease |
| CO | carbon monoxide |
| COX-2 | cyclooxygenase-2 |
| CREB | ATF/cyclic AMP-response element-binding |
| DC | dendritic cell |
| eNOS | endothelial nitric oxide synthase |
| ELK-2 | ETS domain-containing protein 2 |
| ERK | extracellular signal-regulated kinases |
| FITC | fluorescein isothiocyanate |
| GRO-α | growth-regulated oncogene-alpha |
| H&E | hematoxylin and eosin |
| HO-1 | heme oxygenase-1 |
| HUVEC | human umbilical vein endothelial cells |
| IBD | inflammatory bowel disease |
| ICAM | intercellular adhesion molecule |
| IDO | indoleamine 2,3-dioxygenase |
| IEC | intestinal epithelial cell |
| IEL | intraepithelial lymphocyte |
| IFN-γ | interferon-gamma |
| IKK | IκB kinase |
| IL | interleukin |
| ILC | innate lymphoid cell |
| IL-1Ra | IL-1 receptor antagonist |
| iNOS | inducible nitric oxide synthase |
| IRAK-M | interleukin-1 receptor-associated kinase-M |
| JNK | jun N-terminal kinases |
| LFA | lymphocyte function-associated antigen |
| LPS | lipopolysaccharide |
| M | microfold |
| Mac-1 | macrophage 1 antigen |
| MAPK | mitogen-activated protein kinase |
| MCP | monocyte chemoattractant protein |
| MD | myeloid differentiation protein |
| MDA | malondialdehyde |
| MDPI | Multidisciplinary Digital Publishing Institute |
| MHC | major histocompatibility class |
| MIP | macrophage inflammatory protein |
| MKP-1 | mitogen-activated protein kinase phosphatase 1 |
| MMP | matrix metalloproteinase |
| MPO | myeloperoxidase |
| mTOR | mammalian target of rapamycin |
| MyD88 | myeloid differentiation factor 88 |
| MUC | mucin |
| NEC | necrotizing enterocolitis |
| NEMO | NF-κB essential modulator |
| NF-κB | nuclear factor-kappaB |
| NO | nitric oxide |
| Nrf2 | nuclear factor (erythroid-derived 2)-related factor |
| ns | not significant |
| Nur-77 | nerve growth factor 1B |
| PAF | platelet-activating factor |
| PGE2 | prostaglandin E2 |
| PRR | pattern-recognition receptor |
| RA | retinoic acid |
| RIP | receptor-interacting protein |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SARM | selective androgen receptor modulator |
| SCFA | short chain fatty acid |
| SEM | standard error of mean |
| SHIP | inositol polyphosphate 5′-phosphatase D |
| SMAD7 | mothers against decapentaplegic homolog 7 |
| SOD | superoxide dismutase |
| TAB | TAK1-binding |
| TAK | transforming growth factor-beta-activated kinase |
| TBK | TANK-binding kinase |
| TEER | transepithelial electrical resistance |
| TGF-β | transforming growth factor-beta |
| Th | T helper |
| TIMP | tissue inhibitor of metalloproteinase |
| TIRAP | toll/interleukin-1 receptor domain-containing adaptor protein |
| TLR | toll-like receptor |
| TNBS | trinitrobenzene sulfonic acid |
| TNF-α | tumor necrosis factor-alpha |
| TRAF | TNF receptor-associated factor |
| TRAM | translocation associated membrane |
| Treg | regulatory T cell |
| TRIF | TIR-domain-containing adaptor protein inducing interferon-β |
| TSLP | thymic stromal lymphopoietin |
| UC | ulcerative colitis |
| VCAM | vascular cell adhesion molecule |
| VEGF-C | vascular endothelial growth factor-C |
| VLA | very late antigen |
References
- Cho, S.X.; Berger, P.J.; Nold-Petry, C.A.; Nold, M.F. The immunological landscape in necrotising enterocolitis. Expert Rev. Mol. Med. 2016, 18, e12. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Du, X.; Xu, X.; Wang, M.; Li, Z. Monocyte activation and inflammation can exacerbate Treg/Th17 imbalance in infants with neonatal necrotizing enterocolitis. Int. Immunopharmacol. 2018, 59, 354–360. [Google Scholar] [CrossRef]
- Dias, A.M.; Correia, A.; Pereira, M.S.; Almeida, C.R.; Alves, I.; Pinto, V.; Catarino, T.A.; Mendes, N.; Leander, M.; Oliva-Teles, M.T.; et al. Metabolic control of T cell immune response through glycans in inflammatory bowel disease. Proc. Natl. Acad. Sci. USA 2018, 115, E4651–E4660. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, X.; Liu, S.; Zhang, Y.; Zhang, D. Toll-like receptors and inflammatory bowel disease. Front. Immunol. 2018, 9, 72. [Google Scholar] [CrossRef] [PubMed]
- Pallone, F.; Monteleone, G. Mechanisms of tissue damage in inflammatory bowel disease. Curr. Opin. Gastroenterol. 2001, 17, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Lubbad, A.; Oriowo, M.A.; Khan, I. Curcumin attenuates inflammation through inhibition of TLR-4 receptor in experimental colitis. Mol. Cell. Biochem. 2009, 322, 127–135. [Google Scholar] [CrossRef]
- Bischoff, S.C.; Barbara, G.; Buurman, W.; Ockhuizen, T.; Schulzke, J.-D.; Serino, M.; Tilg, H.; Watson, A.; Wells, J.M. Intestinal permeability—A new target for disease prevention and therapy. BMC Gastroenterol. 2014, 14, 189. [Google Scholar] [CrossRef]
- Wang, J.; Ghosh, S.S.; Ghosh, S. Curcumin improves intestinal barrier function: Modulation of intracellular signaling, and organization of tight junctions. Am. J. Physiol. Cell Physiol. 2017, 312, C438–C445. [Google Scholar] [CrossRef]
- Santaolalla, R.; Fukata, M.; Abreu, M.T. Innate immunity in the small intestine. Curr. Opin. Gastroenterol. 2011, 27, 125–131. [Google Scholar] [CrossRef]
- Mara, M.A.; Good, M.; Weitkamp, J.-H. Innate and adaptive immunity in necrotizing enterocolitis. Semin. Fetal Neonatal Med. 2018, 23, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Murai, M.; Turovskaya, O.; Kim, G.; Madan, R.; Karp, C.L.; Cheroutre, H.; Kronenberg, M. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat. Immunol. 2009, 10, 1178–1184. [Google Scholar] [CrossRef] [PubMed]
- Krappmann, D.; Wegener, E.; Sunami, Y.; Esen, M.; Thiel, A.; Mordmuller, B.; Scheidereit, C. The IκB kinase complex and NF-κB act as master regulators of lipopolysaccharide-induced gene expression and control subordinate activation of AP-1. Mol. Cell. Biol. 2004, 24, 6488–6500. [Google Scholar] [CrossRef] [PubMed]
- Le Mandat Schultz, A.; Bonnard, A.; Barreau, F.; Aigrain, Y.; Pierre-Louis, C.; Berrebi, D.; Peuchmaur, M. Expression of TLR-2, TLR-4, NOD2 and pNF-kappaB in a neonatal rat model of necrotizing enterocolitis. PLoS ONE 2007, 2, e1102. [Google Scholar] [CrossRef]
- Swanson, P.A., 2nd; Kumar, A.; Samarin, S.; Vijay-Kumar, M.; Kundu, K.; Murthy, N.; Hansen, J.; Nusrat, A.; Neish, A.S. Enteric commensal bacteria potentiate epithelial restitution via reactive oxygen species-mediated inactivation of focal adhesion kinase phosphatases. Proc. Natl. Acad. Sci. USA 2011, 108, 8803–8808. [Google Scholar] [CrossRef] [PubMed]
- Dowling, D.J.; Levy, O. Ontogeny of early life immunity. Trends Immunol. 2014, 35, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Brandtzaeg, P. The gut as communicator between environment and host: Immunological consequences. Eur. J. Pharmacol. 2011, 668 (Suppl. 1), S16–S32. [Google Scholar] [CrossRef] [PubMed]
- Hering, N.A.; Fromm, M.; Schulzke, J.D. Determinants of colonic barrier function in inflammatory bowel disease and potential therapeutics. J. Physiol. 2012, 590, 1035–1044. [Google Scholar] [CrossRef]
- Neu, J.; Pammi, M. Pathogenesis of NEC: Impact of an altered intestinal microbiome. Semin. Perinatol. 2017, 41, 29–35. [Google Scholar] [CrossRef]
- Afrazi, A.; Sodhi, C.P.; Richardson, W.; Neal, M.; Good, M.; Siggers, R.; Hackam, D.J. New insights into the pathogenesis and treatment of necrotizing enterocolitis: Toll-like receptors and beyond. Pediatr. Res. 2011, 69, 183–188. [Google Scholar] [CrossRef]
- Musemeche, C.A.; Kosloske, A.M.; Bartow, S.A.; Umland, E.T. Comparative effects of ischemia, bacteria, and substrate on the pathogenesis of intestinal necrosis. J. Pediatr. Surg. 1986, 21, 536–538. [Google Scholar] [CrossRef]
- Carlisle, E.M.; Morowitz, M.J. The intestinal microbiome and necrotizing enterocolitis. Curr. Opin. Pediatr. 2013, 25, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hoenig, J.D.; Malin, K.J.; Qamar, S.; Petrof, E.O.; Sun, J.; Antonopoulos, D.A.; Chang, E.B.; Claud, E.C. 16S rRNA gene-based analysis of fecal microbiota from preterm infants with and without necrotizing enterocolitis. ISME J. 2009, 3, 944–954. [Google Scholar] [CrossRef] [PubMed]
- Warner, B.B.; Deych, E.; Zhou, Y.; Hall-Moore, C.; Weinstock, G.M.; Sodergren, E.; Shaikh, N.; Hoffmann, J.A.; Linneman, L.A.; Hamvas, A.; et al. Gut bacteria dysbiosis and necrotising enterocolitis in very low birthweight infants: A prospective case-control study. Lancet 2016, 387, 1928–1936. [Google Scholar] [CrossRef]
- Neu, J.; Walker, W.A. Necrotizing enterocolitis. N. Engl. J. Med. 2011, 364, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Pammi, M.; Cope, J.; Tarr, P.I.; Warner, B.B.; Morrow, A.L.; Mai, V.; Gregory, K.E.; Kroll, J.S.; McMurty, V.; Ferris, M.J.; et al. Intestinal dysbiosis in preterm infants preceding necrotizing enterocolitis: A systematic review and meta-analysis. Microbiome 2017, 5, 31. [Google Scholar] [CrossRef] [PubMed]
- Cotton, C.M.; Taylor, S.; Stoll, B.; Goldberg, R.N.; Hansen, N.I.; Sanchez, P.J.; Ambalavanan, N.; Benjamin, D.K., Jr.; NICHD Neonatal Research Network. Prolonged duration of initial empirical antibiotic treatment is associated with increased rates of necrotizing enterocolitis and death for extremely low birth weight infants. Pediatrics 2009, 123, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Alexander, V.N.; Northrup, V.; Bizzarro, M.J. Antibiotic exposure in the newborn intestive care unit and the risk of necrotizing enterocolitis. J. Pediatr. 2011, 159, 392–397. [Google Scholar] [CrossRef]
- Heida, F.H.; van Zoonen, A.G.J.F.; Hulscher, J.B.F.; Te Kiefte, B.J.C.; Wessels, R.; Kooi, E.M.W.; Bos, A.F.; Harmsen, H.J.M.; de Goffau, M.C. A necrotizing enterocolitis-associated gut microbiota is present in the meconium: Results of a prospective study. Clin. Infect. Dis. 2016, 62, 863–870. [Google Scholar] [CrossRef]
- Terrin, G.; Passariello, A.; De Curtis, M.; Manguso, F.; Salvia, G.; Lega, L.; Messina, F.; Paludetto, R.; Canani, R.B. Ranatidine is associated with infections, necrotizing enterocolitis, and fatal outcome in newborns. Pediatrics 2012, 129, e40–e45. [Google Scholar] [CrossRef]
- Bilali, A.; Galanis, P.; Bartsocas, C.; Sparos, L.; Velonakis, E. H2-blocker therapy and incidence of necrotizing enterocolitis in preterm infants: A case-control study. Pediatr. Neonatol. 2013, 54, 141–142. [Google Scholar] [CrossRef]
- Zivkovic, A.M.; German, J.B.; Lebrilla, C.B.; Mills, D.A. Human milk glycobiome and its impact on the infant gastrointestinal microbiota. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4653–4658. [Google Scholar] [CrossRef]
- Good, M.; Sodhi, C.P.; Hackam, D.J. Evidence-based feeding strategies before and after the development of necrotizing enterocolitis. Expert Rev. Clin. Immunol. 2014, 10, 875–884. [Google Scholar] [CrossRef]
- Sela, D.A. Bifidobacterial utilization of human milk oligosaccharides. Int. J. Food Microbiol. 2011, 149, 58–64. [Google Scholar] [CrossRef]
