Non-Proteasomal UbL-UbA Family of Proteins in Neurodegeneration


Abstract
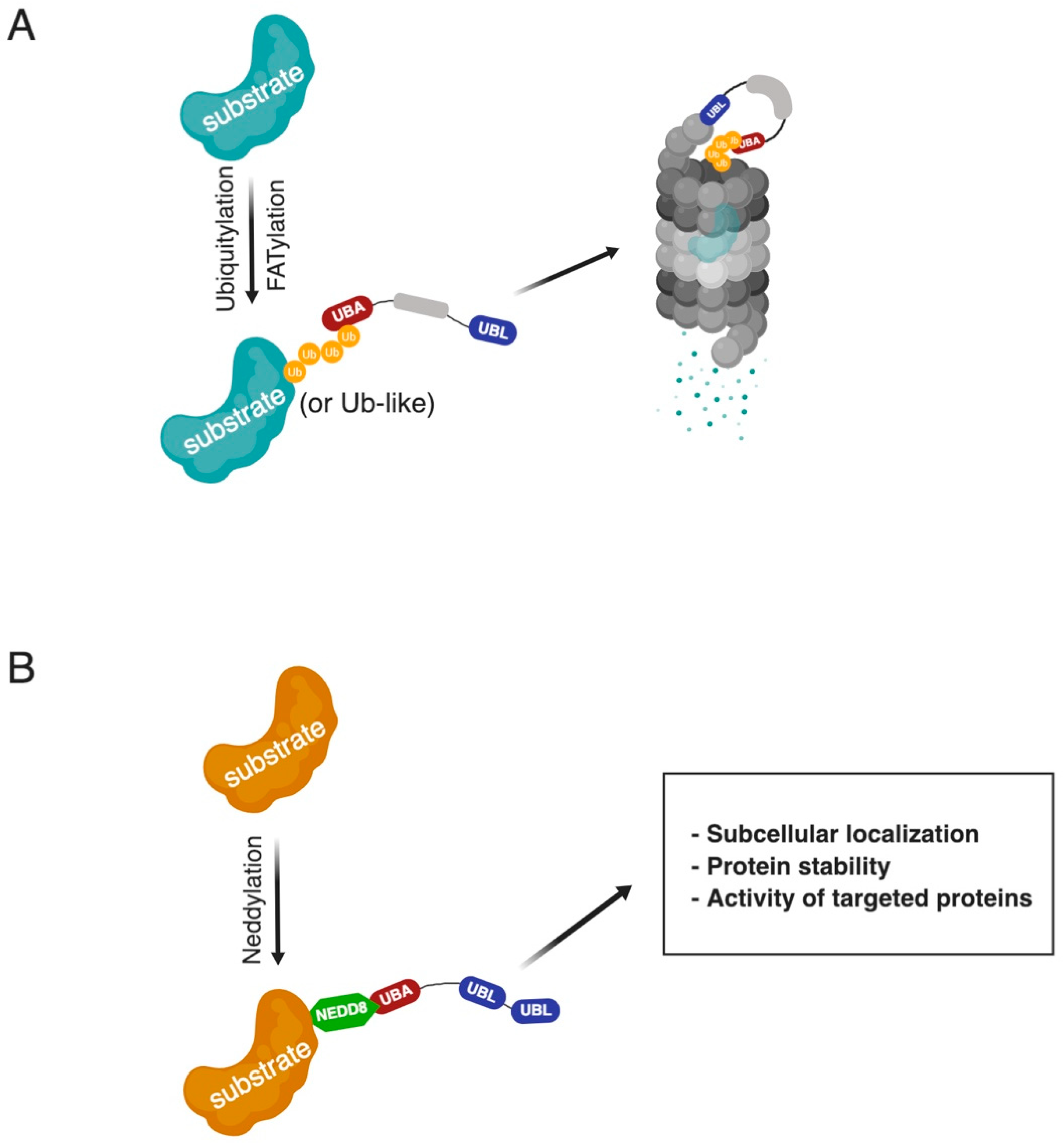
1. Introduction
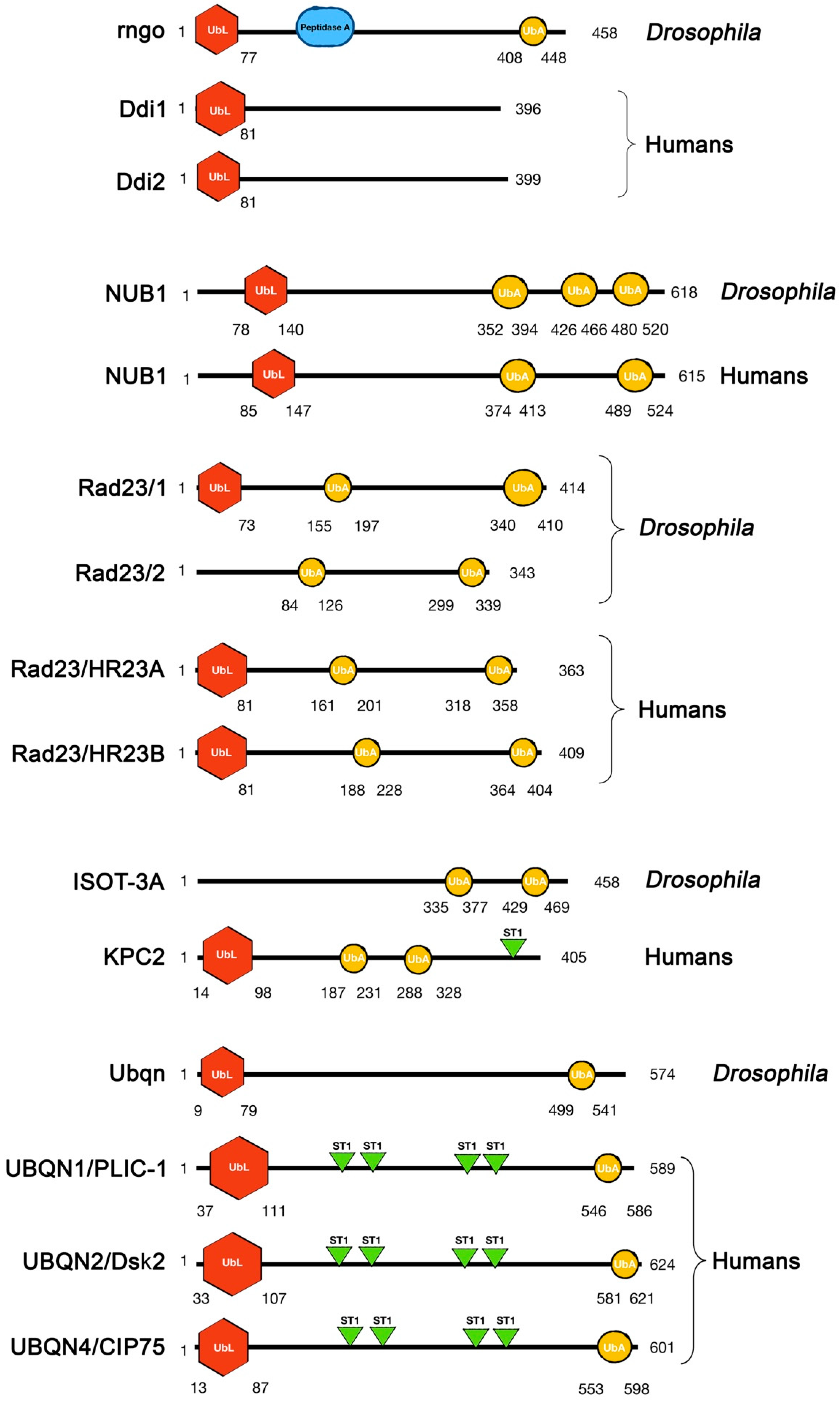
2. Physiological Roles of UbL-UbA Family Proteins and Their Contribution to Diseases
2.1. Ddi
2.2. NUB1
2.3. Rad23
2.4. KPC2
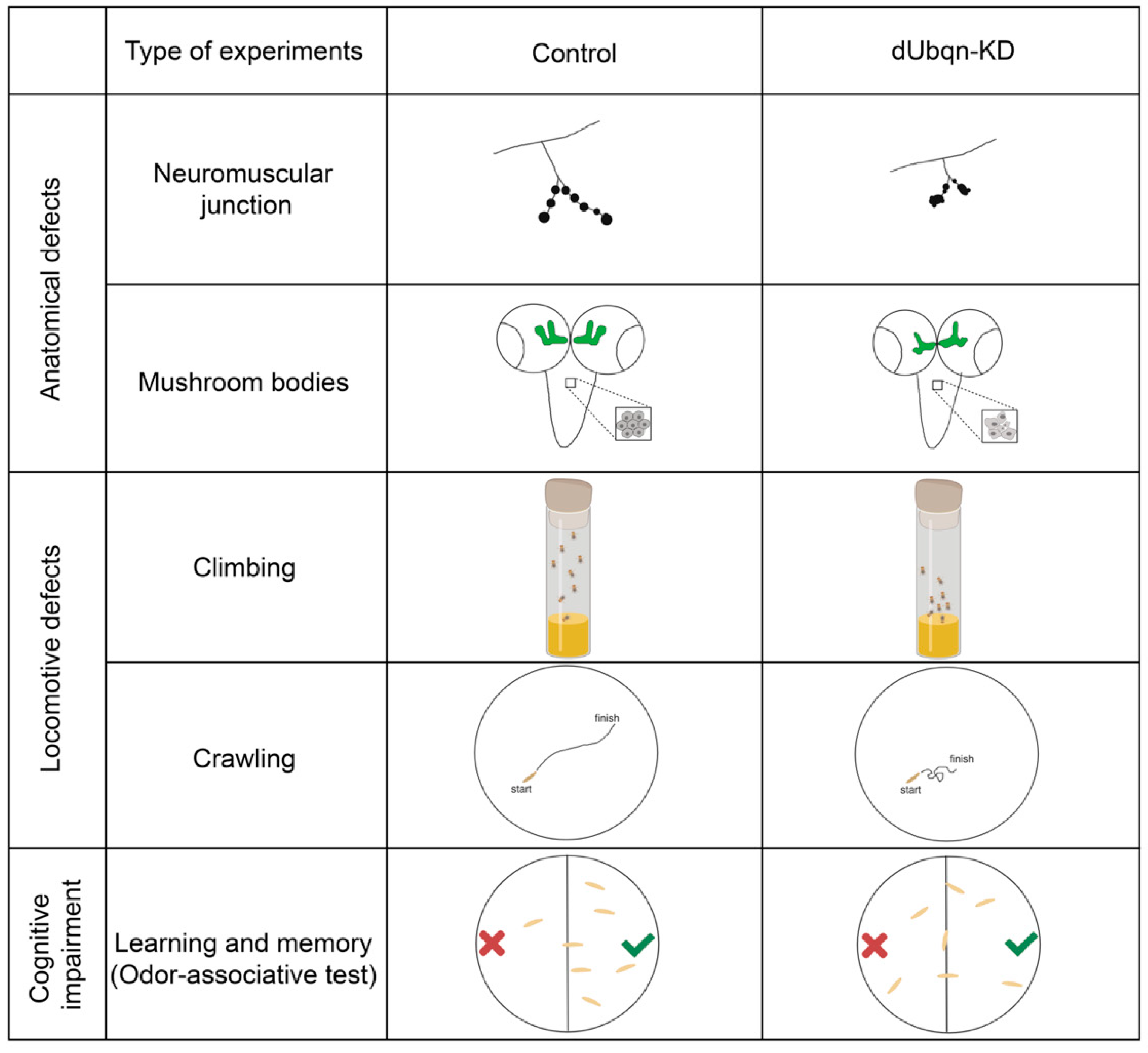
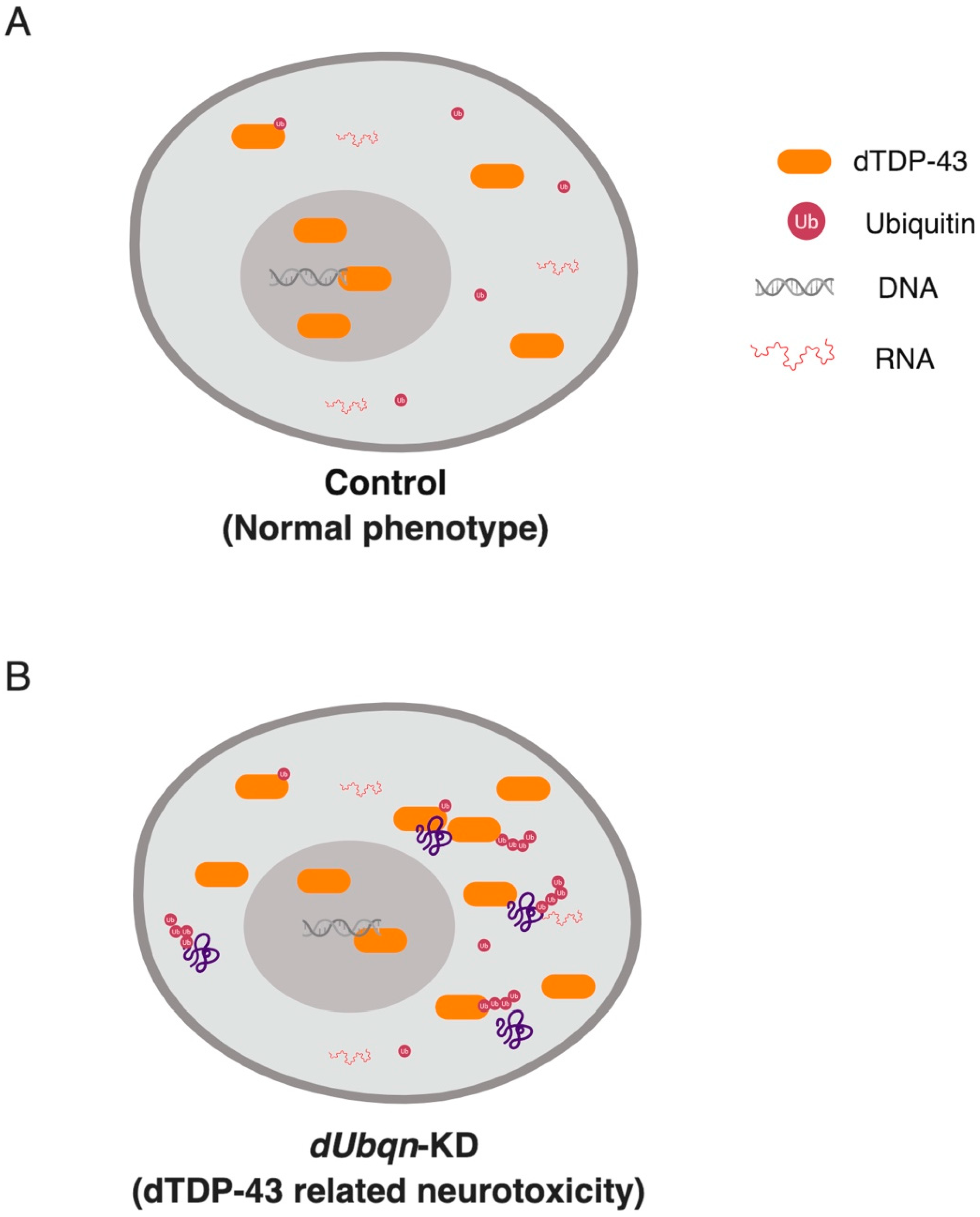
2.5. Ubiquilin (UBQLN)
3. Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Labbadia, J.; Morimoto, R.I. The Biology of Proteostasis in Aging and Disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [PubMed]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef]
- Boland, B.; Yu, W.H.; Corti, O.; Mollereau, B.; Henriques, A.; Bezard, E.; Pastores, G.M.; Rubinsztein, D.C.; Nixon, R.A.; Duchen, M.R.; et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2018, 17, 660–688. [Google Scholar] [CrossRef] [PubMed]