- Niemarkt, H.J.; De Meij, T.G.; van Ganzewinkel, C.-J.; de Boer, N.K.H.; Andriessen, P.; Hutten, M.C.; Kramer, B.W. Necrotizing enterocolitis, gut microbiota, and brain development: Role of the brain-gut axis. Neonatology 2019, 115, 423–431. [Google Scholar] [CrossRef]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chavez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef]
- Ge, X.; Pan, J.; Liu, Y.; Wang, H.; Zhou, W.; Wang, X. Intestinal crosstalk between microbiota and serotonin and its impact on gut motility. Curr. Pharm. Biotechnol. 2018, 19, 190–195. [Google Scholar] [CrossRef]
- Aleksandrova, K.; Romero-Mosquera, B.; Hernandez, V. Diet, gut microbiome and epigenetics: Emerging links with inflammatory bowel diseases and prospects for management and prevention. Nutrients 2017, 9, 962. [Google Scholar] [CrossRef]
- Kostic, A.D.; Xavier, R.J.; Gevers, D. The microbiome in inflammatory bowel disease: Current status and the future ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef]
- Prosberg, M.; Bendtsen, F.; Vind, I.; Petersen, A.M.; Gluud, L.L. The association between the gut microbiota and the inflammatory bowel disease activity: A systematic review and meta-analysis. Scand. J. Gastroenterol. 2016, 51, 1407–1415. [Google Scholar] [CrossRef]
- Wills, E.S.; Jonkers, D.M.A.E.; Savelkoul, P.H.; Masclee, A.A.; Pierik, M.J.; Penders, J. Fecal microbial composition of ulcerative colitis and Crohn’s disease patients in remission and subsequent exacerbation. PLoS ONE 2014, 9, e90981. [Google Scholar] [CrossRef]
- Malavia, D.; Crawford, A.; Wilson, D. Nutritional immunity and fungal pathogenesis: The struggle for micronutrients and the host-pathogen interface. Adv. Microb. Phys. 2017, 70, 85–103. [Google Scholar] [CrossRef]
- Macia, L.; Tan, J.; Vieira, A.T.; Leach, K.; Stanley, D.; Luong, S.; Maruya, M.; Ian McKenzie, C.; Hijikata, A.; Wong, C.; et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 2015, 6, 6734. [Google Scholar] [CrossRef]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Backhed, F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of Clostridia stains from the human microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef]
- Gao, Y.; Huang, Y.; Zhao, Y.; Hu, Y.; Li, Z.; Guo, Q.; Zhao, K.; Lu, N. LL202 protects against dextran sulfate sodium-induced experimental colitis in mice by inhibiting MAPK/AP-1 signaling. Oncotarget 2016, 7, 63981–63994. [Google Scholar] [CrossRef]
- Grishin, A.V.; Wang, J.; Potoka, D.A.; Hackam, D.J.; Upperman, J.S.; Boyle, P.; Zamora, R.; Ford, H.R. Lipopolysaccharide induces cyclooxygenase-2 in intestinal epithelium via a noncanonical p38 MAPK pathway. J. Immunol. 2006, 176, 580–588. [Google Scholar] [CrossRef]
- Nanthakumar, N.N.; Young, C.; Ko, J.S.; Meng, D.; Chen, J.; Buie, T.; Walker, W.A. Glucocorticoid responsiveness in developing human intestine: Possible role in prevention of necrotizing enterocolitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G85–G92. [Google Scholar] [CrossRef][Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target Ther. 2017, 2, e17023. [Google Scholar] [CrossRef]
- Baldwin, A.S. The NF-KB and I-KB proteins: New discoveries and insights. Annu. Rev. Immunol. 1996, 14, 649–681. [Google Scholar] [CrossRef]
- Schreiber, S.; Nikolaus, S.; Hampe, J. Activation of nuclear factor κB in inflammatory bowel disease. Gut 1998, 42, 477–484. [Google Scholar] [CrossRef]
- De Plaen, I.G.; Liu, S.X.; Tian, R.; Neequaye, I.; May, M.J.; Han, X.B.; Hsueh, W.; Jilling, T.; Lu, J.; Caplan, M.S. Inhibition of nuclear factor-kappaB ameliorates bowel injury and prolongs survival in a neonatal rat model of necrotizing enterocolitis. Pediatr. Res. 2007, 62, 716–721. [Google Scholar] [CrossRef]
- Atreya, I.; Atreya, R.; Neurath, M.F. NF-κB in inflammatory bowel disease. J. Intern. Med. 2008, 263, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.; Gerstberger, S.; Carlson, L.; Franzoso, G.; Siebenlist, U. Control of IκB-α proteolysis by site-specific signal induced phosphorylation. Science 1995, 267, 1485–1487. [Google Scholar] [CrossRef]
- Siebenlist, U.; Brown, K.; Claudio, E. Control of lymphocyte development by nuclear factor-κB. Nat. Rev. Immunol. 2005, 5, 435–445. [Google Scholar] [CrossRef]
- Afonina, I.S.; Zhong, Z.; Karin, M.; Beyaert, R. Limiting inflammation—the negative regulation of NF_κB and NLRP3 inflammasome. Nat. Immunol. 2017, 18, 861–869. [Google Scholar] [CrossRef]
- Abreu, M.T. Toll-like receptor signaling in the intestinal epithelium: How bacterial recognition shapes intestinal function. Nat. Rev. Immunol. 2010, 10, 131–144. [Google Scholar] [CrossRef]
- Wullaert, A.; Bonnet, M.C.; Pasparakis, M. NF-κB in the regulation of epithelial homeostasis and inflammation. Cell Res. 2011, 21, 146–158. [Google Scholar] [CrossRef]
- Nenci, A.; Becker, C.; Wullaert, A.; Gareus, R.; van Loo, G.; Danese, S.; Huth, M.; Nikolaev, A.; Neufert, C.; Madison, B.; et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 2007, 446, 557–561. [Google Scholar] [CrossRef]
- Zaph, C.; Troy, A.E.; Taylor, B.C.; Berman-Booty, L.D.; Guild, K.J.; Du, Y.; Yost, E.A.; Gruber, A.D.; May, M.J.; Greten, F.R.; et al. Epithelial-cell-intrinsic IKK-beta expression regulates intestinal immune homeostasis. Nature 2007, 446, 552–556. [Google Scholar] [CrossRef]
- Guha, M.; Mackman, N. LPS induction of gene expression in human monocytes. Cell. Signal. 2001, 13, 85–94. [Google Scholar] [CrossRef]
- Granet, C.; Miossec, P. Combination of the pro-inflammatory cytokines IL-1, TNF-alpha and IL-17 leads to enhanced expression and additional recruitment of AP-1 family members, Egr-1 and NF-κB in osteoblast-like cells. Cytokine 2004, 26, 169–177. [Google Scholar] [CrossRef]
- Dokter, W.H.A.; Koopmans, S.B.; Vellenga, E. Effects of IL-10 and IL-4 on LPS-induced transcription factors (AP-1, NF-IL6 and NF-kappaB) which are involved in IL-6 regulation. Leukemia 1996, 10, 1038–1316. [Google Scholar]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef]
- Karin, M.; Takahashi, T.; Kapahi, P.; Delhase, M.; Chen, Y.; Makris, C.; Rothwarf, D.; Baud, V.; Natoli, G.; Guido, F.; et al. Oxidative stress and gene expression: The AP-1 and NF-κB connections. Biofactors 2001, 15, 87–89. [Google Scholar] [CrossRef]
- Verma, I.M.; Stevenson, J.K.; Schwarz, E.M.; Van Antwerp, D.; Miyamoto, S. Rel/NF-kappa B/I kappa B family: Intimate tales of association and dissociation. Genes Dev. 1995, 9, 2723–2735. [Google Scholar] [CrossRef]
- Fujioka, S.; Niu, J.; Schmidt, C.; Sclabas, G.M.; Peng, B.; Uwagawa, T.; Li, Z.; Evans, D.B.; Abbruzzese, J.L.; Chiao, P.J. NF-κB and AP-1 connection: Mechanism of NF-κB-dependent regulation of AP-1 activity. Mol. Cell Biol. 2004, 24, 7806–7819. [Google Scholar] [CrossRef]
- Angel, P.; Karin, M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta 1991, 1072, 129–157. [Google Scholar] [CrossRef]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef]
- Gazon, H.; Barbeau, B.; Mesnard, J.-M.; Peloponese, J.-M., Jr. Hijacking of the AP-1 signaling pathway during development of ATL. Front. Microbiol. 2018, 8, 2686. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Currie, R.W. Small interfering RNA knocks down heat shock factor-1 (HSF-1) and exacerbates pro-inflammatory activation of NF-kappa B and AP-1 in vascular smooth muscle cells. Cardiovasc. Res. 2006, 69, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Riesenberg, S.; Groetchen, A.; Siddaway, R.; Bald, T.; Reinhardt, J.; Smorra, D.; Kohlmeyer, J.; Renn, M.; Phung, B.; Aymans, P.; et al. MITF and c-Jun antagonism interconnects melanoma dedifferentiation with pro-inflammatory cytokine responsiveness and myeloid cell recruitment. Nat. Commun. 2015, 6, 8755. [Google Scholar] [CrossRef]
- Tremblay, L.; Valenza, F.; Ribeiro, S.P.; Li, J.F.; Slutsky, A.S. Injurious ventilatory strategies increase cytokines and c-fos m-RNA expression in an isolated rat lung model. J. Clin. Investig. 1997, 99, 944–952. [Google Scholar] [CrossRef]
- Roy, A.; Srivastava, M.; Saqib, U.; Liu, D.; Faisal, S.M.; Sugathan, S.; Bishnoi, S.; Baig, M.S. Potential therapeutic targets for inflammation in toll-like receptor 4 (TLR4)-mediated signaling pathways. Int. Immunopharmacol. 2016, 40, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Fanjul-Fernández, M.; Folgueras, A.R.; Cabrera, S.; López-Otín, C. Matrix metalloproteinases: Evolution, gene regulation and functional analysis in mouse models. Cell Res. 2010, 1803, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.K.; Potteti, H.R.; Tamatam, C.R.; Elangovan, I.; Reddy, S.P. c-Jun is required for nuclear factor-κB-dependent, LPS-stimulated Fos-related Antigen-1 transcription in alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 2016, 55, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Cahill, C.M.; Zhu, W.; Oziolor, E.; Yang, Y.-J.; Tam, B.; Rajanala, S.; Rogers, J.T.; Walker, W.A. Differential expression of the activator protein 1 transcription factor regulates interleukin-1β induction of interleukin 6 in the developing enterocyte. PLoS ONE 2016, 11, e0145184. [Google Scholar] [CrossRef]
- Kwon, D.-J.; Ju, S.M.; Youn, G.S.; Choi, S.Y.; Park, J. Suppression of iNOS and COX-2 expression by flavokawain A via blockade of NF-κB and AP-1 activation in RAW 264.7 macrophages. Food Chem. Toxicol. 2013, 58, 479–486. [Google Scholar] [CrossRef]
- Nepelska, M.; Cultrone, A.; Béguet-Crespel, F.; Le Roux, K.; Doré, J.; Arulampalam, V.; Blottière, H.M. Butyrate produced by commensal bacteria potentiates phorbol esters induced AP-1 response in human intestinal epithelial cells. PLoS ONE 2012, 7, e52869. [Google Scholar] [CrossRef]
- Wang, P.-Y.; Wang, S.R.; Xiao, L.; Chen, J.; Wang, J.-Y.; Rao, J.N. c-Jun enhances intestinal epithelial restitution after wounding by increasing phospholipase C-γ1 transcription. Am. J. Physiol. Cell Physiol. 2017, 312, C367–C375. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, Y.; Yamagata, K.; Sakuraba, H.; Munakata, A.; Nakane, A.; Morita, T.; Nishihira, J. Macrophage migration inhibitory factor and activator protein-1 in ulcerative colitis. Ann. N. Y. Acad. Sci. 2006, 1029, 348–349. [Google Scholar] [CrossRef]
- Zingarelli, B.; Yang, Z.; Hake, P.W.; Denenberg, A.; Wong, H.R. Absence of endogenous interleukin-10 enhances early stress response during postischemic injury in mice intestine. Gut 2001, 5, 610–622. [Google Scholar] [CrossRef]
- Nicolaou, F.; Teodoridis, J.M.; Park, H.; Georgakis, A.; Farokhzad, O.C.; Böttinger, E.P.; Da Silva, N.; Rousselot, P.; Chomienne, C.; Ferenczi, K.; et al. CD11c gene expression in hairy cell leukemia is dependent upon activation of the proto-oncogenes ras and junD. Blood 2003, 101, 4033–4041. [Google Scholar] [CrossRef]
- Moriyama, I.; Ishihara, S.; Rumi, M.A.; Aziz, M.D.; Mishima, Y.; Oshima, N.; Kadota, C.; Kadowaki, Y.; Amano, Y.; Kinoshita, Y. Decoy oligodeoxynucleotide targeting activator protein-1 (AP-1) attenuates intestinal inflammation in murine experimental colitis. Lab. Investig. 2008, 88, 652–663. [Google Scholar] [CrossRef] [PubMed]