- Grice, G.L.; Nathan, J.A. The recognition of ubiquitinated proteins by the proteasome. Cell. Mol. Life Sci. 2016, 73, 3497–3506. [Google Scholar] [CrossRef]
- Jang, H.H. Regulation of Protein Degradation by Proteasomes in Cancer. J. Cancer Prev. 2018, 23, 153–161. [Google Scholar] [CrossRef]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef]
- Zhao, J.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. USA 2015, 112, 15790–15797. [Google Scholar] [CrossRef] [PubMed]
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef]
- Livneh, I.; Cohen-Kaplan, V.; Cohen-Rosenzweig, C.; Avni, N.; Ciechanover, A. The life cycle of the 26S proteasome: From birth, through regulation and function, and onto its death. Cell Res. 2016, 26, 869–885. [Google Scholar] [CrossRef]
- Doherty, J.; Baehrecke, E.H. Life, death and autophagy. Nat. Cell Biol. 2018, 20, 1110–1117. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef]
- Xu, P.; Duong, D.M.; Seyfried, N.T.; Cheng, D.; Xie, Y.; Robert, J.; Rush, J.; Hochstrasser, M.; Finley, D.; Peng, J. Quantitative Proteomics Reveals the Function of Unconventional Ubiquitin Chains in Proteasomal Degradation. Cell 2009, 137, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, A.D.; Zhang, N.-Y.; Xu, P.; Han, K.-J.; Noone, S.; Peng, J.; Liu, C.-W. The Lysine 48 and Lysine 63 Ubiquitin Conjugates Are Processed Differently by the 26 S Proteasome. J. Biol. Chem. 2009, 284, 35485–35494. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; Akopian, T.N.; Woo, K.M.; Goldberg, A.L. The sizes of peptides generated from protein by mammalian 26 and 20 S proteasomes. Implications for understanding the degradative mechanism and antigen presentation. J. Biol. Chem. 1999, 274, 3363–3371. [Google Scholar] [CrossRef] [PubMed]