- Boone, D.L.; Ma, A. Connecting the dots from Toll-like receptors to innate immune cells and inflammatory bowel disease. J. Clin. Investig. 2003, 111, 1284–1286. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, C.P.; Neal, M.D.; Siggers, R.; Sho, S.; Ma, C.; Branca, M.F.; Prindle, T., Jr.; Russo, A.M.; Afrazi, A.; Good, M.; et al. Intestinal epithelial Toll-like receptor 4 regulates goblet cell development and is required for necrotizing enterocolitis in mice. Gastroenterology 2012, 143, 708–718. [Google Scholar] [CrossRef]
- Youn, H.S.; Saitoh, S.I.; Miyake, K.; Hwang, D.H. Inhibition of homodimerization of Toll-like receptor 4 by curcumin. Biochem. Pharmacol. 2006, 72, 62–69. [Google Scholar] [CrossRef]
- Park, H.H.; Lo, Y.C.; Lin, S.C.; Wang, L.; Yang, J.K.; Wu, H. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu. Rev. Immunol. 2007, 25, 561–586. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, Y.; Ma, L.; Cao, X.; Xiao, J.; Chen, J.; Jiao, S.; Gao, Y.; Liu, C.; Duan, Z.; et al. Activation of vascular endothelial growth factor receptor-3 in macrophages restrains TLR4-NF-κB signaling and protects against endotoxin shock. Immunity 2014, 40, 501–514. [Google Scholar] [CrossRef]
- Li, L.; Liu, Y.; Chen, H.Z.; Li, F.W.; Wu, J.F.; Zhang, H.K.; He, J.P.; Xing, Y.Z.; Chen, Y.; Wang, W.J.; et al. Impeding the interaction between Nur77 and p38 reduces LPS-induced inflammation. Nat. Chem. Biol. 2015, 11, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Gohda, J.; Matsumura, T.; Inoue, J. Cutting edge: TNFR-associated factor (TRAF) 6 is essential for MyD88-dependent pathway but not toll/IL-1 receptor domain-containing adaptor-inducing IFN-beta (TRIF)-dependent pathway in TLR signaling. J. Immunol. 2004, 173, 2913–2917. [Google Scholar] [CrossRef] [PubMed]
- Neu, J. Gastrointestinal development and meeting the nutritional needs to premature infants. Am. J. Clin. Nutr. 2007, 85, 629S–634S. [Google Scholar] [CrossRef]
- Papillon, S.; Castle, S.L.; Gayer, C.P.; Ford, H.R. Necrotizing enterocolitis: Contemporary management and outcomes. Adv. Pediatr. 2013, 60, 263–279. [Google Scholar] [CrossRef] [PubMed]
- Niño, D.F.; Sodhi, C.P.; Hackam, D.J. Necrotizing enterocolitis: New insights into pathogenesis and mechanisms. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Battersby, A.J.; Gibbons, D.L. The gut mucosal immune system in the neonatal period. Ped. Allergy Immunol. 2013, 24, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Leaphart, C.L.; Cavallo, J.C.; Gribar, S.C.; Cetin, S.; Li, J.; Branca, M.F.; Dubowski, T.D.; Sodhi, C.P.; Hackam, D.J. A critical role for TLR4 in the pathogenesis of necrotizing enterocolitis by modulating intestinal injury and repair. J. Immunol. 2007, 179, 4808–4820. [Google Scholar] [CrossRef] [PubMed]
- Egan, C.E.; Sodhi, C.P.; Good, M.; Lin, J.; Jia, H.; Yamaguchi, Y.; Lu, P.; Ma, C.; Branca, M.F.; Weyandt, S.; et al. Toll-like receptor 4-mediated lymphocyte influx induces neonatal necrotizing enterocolitis. J. Clin. Investig. 2016, 126, 495–508. [Google Scholar] [CrossRef]
- Good, M.; Siggers, R.H.; Sodhi, C.P.; Afrazi, A.; Alkhudari, F.; Egan, C.E.; Neal, M.D.; Yazji, I.; Jia, H.; Lin, J.; et al. Amniotic fluid inhibits Toll-like receptor 4 signaling in the fetal and neonatal intestinal epithelium. Proc. Natl. Acad. Sci. USA 2012, 109, 11330–11335. [Google Scholar] [CrossRef]
- Good, M.; Sodhi, C.P.; Egan, C.E.; Afrazi, A.; Jia, H.; Yamaguchi, Y.; Lu, P.; Branca, M.F.; Ma, C.; Prindle, T., Jr.; et al. Breast milk protects against the development of necrotizing enterocolitis through inhibition of Toll-like receptor 4 in the intestinal epithelium via activation of the epidermal growth factor. Mucosal Immunol. 2015, 8, 1166–1179. [Google Scholar] [CrossRef]
- Neal, M.D.; Sodhi, C.P.; Dyer, M.; Craig, B.T.; Good, M.; Jia, H.; Yazji, I.; Afrazi, A.; Richardson, W.M.; Beer-Stolz, D.; et al. A critical role for TLR4 induction of autophagy in the regulation of enterocyte migration and the pathogenesis of necrotizing enterocolitis. J. Immunol. 2013, 190, 3541–3551. [Google Scholar] [CrossRef]
- Good, M.; Sodhi, C.P.; Yamaguchi, Y.; Jia, H.; Lu, P.; Fulton, W.B.; Martin, L.Y.; Prindle, T.; Nino, D.F.; Zhou, Q.; et al. The human milk oligosaccharide 2′-fucosyllactose attenuates the severity of experimental necrotising enterocolitis by enhancing mesenteric perfusion in the neonatal intestine. Br. J. Nutr. 2016, 116, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Toiyama, Y.; Araki, T.; Yoshiyama, S.; Hiro, J.; Miki, C.; Kusonoki, M. The expression patterns of toll-like receptors in the ileal pouch mucosa of postoperative ulcerative colitis patients. Surg. Today 2006, 36, 287–290. [Google Scholar] [CrossRef]
- Lu, P.; Sodhi, C.P.; Hackam, D.J. Toll-like receptor regulation of intestinal development and inflammation in the pathogenesis of necrotizing enterocolitis. Pathophysiology 2014, 21, 81–93. [Google Scholar] [CrossRef]
- Nanthakumar, N.N.; Meng, D.; Goldstein, A.M.; Zhu, W.; Lu, L.; Uauy, R.; Llanos, A.; Claud, E.C.; Walker, W.A. The mechanism of excessive intestinal inflammation in necrotizing enterocolitis: An immature innate immune response. PLoS ONE 2011, 6, e17776. [Google Scholar] [CrossRef]
- Fusunyan, R.D.; Nanthakumar, N.N.; Baldeon, M.E.; Walker, W.A. Evidence for an innate immune response in the immature human intestine: Toll-like receptors on fetal enterocytes. Pediatr. Res. 2001, 49, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Managlia, E.; Liu, S.X.L.; Yan, X.; Tan, X.-D.; Chou, P.M.; Barrett, T.A.; De Plaen, I.G. Blocking NF-κB activation in Ly6c+ monocytes attenuates necrotizing enterocolitis. Am. J. Pathol. 2019, 189, 604–618. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S.; Locati, M.; Mantovani, A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J. Immunol. 2006, 177, 7303–7311. [Google Scholar] [CrossRef] [PubMed]
- Yazji, I.; Sodhi, C.P.; Lee, E.K.; Good, M.; Egan, C.E.; Afrazi, A.; Neal, M.D.; Jia, H.; Lin, J.; Ma, C.; et al. Endothelial TLR4 activation impairs intestinal microcirculatory perfusion in necrotizing enterocolitis via eNOS-NO-nitrite signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 9451–9456. [Google Scholar] [CrossRef] [PubMed]
- Claud, E.C.; Lu, L.; Anton, P.M.; Savidge, T.; Walker, W.A.; Cherayil, B.J. Developmentallly regulated IkappaB expression in intestinal epithelium and susceptibility to flagellin-induced inflammation. Proc. Natl. Acad. Sci. USA 2004, 101, 7404–7408. [Google Scholar] [CrossRef]
- Nanthakumar, N.N.; Fusunyan, R.D.; Sanderson, I.; Walker, W.A. Inflammation in the developing human intestine: A possible pathophysiologic contribution to necrotizing enterocolitis. Proc. Natl. Acad. Sci. USA 2000, 97, 6043–6048. [Google Scholar] [CrossRef] [PubMed]
- Rentea, R.M.; Welak, S.R.; Fredrich, K.; Donohoe, D.; Pritchard, K.A.; Oldham, K.T.; Gourlay, D.M.; Liedel, J.L. Early enteral stressors in newborns increase inflammatory cytokine expression in a neonatal necrotizing enterocolitis rat model. Eur. J. Ped. Surg. 2013, 23, 39–47. [Google Scholar] [CrossRef][Green Version]
- Jones, C.A.; Holloway, J.A.; Warner, J.O. Phenotype of fetal monocytes and B lymphocytes during the third trimester of pregnancy. J. Reprod. Immunol. 2002, 56, 45–60. [Google Scholar] [CrossRef]
- Hackam, D.J.; Upperman, J.S.; Grishin, A.; Ford, H.R. Disordered enterocyte signaling and intestinal barrier dysfunction in the pathogenesis of necrotizing enterocolitis. Semin. Pediatr. Surg. 2005, 14, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.A.; Doelle, S.M.; Halpern, M.D.; Saunders, T.A.; Holubee, H.; Dvorak, D.; Boitano, S.A.; Dvorak, B. Intestinal barrier failure during experimental necrotizing enterocolitis: Protective effect of EGF treatment. Am. J. Physiol. 2006, 291, G938–G949. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.A.; Mount Patrick, S.K.; Estrada, T.E.; Frisk, H.A.; Rogan, D.T.; Dvorak, B.; Halpern, M.D. Active transport of bile acids decreases mucin 2 in neonatal ileum: Implications for development of necrotizing enterocolitis. PLoS ONE 2011, 6, e27191. [Google Scholar] [CrossRef]
- Nathan, C. Neutrophils and immunity: Challenges and opportunities. Nat. Rev. Immunol. 2006, 6, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Emami, C.N.; Mittal, R.; Wang, L.; Ford, H.R.; Prasadarao, N.V. Role of neutrophils and macrophages in the pathogenesis of necrotizing enterocolitis caused by Cronobacter sakazakii. J. Surg. Res. 2012, 172, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Musemeche, C.; Caplan, M.; Hsueh, W.; Sun, X.; Kelly, A. Experimental necrotizing enterocolitis: The role of polymorphonuclear neutrophils. J. Pediatr. Surg. 1991, 26, 1047–1049. [Google Scholar] [CrossRef]
- Christensen, R.D.; Yoder, B.A.; Baer, V.L.; Snow, G.L.; Butler, A. Early-onset neutropenia in small-for-gestational-age infants. Pediatrics 2015, 136, e1259–e1267. [Google Scholar] [CrossRef]
- Strunk, T.; Temming, P.; Gembruch, U.; Reiss, I.; Bucsky, P.; Schultz, C. Differential maturation of the innate immune response in human fetuses. Ped. Res. 2004, 56, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Maheshwari, A.; Kelly, D.R.; Nicola, T.; Ambalavanan, N.; Jain, S.K.; Murphy-Ullrich, J.; Athar, M.; Shimamura, M.; Bhandari, V.; Aprahamian, C.; et al. TFG-beta2 suppresses macrophage cytokine production and mucosal inflammatory responses in the developing intestine. Gastroenterology 2011, 140, 242–253. [Google Scholar] [CrossRef] [PubMed]
- MohanKumar, K.; Namachivayam, K.; Chapalamadugu, K.C.; Garzon, S.A.; Premkumar, M.H.; Tipparaju, S.M.; Maheshwari, A. Smad7 interrupts TGF-beta signaling in intestinal macrophages and promotes inflammatory activation of these cells during necrotizing enterocolitis. Pediatr. Res. 2016, 79, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Namachivayam, K.; Blanco, C.L.; MohanKumar, K.; Jagadeeswaran, R.; Vasquez, M.; McGill-Vargas, L.; Garzon, S.A.; Jain, S.K.; Gill, R.K.; Freitag, N.E.; et al. Smad7 inhibits autocrine expression of TGF-beta2 in intestinal epithelial cells in baboon necrotizing enterocolitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G167–G180. [Google Scholar] [CrossRef] [PubMed]
- MohanKumar, K.; Kaza, N.; Jagadeeswaran, R.; Garzon, S.A.; Bansal, A.; Kurundkar, A.R.; Namachivayam, K.; Remon, J.I.; Bandepalli, C.R.; Feng, X.; et al. Gut mucosal injury in neonates is marked by macrophage infiltration in contrast to pleomorphic infiltrates in adult: Evidence from an animal model. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G93–G102. [Google Scholar] [CrossRef] [PubMed]
- Emami, C.N.; Mittal, R.; Wang, L.; Ford, H.R.; Prasadarao, N.V. Recruitment of dendritic cells is responsible for intestinal epithelial damage in the pathogenesis of necrotizing enterocolitis by Cronobacter sakazakii. J. Immunol. 2011, 186, 7067–7079. [Google Scholar] [CrossRef] [PubMed]
- Weitkamp, J.H.; Rosen, M.J.; Zhao, Z.; Koyama, T.; Geem, D.; Denning, T.L.; Rock, M.T.; Moore, D.J.; Halpern, M.D.; Matta, P.; et al. Small intestinal intraepithelial TCRgammadelta+ T lymphocytes are present in the premature intestine but selectively reduced in surgical necrotizing enterocolitis. PLoS ONE 2014, 9, e99042. [Google Scholar] [CrossRef]
- Gibbons, D.L.; Haque, S.F.; Silberzahn, T.; Hamilton, K.; Langford, C.; Ellis, P.; Carr, R.; Hayday, A.C. Neonates harbour highly active gammadelta T cells with selective impairments in preterm infants. Eur. J. Immunol. 2009, 39, 1794–1806. [Google Scholar] [CrossRef] [PubMed]
- Ismail, A.S.; Behrendt, C.L.; Hooper, L.V. Reciprocal interactions between commensal bacteria and gamma delta intraepithelial lymphocytes during mucosal injury. J. Immunol. 2009, 182, 3047–3054. [Google Scholar] [CrossRef]
- Weitkamp, J.H.; Koyama, T.; Rock, M.T.; Correa, H.; Goettel, J.A.; Matta, P.; Oswald-Richter, K.; Rosen, M.J.; Engelhardt, B.G.; Moore, D.J.; et al. Necrotising enterocolitis is characterised by disrupted immune regulation and diminished mucosal regulatory (FOXP3)/effector (CD4, CD8) T cell ratios. Gut 2013, 62, 73–82. [Google Scholar] [CrossRef]
- Basha, S.; Surendran, N.; Pichichero, M. Immune responses in neonates. Exp. Rev. Clin. Immunol. 2014, 10, 1171–1184. [Google Scholar] [CrossRef] [PubMed]
- Markel, T.A.; Crisostomo, P.R.; Wairiuko, G.M.; Pitcher, J.; Tsai, B.M.; Meldrum, D.R. Cytokines in necrotizing enterocolitis. Shock 2006, 25, 329–337. [Google Scholar] [CrossRef]
- Kling, K.M.; Kirby, L.; Kwan, K.Y.; Kim, F.; McFadden, D.W. Interleukin-10 inhibits inducible nitric oxide synthase in an animal model of necrotizing enterocolitis. Int. J. Surg. Investig. 1999, 1, 337–342. [Google Scholar] [PubMed]
- Baud, V.; Karin, M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001, 11, 372–377. [Google Scholar] [CrossRef]
- Tracey, K.J.; Fong, Y.; Hesse, D.G.; Manogue, K.R.; Lee, A.T.; Kuo, G.C.; Lowry, S.F.; Cerami, A. Anti-cachectin/THF monoclonal antibodies prevent septic shock during lethal bacteremia. Nature 1987, 330, 662–664. [Google Scholar] [CrossRef]
- Baregamian, N.; Song, J.; Bailey, C.E.; Papaconstantinou, J.; Evers, B.M.; Chung, D.H. Tumor necrosis factor-alpha and apoptosis signal-regulating kinase 1 control reactive oxygen species release, mitochondrial autophagy, and C-Jun N-terminal kinase/p38 phosphorylation during necrotizing enterocolitis. Oxid. Med. Cell. Longev. 2009, 2, 297–306. [Google Scholar] [CrossRef]
- Caplan, M.S.; Hsueh, W. Necrotizing enterocolitis: Role of platelet activating factor, endotoxin, and tumor necrosis factor. J. Pediatr. 1990, 117, S47–S51. [Google Scholar] [CrossRef]
- Harris, M.C.; Costarino, A.T., Jr.; Sullivan, J.S.; Dulkerian, S.; McCawley, L.; Corcoran, L.; Butler, S.; Kilpatrick, L. Cytokine elevations in critically ill infants with sepsis and necrotizing enterocolitis. J. Pediatr. 1994, 124, 105–111. [Google Scholar] [CrossRef]
- Morecroft, J.A.; Spitz, L.; Hamilton, P.A.; Holmes, S.J. Plasma cytokine levels in necrotizing enterocolitis. Acta Paediatr. Suppl. 1994, 396, 18–20. [Google Scholar] [CrossRef]
- Churg, A.; Wang, R.D.; Tai, H.; Wang, X.; Xie, C.; Dai, J.; Shapiro, S.D.; Wright, J.L. Macrophage metalloelastase mediates acute cigarette smoke-induced inflammation via tumor necrosis factor-alpha release. Am. J. Respir. Crit. Care Med. 2003, 167, 1083–1089. [Google Scholar] [CrossRef]
- Saren, P.; Welgus, H.G.; Kovanen, P.T. TNF-α and IL-1β selectively induce expression of 92 kDa gelatinase by human macrophages. J. Immunol. 1996, 157, 4159–4165. [Google Scholar] [PubMed]
- Impola, U.; Toriseva, M.; Suomela, S.; Jeskanen, L.; Hieta, N.; Jahkola, T.; Grenman, R.; Kähäri, V.; Saarialho-Kere, U. Matrix metalloproteinase-19 is expressed by proliferating epithelium but disappears with neoplastic dedifferentiation. Int. J. Cancer 2003, 103, 709–716. [Google Scholar] [CrossRef]
- Pender, S.L.; Braegger, C.; Gunther, C.; Monteleone, G.; Meuli, M.; Schuppan, D.; MacDonald, T.T. Matrix metalloproteinases in necrotising enterocolitis. Pediatr. Res. 2003, 54, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Birkedal-Hansen, H.; Moore, W.G.; Bodden, M.K.; Windsor, L.J.; Birkedal-Hansen, B.; DeCarlo, A.; Engler, J.A. Matrix metalloproteinases: A review. Crit. Rev. Oral Biol. Med. 1993, 4, 197–250. [Google Scholar] [CrossRef]
- Male, D. Immunology: An Illustrated Outline, 3rd ed.; Mosby: London, UK, 1998; pp. 1–146. [Google Scholar]
- Minekawa, R.; Takeda, T.; Sakata, M.; Hayashi, M.; Isobe, A.; Yamamoto, T.; Tasaka, K.; Murata, Y. Human breast milk suppresses the transcriptional regulation of IL-1β induced NF-κB signaling in human intestinal cells. Am. J. Physiol. Cell Physiol. 2004, 287, C1401–C1411. [Google Scholar] [CrossRef] [PubMed]
- Viscardi, R.M.; Lyon, N.H.; Sun, C.C.; Hebel, J.R.; Hasday, J.D. Inflammatory cytokine mRNAs in surgical specimens of necrotizing enterocolitis and normal newborn intestine. Pediatr. Pathol. Lab. Med. 1997, 17, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Edelson, M.B.; Bagwell, C.E.; Rozycki, H.J. Circulating pro- and counterinflammatory cytokine levels and severity in necrotizing enterocolitis. Pediatrics 1999, 103, 766–771. [Google Scholar] [CrossRef]
- Romagnoli, C.; Freeza, S.; Cingolani, A.; De Luca, A.; Puopolo, M.; De Carolis, M.P.; Vento, G.; Antinori, A.; Tortorolo, G. Plasma levels of interleukin-6 and interleukin-10 in preterm neonates evaluated for sepsis. Eur. J. Pediatr. 2001, 160, 345–350. [Google Scholar] [CrossRef]
- Ren, Y.; Lin, C.L.; Chen, X.Y.; Huang, X.; Lui, V.; Nicholls, J.; Lan, H.Y.; Tam, P.K. Up-regulation of macrophage migration inhibitory factor in infants with acute neonatal necrotizing enterocolitis. Histopathology 2005, 46, 659–667. [Google Scholar] [CrossRef]
- Goepfert, A.R.; Andrews, W.W.; Waldemar, C.; Ramsey, P.S.; Cliver, S.P.; Goldenber, R.L.; Hauth, J.C. Umbilical cord plasma interleukin-6 concentrations in preterm infants and risk of neonatal morbidity. Am. J. Obstet. Gynecol. 2004, 191, 1375–1381. [Google Scholar] [CrossRef]
- Luster, A.D. Chemokines—Chemotactic cytokines that mediate inflammation. N. Engl. J. Med. 1998, 338, 436–445. [Google Scholar] [CrossRef]
- Chikano, S.; Sawada, K.; Shimoyama, T.; Kashiwamura, S.I.; Sugihara, A.; Sekikawa, K.; Terada, N.; Nakanishi, K.; Okamura, H. IL-18 and IL-12 induce intestinal inflammation and fatty liver in mice in an IFN-gamma dependent manner. Gut 2000, 47, 779–786. [Google Scholar] [CrossRef]
- Kashiwamura, S.; Ueda, H.; Okamura, H. Roles of interleukin-18 in tissue destruction and compensatory reactions. J. Immunol. 2002, 25 (Suppl. 1), S4–S11. [Google Scholar] [CrossRef]
- Halpern, M.D.; Holubec, H.; Dominguez, J.A.; Williams, C.S.; Meza, Y.G.; McWilliam, D.L.; Payne, C.M.; McCuskey, R.S.; Besselsen, D.G.; Dvorak, B. Upregulation of IL-18 and Il-12 in the ileum of neonatal rats with necrotizing enterocolitis. Pediatr. Res. 2002, 51, 733–739. [Google Scholar] [CrossRef]
- Nadler, E.P.; Dickinson, E.; Knisely, A.; Zhang, X.R.; Boyle, P.; Beer-Stolz, D.; Watkins, S.C.; Ford, H.R. Expression of inducible nitric oxide synthase and interleukin 12 in experimental necrotizing enterocolitis. J. Surg. Res. 2000, 92, 71–77. [Google Scholar] [CrossRef]
- Ford, H.R.; Sorrells, D.L.; Knisely, A.S. Inflammatory cytokines, nitric oxide, and necrotizing enterocolitis. Semin. Pediatr. Surg. 1996, 5, 155–159. [Google Scholar]
- Benkoe, T.; Baumann, S.; Weninger, M.; Pones, M.; Reck, C.; Rebhandl, W.; Oehler, R. Comprehensive evaluation of 11 cytokines in premature infants with surgical necrotizing enterocolitis. PLoS ONE 2013, 8, e58720. [Google Scholar] [CrossRef]
- Maheshwari, A.; Schelonka, R.L.; Dimmitt, R.A.; Carlo, W.A.; Munoz-Hernandez, B.; Das, A.; McDonald, S.A.; Thorsen, P.; Skogstrand, K.; Hougaard, D.M.; et al. Cytokines associated with necrotizing enterocolitis in extremely-low-birth-weight infants. Pediatr. Res. 2014, 76, 100–108. [Google Scholar] [CrossRef]
- Fiocchi, C. Inflammatory bowel disease: Etiology and pathogenesis. Gastroenterology 1998, 115, 182–205. [Google Scholar] [CrossRef]
- Uhlig, H.H.; Powrie, F. Translating immunology into therapeutic concepts for inflammatory bowel disease. Annu. Rev. Immunol. 2018, 36, 755–781. [Google Scholar] [CrossRef]
- Zeissig, S.; Bürgel, N.; Günzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Changes in expression and distribution of claudins 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Vezza, T.; Rodríguez-Nogales, A.; Algieri, F.; Utrilla, M.P.; Rodriguez-Cabezas, M.E.; Galvez, J. Flavonoids in inflammatory bowel disease: A review. Nutrients 2016, 8, 211. [Google Scholar] [CrossRef]
- Cao, Y.; Shen, J.; Ran, Z.H. Association between Faecalibacterium prausnitzii reduction and inflammatory bowel disease: A meta-analyis and systematic review of the literature. Gastroenterol. Res. Pract. 2014, 2014, 872725. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Kolodziejezyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.R.; Turner, J.R. Inflammatory bowel disease: Is it really just another break in the wall? Gut 2007, 56, 6–8. [Google Scholar] [CrossRef] [PubMed]
- Hollander, D. Crohn’s disease—A permeability disorder of the tight junction? Gut 1988, 29, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef]
- Arnott, I.D.; Kingstone, K.; Ghosh, S. Abnormal intestinal permeability predicts relapse in inactive Crohn disease. Scand. J. Gastroenterol. 2000, 35, 1163–1170. [Google Scholar]
- Wyatt, J.; Vogelsang, H.; Hubl, W.; Waldhoer, T.; Lochs, H. Intestinal permeability and the prediction of relapse in Crohn’s disease. Lancet 1993, 341, 1437–1439. [Google Scholar] [CrossRef]
- Katz, K.D.; Hollander, D.; Vadheim, C.M.; McElree, C.; Delahunty, T.; Dadufalza, V.D.; Krugliak, P.; Rotter, J.I. Intestinal permeability in patients with Crohn’s disease and their healthy relatives. Gastroenterology 1989, 97, 927–931. [Google Scholar] [CrossRef]
- Peeters, M.; Geypens, B.; Claus, D.; Nevens, H.; Ghoos, Y.; Verbeke, G.; Baert, F.; Vermeire, S.; Vlietinck, R.; Rutgeerts, P. Clustering of increased small intestinal permeability in families with Crohn’s disease. Gastroenterology 1997, 113, 802–807. [Google Scholar] [CrossRef]
- Smith, P.D.; Smythies, L.E.; Shen, R.; Greenwell-Wild, T.; Gliozzi, M.; Wahl, S.M. Intestinal macrophages and response to microbial environment. Mucosal Immunol. 2011, 4, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.; Heinig, T.; Panzer, U.; Reinking, R.; Bouchard, A.; Stahl, P.D.; Raedler, A. Impaired response of activated mononuclear phagocytes to interleukin-4 in inflammatory bowel disease. Gastroenterology 1995, 108, 21–33. [Google Scholar] [CrossRef]
- Podolsky, D.K. Inflammatory bowel disease. N. Engl. J. Med. 2002, 347, 417–429. [Google Scholar] [CrossRef]
- Holtmann, M.H.; Neurath, M.F. Differential TNF-signaling in chronic inflammatory disorders. Curr. Mol. Med. 2004, 4, 439–444. [Google Scholar] [CrossRef]
- Sartor, R.B. Mechanisms of disease: Pathogenesis of Crohn’s disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 390–407. [Google Scholar] [CrossRef]
- Shih, D.Q.; Targan, S.R. Insights into IBD pathogenesis. Curr. Gastroenterol. Rep. 2009, 11, 473–480. [Google Scholar] [CrossRef]
- Strober, W.; Fuss, I.; Mannon, P. The fundamental basis of inflammatory bowel disease. J. Clin. Investig. 2007, 117, 514–521. [Google Scholar] [CrossRef]
- Cerovic, V.; Houston, S.A.; Scott, C.L.; Aumeunier, A.; Yrlid, U.; Mowat, A.M.; Milling, S.W. Intestinal cd103(-) dendritic cells migrate in lymph and prime effector T cells. Mucosal Immunol. 2013, 6, 104–113. [Google Scholar] [CrossRef]
- Laing, K.J.; Secombes, C.J. Chemokines. Dev. Comp. Immunol. 2004, 28, 443–460. [Google Scholar] [CrossRef]
- Kim, J.M.; Jung, H.Y.; Lee, J.Y.; Youn, J.; Lee, C.H.; Kim, K.H. Mitogen-activated protein kinase and activator protein-1 dependent signals are essential for Bacteroides fragilis enterotoxin-induced enteritis. Eur. J. Immunol. 2005, 35, 2648–2657. [Google Scholar] [CrossRef] [PubMed]
- Vainer, B. Intercellular adhesion molecule-1 (ICAM-1) in ulcerative colitis: Presence, visualization, and significance. APMIS Suppl. 2010, 129, 1–43. [Google Scholar] [CrossRef]
- Steidler, L.; Hans, W.; Schotte, L.; Neirynck, S.; Obermeier, F.; Falk, W.; Fiers, W.; Remaut, E. Treatment of murine colitis by Lactococcus lactis secreting interleukin-10. Science 2000, 289, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Kitani, A.; Fuss, U.; Nakamura, K.; Schwartz, O.M.; Usui, T.; Strober, W. Treatment of experimental (trinitrobenzene sulfonic acid) colitis by intranasal administration of transforming growth factor (TGF)-β1 plasmid: TGF-β1-mediated suppression of T helper cell type 1 response occurs by interleukin (IL)-10 induction and IL-12 receptor β2 chain downregulation. J. Exp. Med. 2000, 192, 41–52. [Google Scholar] [PubMed]
- Fukata, M.; Chen, A.; Vamadevan, A.S.; Cohen, J.; Breglio, K.; Krishnareddy, S.; Hsu, D.; Xu, R.; Harpaz, N.; Dannenberg, A.J.; et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology 2007, 133, 1869–1881. [Google Scholar] [CrossRef] [PubMed]
- Vlantis, K.; Wullaert, A.; Sasaki, Y.; Schmidt-Supprian, M.; Rajewsky, K.; Roskams, T.; Pasparakis, M. Constitutive IKK2 activation in intestinal epithelial cells induces intestinal tumors in mice. J. Clin. Investig. 2011, 121, 2781–2793. [Google Scholar] [CrossRef]
- Cario, E.; Podolsky, D.K. Differential alteration in intestinal epithelial cells expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect. Immun. 2000, 68, 7010–7017. [Google Scholar] [CrossRef] [PubMed]
- Ortegea-Cava, C.F.; Ishihara, S.; Rumi, M.A.; Kawashima, K.; Ishimura, N.; Kazumori, H.; Udagawa, J.; Kadowaki, Y.; Kinoshita, Y. Strategic compartmentalization of Toll-like receptor 4 in the mouse gut. J. Immunol. 2003, 170, 3977–3985. [Google Scholar] [CrossRef]
- Rogler, G.; Brand, K.; Vogl, D.; Page, S.; Hofmeister, R.; Andus, T.; Knuechel, R.; Baeuerle, P.A.; Scholmerich, J.; Gross, V. Nuclear factor kappaB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology 1998, 115, 357–369. [Google Scholar] [CrossRef]
- Neurath, M.F.; Petterson, S.; Meyer zum Büschenfelde, K.H.; Strober, W. Local administration of antisense phosphorothioate oligonucleotides to the p65 subunit of NF-κB abrogates established experimental colitis in mice. Nat. Med. 1996, 2, 998–1004. [Google Scholar] [CrossRef]
- Waetzig, G.H.; Seegert, D.; Rosenstiel, P.; Nikolaus, S.; Schreiber, S. p38 mitogen-activated protein kinase is activated and linked to TNF-alpha signaling in inflammatory bowel disease. J. Immunol. 2002, 168, 5342–5351. [Google Scholar] [CrossRef]
- Abreu-Martin, M.T.; Palladino, A.A.; Faris, M.; Carramanzana, N.M.; Nel, A.E.; Targan, S.R. Fas activates the JNK pathway in human colonic epithelial cells: Lack of a direct role in apoptosis. Am. J. Physiol. 1999, 276, G599–G605. [Google Scholar] [CrossRef] [PubMed]
- Bantel, H.; Schmitz, M.L.; Raible, A.; Gregor, M.; Schulze-Osthoff, K. Critical role of NF-κB and stress-activated protein kinases in steroid unresponsiveness. FASEB J. 2002, 16, 1832–1834. [Google Scholar] [CrossRef]
- Salzman, N.H.; Underwood, M.A.; Bevins, C.L. Paneth cells, defensins, and the commensal microbiota: A hypothesis on intimate interplay at the intestinal mucosa. Semin. Immunol. 2007, 19, 70–83. [Google Scholar] [CrossRef]
- Iida, T.; Onodera, K.; Nakase, H. Role of autophagy in the pathogenesis of inflammatory bowel disease. World J. Gastroenterol. 2017, 23, 1944–1953. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, K.R.; Crockard, A.D.; Halliday, M.I.; Rowlands, B.J. Class II major histocompatibility complex antigen expression on peripheral blood monocytes in patients with inflammatory bowel disease. Gut 1994, 35, 511–516. [Google Scholar] [CrossRef] [PubMed][Green Version]
- MacDonald, T.T.; Monteleone, G.; Pender, S.L.F. Recent developments in the immunology of inflammatory bowel disease. Scand. J. Immunol. 2000, 51, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Atreya, R.; Mudter, J.; Finotto, S.; Müllberg, J.; Jostock, T.; Wirtz, S.; Schütz, M.; Bartsch, B.; Holtmann, M.; Becker, C.; et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: Evidence in Crohn’s disease and experimental colitis in vivo. Nat. Med. 2000, 6, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Fuss, I.J.; Neurath, M.; Boirivant, M.; Klein, J.S.; de la Motte, C.; Strong, S.A.; Fiocchi, C.; Strober, W. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J. Immunol. 1996, 157, 1261–1270. [Google Scholar]
- Parronchi, P.; Romagnani, P.; Annunziato, F.; Sampognaro, S.; Becchio, A.; Giannarini, L.; Maggi, E.; Pupilli, C.; Tonelli, F.; Romagnani, S. Type 1 T-helper cell predominance and interleukin-12 expression in the gut of patients with Crohn’s disease. Am. J. Pathol. 1997, 150, 823–832. [Google Scholar] [PubMed]
- Romagnani, S. Lymphokine production by human T cells in disease states. Annu. Rev. Immunol. 1994, 12, 227–257. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimpour, S.; Shahbazi, M.; Khalili, A.; Tahoori, M.T.; Zavaran Hosseini, A.; Amari, A.; Aghili, B.; Abediankenari, S.; Mohammadizad, H.; Mohammadnia-Afrouzi, M. Elevated levels of IL-2 and IL-21 produced by CD4+ T cells in inflammatory bowel disease. J. Biol. Regul. Homeost. Agents 2017, 31, 279–287. [Google Scholar]
- Breese, E.; Braegger, C.P.; Corrigan, C.J.; Walker-Smith, J.A.; MacDonald, T.T. Interleukin-2 and interferon-γ secreting T cells in normal and diseased human intestinal mucosa. Immunology 1993, 78, 127–131. [Google Scholar] [PubMed]
- Mariani, P.; Bachetoni, A.; D’Alessandro, M.; Lomanto, D.; Mazzocchi, P.; Speranza, V. Effector Th-1 cells with cytotoxic function in the intestinal lamina propria of patients with Crohn’s disease. Dig. Dis. Sci. 2000, 45, 2029–2035. [Google Scholar] [CrossRef] [PubMed]
- Fuss, I.J.; Marth, T.; Neurath, M.F.; Pearlstein, G.R.; Jain, A.; Strober, W. Anti-interleukin 12 treatment regulates apoptosis of Th1 cells in experimental colitis in mice. Gastroenterology 1999, 117, 1078–1088. [Google Scholar] [CrossRef]
- Stockinger, B.; Veldhoen, M. Differentiation and function of Th17 T cells. Curr. Opin. Immunol. 2007, 19, 281–286. [Google Scholar] [CrossRef]
- Schmidt, C.; Giese, T.; Ludwig, B.; Mueller-Molaian, I.; Marth, T.; Zeuzem, S.; Meuer, S.C.; Stallmach, A. Expression of interleukin-12-related cytokine transcripts in inflammatory bowel disease: Elevated interleukin-23p19 and interleukin-27p28 in Crohn’s disease but not in ulcerative colitis. Inflamm. Bowel Dis. 2005, 11, 16–23. [Google Scholar] [CrossRef]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef]
- O’Connor, W., Jr.; Kamanaka, M.; Booth, C.J.; Town, T.; Nakae, S.; Iwakura, Y.; Kolls, J.K.; Flavell, R.A. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat. Immunol. 2009, 10, 603–609. [Google Scholar] [CrossRef]
- Song, X.; Dai, D.; He, X.; Zhu, S.; Yao, Y.; Gao, H.; Wang, J.; Qu, F.; Qiu, J.; Wang, H.; et al. Growth factor FGF2 cooperates with interleukin-17 to repair intestinal epithelial damage. Immunity 2015, 43, 488–501. [Google Scholar] [CrossRef]
- Yen, D.; Cheung, J.; Scheerens, H.; Poulet, F.; McClanahan, T.; McKenzie, B.; Kleinschek, M.A.; Owyang, A.; Mattson, J.; Blumenschein, W.; et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Investig. 2006, 116, 1310–1316. [Google Scholar] [CrossRef] [PubMed]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Mazieiro, R.; Frizon, R.R.; Barbalho, S.M.; de Alvares Goulart, R. Is curcumin a possibility to treat inflammatory bowel diseases? J. Med. Food 2018, 21, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Bamias, G.; Cominelli, F. Role of Th2 immunity in intestinal inflammation. Curr. Opin. Gastroenterol. 2015, 31, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Shohan, M.; Elahi, S.; Shirzad, H.; Rafieian-Kopaei, M.; Bagheri, N.; Soltani, E. Th9 cells: Probably players in ulcerative colitis pathogenesis. Int. Rev. Immunol. 2018, 37, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Fina, D.; Caruso, R.; Pallone, F.; Monteleone, G. Interleukin-21 (IL-21) controls inflammatory pathways in the gut. Endocr. Metab. Immune Disord. Drug Targets 2007, 7, 288–291. [Google Scholar] [CrossRef]
- Rivas, M.N.; Koh, Y.T.; Chen, A.; Nguyen, A.; Lee, Y.H.; Lawson, G.; Chatila, T.A. MyD88 is critically involved in immune tolerance breakdown at environmental interfaces of Foxp3-deficient mice. J. Clin. Investig. 2012, 122, 1933–1947. [Google Scholar] [CrossRef]
- Mayne, C.G.; Williams, C.B. Induced and natural regulatory T cells in the development of inflammatory bowel disease. Inflamm. Bowel Dis. 2013, 19, 1772–1788. [Google Scholar] [CrossRef]
- De Souza, H.S.; Fiocchi, C. Immunopathogenesis of IBD: Current state of the art. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 13–27. [Google Scholar] [CrossRef]
- Veltkamp, C.; Anstaett, M.; Wahl, K.; Möller, S.; Gangl, S.; Bachmann, O.; Hardtke-Wolenski, M.; Länger, F.; Stremmel, W.; Manns, M.P.; et al. Apoptosis of regulatory T lymphocytes is increased in chronic inflammatory bowel disease and reversed by anti-TNFα treatment. Gut 2011, 60, 1345–1353. [Google Scholar] [CrossRef]
- Liu, Z.; Geboes, K.; Colpaert, S.; Overbergh, L.; Mathieu, C.; Heremans, H.; de Boer, M.; Boon, L.; D’Haens, G.; Rutgeerts, P.; et al. Prevention of experimental colitis in SCID mice reconstituted with CD45RBhigh CD4+ T cells by blocking the CD40-CD154 interactions. J. Immunol. 2000, 164, 6005–6014. [Google Scholar] [CrossRef]
- Harrison, O.J.; Srinivasan, N.; Pott, J.; Schiering, C.; Krausgruber, T.; Ilott, N.E.; Maloy, K.J. Epithelial-derived IL-18 regulates Th17 cell differentiation and Foxp3+ Treg cell function in the intestine. Mucosal Immunol. 2015, 8, 1226–12236. [Google Scholar] [CrossRef]
- Nowarski, R.; Jackson, R.; Gagliani, N.; de Zoete, M.R.; Palm, N.W.; Bailis, W.; Low, J.S.; Harman, C.C.; Graham, M.; Elinav, E.; et al. Epithelial IL-18 equilibrium controls barrier function in colitis. Cell 2015, 163, 1444–14456. [Google Scholar] [CrossRef]
- Ludwiczek, O.; Kaser, A.; Novick, D.; Dinarello, C.A.; Rubinstein, M.; Tilg, H. Elevated systemic levels of free interleukin-18 (IL-18) in patients with Crohn’s disease. Eur. Cytokine Netw. 2005, 16, 27–33. [Google Scholar]
- Artis, D.; Spits, H. The biology of innate lymphoid cells. Nature 2015, 517, 293–301. [Google Scholar] [CrossRef]
- Elemam, N.M.; Hannawi, S.; Maghazachi, A.A. Innate lymphoid cells (ILCs) as mediators of inflammation, release of cytokines and lytic molecules. Toxins 2017, 9, 398. [Google Scholar] [CrossRef]
- Bernink, J.H.; Peters, C.P.; Munneke, M.; te Velde, A.A.; Meijer, S.L.; Weijer, K.; Hreggvidsdottir, H.S.; Heinsbroek, S.E.; Legrand, N.; Buskens, C.J.; et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 2013, 14, 221–229. [Google Scholar] [CrossRef]
- Fuchs, A.; Vermi, W.; Lee, J.S.; Lonardi, S.; Gilfillan, S.; Newberry, R.D.; Cella, M.; Colonna, M. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity 2013, 38, 769–781. [Google Scholar] [CrossRef]
- Sugimoto, K.; Ogawa, A.; Mizoguchi, E.; Shimomura, Y.; Andoh, A.; Bhan, A.K.; Blumberg, R.S.; Xavier, R.J.; Mizoguchi, A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Investig. 2008, 118, 534–544. [Google Scholar] [CrossRef]
- Zenewicz, L.A.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.J.; Stevens, S.; Flavell, R.A. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 2008, 29, 947–957. [Google Scholar] [CrossRef]
- Pelczar, P.; Witkowski, M.; Perez, L.G.; Kempski, J.; Hammel, A.G.; Brockmann, L.; Kleinschmidt, D.; Wende, S.; Haueis, C.; Bedke, T.; et al. A pathogenic role for T cell-derived IL-22BP in inflammatory bowel disease. Science 2016, 354, 358–362. [Google Scholar] [CrossRef]
- Van Deventer, S.J.H. Tumour necrosis factor and Crohn’s disease. Gut 1997, 40, 443–448. [Google Scholar] [CrossRef]
- Keates, A.C.; Castagnuolo, I.; Crickshank, W.W.; Qiu, B.; Arseneau, K.O.; Brazer, W.; Kelly, C.P. Interleukin 16 is upregulated in Crohn’s disease and participates in TNBS colitis in mice. Gastroenterology 2000, 119, 972–982. [Google Scholar] [CrossRef]
- Prasad, S.; Mingrino, R.; Kaukinen, K.; Hayes, K.L.; Powell, R.M.; MacDonald, T.T.; Collins, J.E. Inflammatory processes have differential effects on claudins 2, 3 and 4 in colonic epithelial cells. Lab. Investig. 2005, 85, 1139–1162. [Google Scholar] [CrossRef]
- McCabe, R.P.; Secrist, H.; Botney, M.; Egan, M.; Peters, M.G. Cytokine mRNA expression in intestine from normal and inflammatory bowel disease patients. Clin. Immunol. Immunopathol. 1993, 66, 52–58. [Google Scholar] [CrossRef]
- Nakamura, M.; Saito, H.; Kasanuki, J.; Tamura, Y.; Yoshida, S. Cytokine production in patients with inflammatory bowel disease. Gut 1992, 33, 55–58. [Google Scholar] [CrossRef]
- Arend, W.P. The balance between IL-1 and IL-1Ra in disease. Cytokine Growth Factor Rev. 2002, 13, 323–340. [Google Scholar] [CrossRef]
- Maeda, S.; Ohno, K.; Nakamura, K.; Uchida, K.; Nakashima, K.; Fukushima, K.; Tsukamoto, A.; Goto-Koshino, Y.; Fujino, Y.; Tsujimoto, H. Mucosal imbalance of interleukin-1β and interleukin-1 receptor antagonist in canine inflammatory bowel disease. Vet. J. 2012, 194, 66–70. [Google Scholar] [CrossRef]
- Casini-Raggi, V.; Kam, L.; Chong, Y.J.T.; Fiocchi, C.; Pizarro, T.T.; Cominelli, F. Mucosal imbalance of IL-1 and IL-1 receptor antagonist in inflammatory bowel disease. A novel mechanism of chronic intestinal inflammation. J. Immunol. 1995, 154, 2434–2440. [Google Scholar]
- Ngoh, E.N.; Weisser, S.B.; Lo, Y.; Kozicky, L.K.; Jen, R.; Brugger, H.K.; Menzies, S.C.; McLarren, K.W.; Nackiewicz, D.; van Rooijen, N.; et al. Activity of SHIP, which prevents expression of interleukin Iβ, is reduced in patients with Crohn’s disease. Gastroenterology 2016, 150, 465–476. [Google Scholar] [CrossRef]
- MacDonald, T.T.; Bajaj-Elliott, M.; Pender, S.L.F. T cells orchestrate intestinal mucosal shape and integrity. Immunol. Today 1999, 20, 505–510. [Google Scholar] [CrossRef]
- Louis, E.; Ribberns, C.; Godon, A.; I Franchimont, D.; De Groote, D.; Hardy, N.; Boniver, J.; Belaiche, J.; Malaise, M. Increased production of matrix metalloproteinase-3 and tissue inhibitor of metalloproteinase-1 by inflamed mucosa in inflammatory bowel disease. Clin. Exp. Immunol. 2000, 120, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Heuschkel, R.B.; MacDonald, T.T.; Monteleone, G.; Bajaj-Elliott, M.; Smith, J.A.; Pender, S.L. Imbalance of stromelysin-1 and TIMP-1 in the mucosal lesions of children with inflammatory bowel disease. Gut 2000, 47, 57–62. [Google Scholar] [CrossRef][Green Version]
- Saarialho-Kere, U.; Vaalamo, M.; Puolakkainen, P.; Airola, K.; Parks, W.C.; Karjalainen-Lindsberg, M.L. Enhanced expression of matrilysin, collagenase, and stromelysin-1 in gastrointestinal ulcers. Am. J. Pathol. 1996, 148, 519–526. [Google Scholar]
- Monteleone, G.; Kumberova, A.; Croft, N.M.; McKenzie, C.; Steer, H.W.; MacDonald, T.T. Blocking Smad7 restores TGF-β1 signaling in chronic inflammatory bowel disease. J. Clin. Investig. 2001, 108, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Heller, F.; Florian, P.; Bojarski, C.; Richter, J.; Christ, M.; Hillenbrand, B.; Mankertz, J.; Gitter, A.H.; Bürgel, N.; Fromm, M.; et al. Interleukin-13 is the key effector th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 2005, 129, 550–564. [Google Scholar] [CrossRef] [PubMed]
- Pari, L.; Tewas, D.; Eckel, J. Role of curcumin in health and disease. Arch. Physiol. Biochem. 2008, 114, 127–149. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Patchva, S.; Aggarwal, B.B. Therapeutic roles of curcumin: Lessons learned from clinical trials. AAPS J. 2013, 15, 195–218. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.T.; Vaughn, A.R.; Sharma, V.; Chopra, D.; Mills, P.J.; Peterson, S.N.; Sivamani, R.K. Effects of turmeric and curcumin dietary supplementation on human gut microbiota: A double-blind, randomized, placebo-controlled pilot study. J. Evid. Based Integr. Med. 2018, 23, 1–8. [Google Scholar] [CrossRef]
- Lao, C.D.; Ruffin, M.T.; Normolle, D.; Heath, D.D.; Murray, S.I.; Bailey, J.M.; Boggs, M.E.; Crowell, J.; Rock, C.L.; Brenner, D.E. Dose escalation of a curcuminoid formulation. BMC Complement. Altern. Med. 2006, 6, 10. [Google Scholar] [CrossRef]
- De, R.; Kundu, P.; Swarnakar, S.; Ramamurthy, T.; Chowdhury, A.; Nair, G.B.; Mukhopadhyay, A.K. Antimicrobial activity of curcumin against Helicobacter pylori isolates from India and during infections in mice. Antimicrob. Agents Chemother. 2009, 53, 1592–1597. [Google Scholar] [CrossRef] [PubMed]
- Rai, D.; Singh, J.K.; Roy, N.; Panda, D. Curcumin inhibits FtsZ assembly: An attractive mechanism for its antibacterial activity. Biochem. J. 2008, 410, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Niamsa, N.; Sittiwet, C. Antimicrobial activity of Curcuma longa aqueous extract. J. Pharmacol. Toxicol. 2009, 4, 173–177. [Google Scholar]
- Patole, S. Microbiota and necrotizing enterocolitis. Nestle Nutr. Inst. Workshop Ser. 2017, 88, 81–94. [Google Scholar] [CrossRef]
- Chassaing, B.; Darfeuille-Michaud, A. The commensal microbiota and enteropathogens in the pathogenesis of inflammatory bowel diseases. Gastroenterology 2011, 140, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.; Tang, H.; Mazmanian, S.K. Pathobionts of the gastrointestinal microbiota and inflammatory disease. Curr. Opin. Immunol. 2011, 23, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Liu, L.; Ji, H.-F. Regulative effects of curcumin spice administration on gut microbiota and its pharmacological implications. Food Nutr. Res. 2017, 61, 1361780. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Wang, H.; Zhang, P.; Gao, C.; Tao, J.; Ge, Z.; Zhu, D.; Bi, Y. Modulation of gut microbiota contributes to curcumin-mediated attenuation of hepatic steatosis in rats. Biochim. Biophys. Acta 2017, 1861, 1801–1812. [Google Scholar] [CrossRef]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, Y.; Xiang, L.; Wang, Z.; Xiao, G.C.; Hu, J. Effect of curcumin on the diversity of gut microbiota in ovariectomized rats. Nutrients 2017, 9, 1146. [Google Scholar] [CrossRef] [PubMed]
- Ohno, M.; Nishida, A.; Sugitani, Y.; Nishino, K.; Inatomi, O.; Sugimoto, M.; Kawahara, M.; Andoh, A. Nanoparticle curcumin ameliorates experimental colitis via modulation of gut microbiota and induction of regulatory T cells. PLoS ONE 2017, 12, e0185999. [Google Scholar] [CrossRef] [PubMed]
- Law, I.K.; Bakirtzi, K.; Polytarchou, C.; Oikonomopoulos, A.; Hommes, D.; Iliopoulos, D.; Pothoulakis, C. Neurotensin—regulated miR-133alpha is involved in proinflammatory signaling in human colonic epithelial cells and in experimental colitis. Gut 2015, 64, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Narushima, S.; Sugiura, Y.; Oshima, K.; Atarashi, K.; Hattori, M.; Suematsu, M.; Honda, K. Characterization of the 17 strains of regulatory T cell-inducing human-derived Clostridia. Gut Microbes 2014, 5, 333–339. [Google Scholar] [CrossRef]
- McFadden, R.M.; Larmonier, C.B.; Shehab, K.W.; Midura-Kiela, M.; Ramalingam, R.; Harrison, C.A.; Besselsen, D.G.; Chase, J.H.; Caporaso, J.G.; Jobin, C.; et al. The role of curcumin in modulating colonic microbiota during colitis and colon cancer prevention. Inflamm. Bowel Dis. 2015, 21, 2483–2494. [Google Scholar] [CrossRef]
- Burapan, S.; Kim, M.; Han, J. Curcuminoid demethylation as an alternative metabolism by human intestinal microbiota. J. Agric. Food Chem. 2017, 65, 3305–3310. [Google Scholar] [CrossRef]
- Hwang, S.W.; Kim, J.H.; Lee, C.; Im, I.P.; Kim, J.S. Intestinal alkaline phosphatase ameliorates experimental colitis via toll-like receptor 4-dependent pathway. Eur. J. Pharmacol. 2018, 820, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Boozari, M.; Butler, A.E.; Sahebkar, A. Impact of curcumin on toll-like receptors. J. Cell. Physiol. 2019. Preprint. [Google Scholar] [CrossRef] [PubMed]
- Gradišar, H.; Keber, M.M.; Pristovšek, P.; Jerala, R. MD-2 as the target of curcumin in the inhibition of response to LPS. J. Leukoc. Biol. 2007, 82, 968–974. [Google Scholar] [CrossRef]
- Peng, L.; Li, X.; Song, S.; Wang, Y.; Xu, L. Effects of curcumin on mRNA expression of cytokines related to toll-like receptor 4 signaling in THP-1 cells. Chin. J. Dermatol. 2010, 43, 493–496. [Google Scholar]
- Baliga, M.S.; Joseph, N.; Venkataranganna, M.V.; Saxena, A.; Ponemone, V.; Fayad, R. Curcumin, an active component of turmeric in the prevention and treatment of ulcerative colitis: Preclinical and clinical observations. Food Funct. 2012, 3, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Eckert, J.; Scott, B.; Lawrence, S.M.; Ihnat, M.; Chaaban, H. FLLL32, a curcumin analog, ameliorates intestinal injury in necrotizing enterocolitis. J. Inflamm. Res. 2017, 10, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Murano, M.; Maemura, K.; Hirata, I.; Toshina, K.; Nishikawa, T.; Hamamoto, N.; Sasaki, S.; Saitoh, O.; Katsu, K. Therapeutic effect of intracolonically administered nuclear factor kappa B (p65) antisense oligonucleotide on mouse dextran sulphate sodium (DSS)-induced colitis. Clin. Exp. Immunol. 2000, 120, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.T.; Chen, Y.Q.; Ouyang, Q. Sulfasalazine inhibits activation of nuclear factor-kappaB in patients with ulcerative colitis. J. Gastroenterol. Hepatol. 2005, 20, 1016–1024. [Google Scholar] [CrossRef]
- Hommes, D.; van den Blink, B.; Plasse, T.; Bartelsman, J.; Xu, C.; Macpherson, B.; Tytgat, G.; Peppelenbosch, M.; Van Deventer, S. Inhibition of stress-activated MAP kinases induces clinical improvement in moderate to severe Crohn’s disease. Gastroenterology 2002, 122, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.H.; Lin-Shiau, S.Y.; Lin, J.K. Comparative studies on the suppression of nitric oxide synthase by curcumin and its hydrogenated metabolites through down-regulation of IkappaB kinase and NFkappaB activation in macrophages. Biochem. Pharmacol. 2000, 60, 1665–1676. [Google Scholar] [CrossRef]
- Jobin, C.; Bradham, C.A.; Russo, M.P.; Juma, B.; Narula, A.S.; Brenner, D.A.; Sartor, R.B. Curcumin blocks cytokine-mediated NF-κB activation and proinflammatory gene expression by inhibiting inhibitory factor I-κB kinase activity. J. Immunol. 1999, 163, 3474–3483. [Google Scholar]
- Shishodia, S.; Singh, T.; Chaturvedi, M.M. Modulation of transcription factors by curcumin. Adv. Exp. Med. Biol. 2007, 595, 127–148. [Google Scholar] [CrossRef]
- Hahm, E.R.; Cheon, G.; Lee, J.; Kim, B.; Park, C.; Yang, C.H. New and known symmetrical curcumin derivatives inhibit the formation of Fos-Jun-DNA complex. Cancer Lett. 2002, 184, 89–96. [Google Scholar] [CrossRef]
- Huang, T.S.; Lee, S.C.; Lin, J.K. Suppression of c-Jun/AP-1 activation by an inhibitor of tumor promotion in mouse fibroblast cells. Proc. Natl. Acad. Sci. USA 1991, 88, 5292–5296. [Google Scholar] [CrossRef]
- Huang, M.T.; Smart, R.C.; Wong, C.Q.; Conney, A.H. Inhibitory effect of curcumin, chlorogenic acid, caffeic acid, and ferulic acid on tumor promotion in mouse skin by 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 1988, 48, 5941–5946. [Google Scholar] [PubMed]
- Ukil, A.; Maity, S.; Karmakar, S.; Datta, N.; Vedasiromoni, J.R.; Das, P.K. Curcumin, the major component of food flavour turmeric, reduces mucosal injury in trinitrobenzene sulphonic acid-induced colitis. Br. J. Pharmacol. 2003, 139, 209–2018. [Google Scholar] [CrossRef] [PubMed]
- Salh, B.; Assi, K.; Templeman, V.; Parhar, K.; Owen, D.; Gomez-Munoz, A.; Jacobson, K. Curcumin attenuates DNB-induced murine colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, G235–G243. [Google Scholar] [CrossRef] [PubMed]
- Holt, P.R.; Katz, S.; Kirshoff, R. Curcumin therapy in inflammatory bowel disease: A pilot study. Dig. Dis. Sci. 2005, 50, 2191–2193. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.O.; Jin, C.Y.; Lee, J.D.; Choi, Y.H.; Ahn, S.C.; Lee, C.M.; Jeong, S.C.; Park, Y.M.; Kim, G.Y. Curcumin decreases binding of Shiga-like toxin-1B on human intestinal epithelial cell line HT29 stimulated with TNF-alpha and Il-1beta: Suppression of p38, JNK and NF-kappaB p65 as potential targets. Biol. Pharm. Bull. 2006, 29, 1470–1475. [Google Scholar] [CrossRef]
- Jian, Y.T.; Mai, G.F.; Wang, J.D.; Zhang, Y.L.; Luo, R.C.; Fang, Y.X. Preventive and therapeutic effects of NF-κB inhibitor curcumin in rats colitis induced by trinitrobenzene sulfonic acid. World J. Gastroenterol. 2005, 11, 1747–1752. [Google Scholar] [CrossRef] [PubMed]
- Camacho-Barquero, L.; Villegas, I.; Sánchez-Fidalgo, S.; Motilva, V.; de la Lastra, C.A. Curcumin, a Curcuma longa constituent, acts on MAPK p38 pathway modulating COX-2 and iNOS expression in chronic experimental colitis. Int. Immunopharmacol. 2007, 7, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Hanai, H.; Tozawa, K.; Aoshi, T.; Uchijima, M.; Nagata, T.; Koide, Y. Curcumin prevents and ameliorates trinitrobenzene sulfonic acid-induced colitis in mice. Gastroenterology 2002, 123, 1912–1922. [Google Scholar] [CrossRef]
- Kim, G.Y.; Kim, K.H.; Lee, S.H.; Yoon, M.S.; Lee, H.J.; Moon, D.O.; Lee, C.M.; Ahn, S.C.; Park, Y.C.; Park, Y.M. Curcumin inhibits immunostimulatory function of dendritic cells: MAPKs and translocation of NF-kappa B as potential targets. J. Immunol. 2005, 174, 8116–8124. [Google Scholar] [CrossRef]
- Cong, Y.; Wang, L.; Konrad, A.; Schoeb, T.; Elson, C.O. Curcumin induces the tolerogenic dendritic cell that promotes differentiation of intestine-protective regulatory T cells. Eur. J. Immunol. 2009, 39, 3134–3146. [Google Scholar] [CrossRef]
- Abdollahi, E.; Momtazi, A.A.; Johnston, T.P.; Sahebkar, A. Therapeutic effects of curcumin in inflammatory and immune-mediated diseases: A nature-made jack-of-all-trades? J. Cell Physiol. 2018, 233, 830–848. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.D.; Jeong, Y.-I.; Lee, C.-M.; Noh, K.T.; Jeong, S.K.; Chun, S.H.; Choi, O.H.; Park, W.S.; Han, J.; Shin, Y.K.; et al. COX-2 and PGE2 signaling is essential for the regulation of IDO expression by curcumin in murine bone marrow-derived dendritic cells. Int. Immunopharmacol. 2010, 10, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Kuo, J.; Jiang, H.; Deeb, D.; Liu, Y.; Divine, G.; Chapman, R.A.; Dulchavsky, S.A.; Gautam, S.C. Immunomodulatory activity of curcumin: Suppression of lymphocyte proliferation, development of cell-mediated cytotoxicity, and cytokine production in vitro. Biochem. Pharmacol. 2004, 68, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.S.; Mishra, K.P.; Singh, D.P.; Mehrota, S.; Singh, V.K. Immunomodulatory effects of curcumin. Immunopharmacol. Immunotoxicol. 2005, 27, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Jagetia, G.C.; Aggarwal, B.B. “Spicing up” of the immune system by curcumin. J. Clin. Immunol. 2007, 27, 19–35. [Google Scholar] [CrossRef]
- Kang, B.Y.; Chung, S.W.; Chung, W.-J.; Im, S.-Y.; Hwang, S.Y.; Kim, T.S. Inhibition of interleukin-12 production in lipopolysaccharide-activated macrophages by curcumin. Eur. J. Pharmacol. 1999, 384, 191–195. [Google Scholar] [CrossRef]
- Zhang, M.; Deng, C.S.; Zheng, J.J.; Xia, J. Curcumin regulated shift from Th1 to Th2 in trinitrobenzene sulphonic acid-induced chronic colitis. Acta Pharmacol. 2006, 27, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Bakır, B.; Yetkin Ay, Z.; Büyükbayram, H.İ.; Kumbul Doğuç, D.; Bayram, D.; Candan, I.A.; Uskun, E. Effect of curcumin on systemic T helper 17 cell response: Gingival expression of interleukin-17 and retinoic acid receptor-related orphan receptor γt and alveolar bone loss in experimental periodontitis. J. Periodontol. 2016, 87, e183–e191. [Google Scholar] [CrossRef]
- Brouet, I.; Ohshima, H. Curcumin, an anti-tumour promoter and anti-inflammatory agent, inhibits induction of nitric oxide synthase in activated macrophages. Biochem. Biophys. Res. Commun. 1995, 206, 533–540. [Google Scholar] [CrossRef]
- Mani, H.; Sidhu, G.S.; Kumari, R.; Gaddipati, J.P.; Seth, P.; Maheshwari, R.K. Curcumin differentially regulates TGF-β1, its receptors and nitrix oxide synthase during impaired wound healing. BioFactors 2002, 16, 29–43. [Google Scholar] [CrossRef]
- Joe, B.; Lokesh, B.R. Role of capsaicin, curcumin and dietary n-3 fatty acids in lowering the generation of reactive oxygen species in rat peritoneal macrophages. Biochim. Biophys. Acta 1994, 1224, 255–263. [Google Scholar] [CrossRef]
- Joe, B.; Lokesh, B.R. Dietary n-3 fatty acids, curcumin and capsaicin lower the release of lysosomal enzymes and eicosanoids in rat peritoneal macrophages. Mol. Cell. Biochem. 2000, 203, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Billerey-Larmonier, C.; Uno, J.K.; Larmonier, N.; Midura, A.J.; Timmermann, B.; Ghishan, F.K.; Kiela, P.R. Protective effects of dietary curcumin in mouse model of chemically induced colitis are strain dependent. Inflamm. Bowel Dis. 2008, 14, 780–793. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.M.; Huang, H.I.; Fenton, M.R.; Fong, D. In vivo inhibition of nitric oxide synthase gene expression by curcumin, a cancer preventive natural produce with anti-inflammatory properties. Biochem. Pharmacol. 1998, 55, 1955–1962. [Google Scholar] [CrossRef]
- Li, X.; Liu, X. Effect of curcumin on immune function of mice. J. Huazhong Univ. Sci. Technol. Med. Sci. 2005, 25, 137–140. [Google Scholar] [PubMed]
- Antony, S.; Kuttan, R.; Kuttan, G. Immunomodulatory activity of curcumin. Immunol. Investig. 1999, 28, 291–303. [Google Scholar] [CrossRef]
- Bai, X.; Oberley-Deegan, R.E.; Bai, A.; Ovrutsky, A.R.; Kinney, W.H.; Weaver, M.; Zhang, G.; Honda, J.R.; Chan, E.D. Curcumin enhances human macrophage control of Mycobacterium tuberculosis infection. Respirology 2016, 21, 951–957. [Google Scholar] [CrossRef]
- Mimche, P.N.; Thompson, E.; Taramelli, D.; Vivas, L. Curcumin enhances non-opsonic phagocytosis of Plasmodium falciparum through up-regulation of CD36 surface expression on monocytes/macrophages. J. Antimicrob. Chemother. 2012, 67, 1895–1904. [Google Scholar] [CrossRef]
- Martin, A.R.; Villegas, I.; La Casa, C.; de la Lastra, C.A. Resveratrol, a polyphenol found in grapes, suppresses oxidative damage and stimulates apoptosis during early colonic inflammation in rats. Biochem. Pharmacol. 2004, 67, 1399–1410. [Google Scholar] [CrossRef]
- Jiang, H.; Deng, C.S.; Zhang, M.; Xia, J. Curcumin-attenuated trinitrobenzene sulphonic acid induces chronic colitis by inhibiting expression of cyclooxygenase-2. World J. Gastrolenterol. 2006, 12, 3848–3853. [Google Scholar] [CrossRef]
- Zhang, M.; Denc, C.; Zheng, J.; Xia, J.; Sheng, D. Curcumin inhibits trinitrobenzene sulphonic acid-induced colitis in rats by activation of peroxisome proliferator-activated receptor gamma. Int. Immunopharmacol. 2006, 6, 1233–1242. [Google Scholar] [CrossRef]
- Larmonier, C.B.; Midura-Kiela, M.T.; Ramalingam, R.; Laubitz, D.; Janikashvili, N.; Larmonier, N.; Ghishan, F.K.; Kiela, P.R. Modulation of neutrophil motility by curcumin: Implications for inflammatory bowel disease. Inflamm. Bowel Dis. 2011, 17, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Larmonier, C.B.; Uno, J.K.; Lee, K.M.; Karrasch, T.; Laubitz, D.; Thurston, R.; Midura-Kiela, M.T.; Ghishan, F.K.; Sartor, R.B.; Jobin, C.; et al. Limited effects of dietary curcumin on Th-1 driven colitis in IL-10 deficient mice suggest an IL-10-dependent mechanism of protection. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G1079–G1091. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R. Inhibition of neutrophil response by curcumin. Agents Actions 1989, 28, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, Y.; Han, Y.; Kayahara, M.; Watanabe, T.; Arishige, H.; Kato, N. Consumption of curcumin elevates fecal immunoglobulin A, an index of intestinal immune function, in rats fed a high-fat diet. J. Nutr. Sci. Vitaminol. 2010, 56, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Churchill, M.; Chadburn, A.; Bilinski, R.T.; Bertagnolli, M.M. Inhibition of intestinal tumors by curcumin is associated with changes in the intestinal immune cell profile. J. Surg. Res. 2000, 89, 169–175. [Google Scholar] [CrossRef]
- Decoté-Ricardo, D.; Chagas, K.; Rocha, J.; Redner, P.; Lopes, U.; Cambier, J.; Pecanha, L. Modulation of in vitro murine B-lymphocyte response by curcumin. Phytomedicine 2009, 16, 982–988. [Google Scholar] [CrossRef]
- Timmermans, W.M.C.; van Laar, J.A.M.; van der Houwen, T.B.; Kamphuis, L.S.J.; Bartol, S.J.W.; Lam, K.H.; Ouwendijk, R.J.; Sparrow, M.P.; Gibson, P.R.; van Hagen, P.M.; et al. B-cell dysregulation in Crohn’s disease is partially restored with infliximab therapy. PLoS ONE 2016, 11, e0160103. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.; Srikanth, C.V.; Antony, R.; Shi, H.N.; Cherayil, B.J. A role for natural killer cells in intestinal inflammation caused by infection with Salmonella enterica serovar Typhimurium. FEMS Immunol. Med. Microbiol. 2007, 51, 372–380. [Google Scholar] [CrossRef]
- Mouzaoui, S.; Rahim, I.; Djerdjouri, B. Aminoguanidine and curcumin attenuated tumor necrosis factor (TNF)-alpha induced oxidative stress, colitis and hepatotoxicity in mice. Int. Immunopharmacol. 2012, 12, 302–311. [Google Scholar] [CrossRef]
- Midura-Kiela, M.T.; Radhakrishnan, V.M.; Larmonier, C.B.; Laubitz, D.; Ghishan, F.K.; Kiela, P.R. Curcumin inhibits interferon-γ signaling in colonic epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G85–G96. [Google Scholar] [CrossRef]
- Kim, Y.S.; Young, M.R.; Bobe, G.; Colburn, N.H.; Milner, J.A. Bioactive food components, inflammatory targets, and cancer prevention. Cancer Prev. Res. 2009, 2, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Song, W.-B.; Wang, Y.-Y.; Meng, F.-S.; Zhang, Q.-H.; Zeng, J.-Y.; Xiao, L.-P.; Yu, X.-P.; Peng, D.-D.; Su, L.; Xiao, B.; et al. Curcumin protects intestinal mucosal barrier function of rat enteritis via activation of MKP-1 and attenuation of p38 and NF-κB activation. PLoS ONE 2010, 5, e12969. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, J.; Ye, B.; Wang, Q.; Xie, X.; Shen, H. Protective effect of curcumin on TNBS-induced intestinal inflammation is mediated through the JAK/STAT pathway. BMC Complement. Altern. Med. 2016, 16, 299. [Google Scholar] [CrossRef]
- Kang, G.; Kong, P.J.; Yuh, Y.J.; Lim, S.Y.; Yim, S.V.; Chun, W.; Kim, S.S. Curcumin suppresses lipopolysaccharide-induced cyclooxygenase-2 expression by inhibiting activator protein 1 and nuclear factor kappaB bindings in BV2 microglial cells. J. Pharmacol. Sci. 2004, 94, 325–328. [Google Scholar] [CrossRef]
- Plummer, S.M.; Holloway, K.A.; Manson, M.M.; Munks, R.J.; Kaptein, A.; Farrow, S.; Howells, L. Inhibition of cyclo-oxygenase 2 expression in colon cells by the chemoprotective agent curcumin involves inhibition of NF-kappaB activation via the NIK/IKK signaling complex. Oncogene 1999, 18, 6013–6020. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Boland, C.R.; Chauhan, D.P. Specific inhibition of cyclooxygenase-2 (COX-2) expression by dietary curcumin in HT-29 human colon cancer cells. Cancer Lett. 2001, 172, 111–118. [Google Scholar] [CrossRef]
- Hanai, H.; Iida, T.; Takeuchi, K.; Watanabe, F.; Maruyama, Y.; Andoh, A.; Tsujikawa, T.; Fujiyama, Y.; Mitsuyama, K.; Sata, M.; et al. Curcumin maintenance therapy for ulcerative colitis: Randomized, multicenter, double-blind, placebo-controlled trial. Clin. Gastroenterol. Hepatol. 2006, 4, 1502–1506. [Google Scholar] [CrossRef]
- Lang, A.; Salomon, N.; Wu, J.C.Y.; Kopylov, U.; Lahat, A.; Har-Noy, O.; Ching, J.Y.L.; Cheong, P.K.; Avidan, B.; Gamus, D.; et al. Curcumin in combination with mesalamine induces remission in patients with mild-to-moderate ulcerative colitis in randomized controlled trial. Clin. Gastroenterol. Hepatol. 2015, 13, 1444–1449. [Google Scholar] [CrossRef] [PubMed]
- Babbs, C.F. Oxygen radicals in ulcerative colitis. Free Radic. Biol. Med. 1992, 13, 169–181. [Google Scholar] [CrossRef]
- Grisham, M.B. Oxidants and free radicals in inflammatory bowel disease. Lancet 1994, 344, 859–861. [Google Scholar] [CrossRef]
- Aceti, A.; Beghetti, I.; Martini, S.; Faldella, G.; Corvaglia, L. Oxidative stress and necrotizing enterocolitis: Pathogenetic mechanisms, opportunities for intervention, and the role of human milk. Oxid. Med. Cell. Longev. 2018, 2018, 7397659. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.L.; Ge, Z.; Fox, J.G.; Schauer, D.B. Disruption of tight junctions and induction of proinflammatory cytokine responses in colonic epithelial cells by Campylobacter jejuni. Infect. Immun. 2006, 74, 6581–6589. [Google Scholar] [CrossRef] [PubMed]
- Tanida, S.; Mizoshita, T.; Mizushima, T.; Sasaki, M.; Shimura, T.; Kamiya, T.; Kataoka, H.; Joh, T. Involvement of oxidative stress and mucosal address in cell adhesion molecule-1 (MAdCAM-1) in inflammatory bowel disease. J. Clin. Biochem. Nutr. 2011, 48, 112–116. [Google Scholar] [CrossRef]
- Basuroy, S.; Seth, A.; Elias, B.; Naren, A.P.; Rao, R. MAPK interacts with occluding and mediates EGF-induced prevention of tight junction disruption by hydrogen peroxide. Biochem. J. 2006, 393 (Pt 1), 69–77. [Google Scholar] [CrossRef]
- Grisham, M.B.; Yamada, T. Neutrophils, nitrogen oxides, and inflammatory bowel disease. Ann. N. Y. Acad. Sci. 1992, 664, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, J.O.; Hellstrom, P.M.; Lundberg, J.M.; Alving, K. Greatly increased luminal nitric oxide in ulcerative colitis. Lancet 1994, 344, 1673–1674. [Google Scholar] [CrossRef]
- Wallace, J.L.; Miller, M.J.S. Nitric oxide in mucosal defense: A little goes a long way. Gastroenterology 2000, 119, 512–520. [Google Scholar] [CrossRef]
- Stroes, E.; Hijmering, M.; van Zandvoort, M.; Wever, R.; Rabelink, T.J.; Van Faassen, E.E. Origin of superoxide production by endothelial nitric oxide synthase. FEBS Lett. 1998, 438, 161–164. [Google Scholar] [CrossRef]
- Cross, R.K.; Wilson, K.T. Nitric oxide in inflammatory bowel disease. Inflamm. Bowel Dis. 2003, 9, 179–189. [Google Scholar] [CrossRef]
- Sreejayan; Rao, M.N. Curcuminoids as potent inhibitors of lipid peroxidation. J. Pharm. Pharmacol. 1994, 46, 1013–1016. [Google Scholar] [CrossRef] [PubMed]
- Onoda, M.; Inano, H. Effect of curcumin on the production of nitric oxide by cultured rat mammary gland. Nitric Oxide 2000, 4, 505–515. [Google Scholar] [CrossRef]
- Venkataranganna, M.V.; Rafiq, M.; Gopumadhavan, S.; Peer, G.; Babu, U.V.; Mitra, S.K. NCB-02 (standardized Curcumin preparation) protects dinitrochlorobenzene-induced colitis through down-regulation of NFkappa-B and iNOS. World J. Gastroenterol. 2007, 13, 1103–1107. [Google Scholar] [CrossRef]
- Sanchez-Munoz, F.; Dominguez-Lopez, A.; Yamamoto-Furusho, J.K. Role of cytokines in inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 4280–4288. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, G.; Saermark, T.; Kirkegaard, T.; Brynskov, J. Spontaneous and cytokine induced expression and activity of matrix metalloproteinases in human colonic epithelium. Clin. Exp. Immunol. 2009, 155, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Rath, T.; Roderfeld, M.; Halwe, J.M.; Tschuschner, A.; Roeb, E.; Graf, J. Cellular sources of MMP-7, MMP-13 and MMP-28 in ulcerative colitis. Scand. J. Gastroenterol. 2010, 45, 1186–1196. [Google Scholar] [CrossRef]
- Koelink, P.J.; Overbeek, S.A.; Braber, S.; Morgan, M.E.; Henricks, P.A.; Abdul Roda, M.; Verspaget, H.W.; Wolfkamp, S.C.; te Velde, A.A.; Jones, C.W.; et al. Collagen degradation and neutrophilic infiltration: A vicious circle in inflammatory bowel disease. Gut 2014, 63, 578–587. [Google Scholar] [CrossRef]
- Vandooren, J.; van den Steen, P.E.; Opdenakker, G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): The next decade. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 222–272. [Google Scholar] [CrossRef]
- Thaloor, D.; Singh, A.K.; Sidhu, G.S.; Prasad, P.V.; Kleinman, H.K.; Maheshwari, R.K. Inhibition of angiogenic differentiation of human umbilical vein endothelial cells by curcumin. Cell Growth Differ. 1998, 9, 305–312. [Google Scholar]
- Clutterbuck, A.L.; Allaway, D.; Harris, P.; Mobasheri, A. Curcumin reduces prostaglandin E2, matrix metalloproteinase-3 and proteoglycan release in the secretome of interleukin 1β-treated articular cartilage. F1000 Res. 2013, 2, 147. [Google Scholar] [CrossRef]
- Hwang, B.M.; Noh, E.M.; Kim, J.S.; Kim, J.M.; You, Y.O.; Hwang, J.K.; Kwon, K.B.; Lee, Y.R. Curcumin inhibits UVB-induced matrix metalloproteinase-1/3 expression by suppressing the MAPK-p38/JNK pathways in human dermal fibroblasts. Exp. Dermatol. 2013, 22, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Murakami, A.; Ohigashi, H. Modifying effects of dietary factors on (-)-epigallocatechin-3-galalte-induced pro-matrix metalloproteinase-7 production in HT-29 human colorectal cancer cells. Biosci. Biotechnol. Biochem. 2007, 71, 2442–2450. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Kong, A.N. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol. Carcinog. 2009, 48, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Motterlini, R.; Foresti, R.; Bassi, R.; Green, C.J. Curcumin, an antioxidant and anti-inflammatory agent, induces heme oxygenase-1 and protects endothelial cells against oxidative stress. Free Radic. Biol. Med. 2000, 28, 1303–1312. [Google Scholar] [CrossRef]
- McNally, S.J.; Harrison, E.M.; Ross, J.A.; Garden, O.J.; Wigmore, S.J. Curcumin induces heme oxygenase 1 through generation of reactive oxygen species, p38 activation and phosphatase inhibition. Int. J. Mol. Med. 2007, 19, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, C.B.; Gregersen, N. Stress response profiles in human fibroblasts exposed to heat shock or oxidative stress. Methods Mol. Biol. 2010, 648, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Dou, W.; Zheng, Y.; Wen, Q.; Qin, M.; Wang, X.; Tang, H.; Zhang, R.; Lv, D.; Wang, J.; Zhao, S. Curcumin upregulates Nrf2 translocation and protects rat hepatic stellate cells against oxidative stress. Mol. Med. Rep. 2016, 13, 1717–1724. [Google Scholar] [CrossRef]
- Wang, N.; Wang, G.; Hao, J.; Ma, J.; Wang, Y.; Jiang, X.; Jiang, H. Curcumin ameliorates hydrogen peroxide-induced epithelial barrier disruption by upregulating heme oxygenase-1 expression in human intestinal epithelial cells. Dig. Dis. Sci. 2012, 57, 1792–1801. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burge, K.; Gunasekaran, A.; Eckert, J.; Chaaban, H. Curcumin and Intestinal Inflammatory Diseases: Molecular Mechanisms of Protection. Int. J. Mol. Sci. 2019, 20, 1912. https://doi.org/10.3390/ijms20081912
Burge K, Gunasekaran A, Eckert J, Chaaban H. Curcumin and Intestinal Inflammatory Diseases: Molecular Mechanisms of Protection. International Journal of Molecular Sciences. 2019; 20(8):1912. https://doi.org/10.3390/ijms20081912
Chicago/Turabian StyleBurge, Kathryn, Aarthi Gunasekaran, Jeffrey Eckert, and Hala Chaaban. 2019. "Curcumin and Intestinal Inflammatory Diseases: Molecular Mechanisms of Protection" International Journal of Molecular Sciences 20, no. 8: 1912. https://doi.org/10.3390/ijms20081912
APA StyleBurge, K., Gunasekaran, A., Eckert, J., & Chaaban, H. (2019). Curcumin and Intestinal Inflammatory Diseases: Molecular Mechanisms of Protection. International Journal of Molecular Sciences, 20(8), 1912. https://doi.org/10.3390/ijms20081912