- Palombella, V.J.; Rando, O.J.; Goldberg, A.L.; Maniatis, T. The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell 1994, 78, 773–785. [Google Scholar] [CrossRef]
- Lin, L.; Ghosh, S. A glycine-rich region in NF-kappaB p105 functions as a processing signal for the generation of the p50 subunit. Mol. Cell. Biol. 1996, 16, 2248–2254. [Google Scholar] [CrossRef] [PubMed]
- Rape, M.; Jentsch, S. Taking a bite: Proteasomal protein processing. Nat. Cell Biol. 2002, 4, E113–E116. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, T.; Matuschewski, K.; Rape, M.; Schlenker, S.; Ulrich, H.D.; Jentsch, S. Activation of a membrane-bound transcription factor by regulated ubiquitin/proteasome-dependent processing. Cell 2000, 102, 577–586. [Google Scholar] [CrossRef]
- Pan, Y.; Bai, C.B.; Joyner, A.L.; Wang, B. Sonic hedgehog Signaling Regulates Gli2 Transcriptional Activity by Suppressing Its Processing and Degradation. Mol. Cell. Biol. 2006, 26, 3365–3377. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, J.; Ma, D.; Zhang, M.; Hu, S.; Shao, S.; Gong, C.-X. Subcutaneous administration of liraglutide ameliorates Alzheimer-associated tau hyperphosphorylation in rats with type 2 diabetes. J. Alzheimers Dis. 2013, 37, 637–648. [Google Scholar] [CrossRef]
- Kraut, D.A. Slippery substrates impair ATP-dependent protease function by slowing unfolding. J. Biol. Chem. 2013, 288, 34729–34735. [Google Scholar] [CrossRef] [PubMed]
- Keren-Kaplan, T.; Zeev Peters, L.; Levin-Kravets, O.; Attali, I.; Kleifeld, O.; Shohat, N.; Artzi, S.; Zucker, O.; Pilzer, I.; Reis, N.; et al. Structure of ubiquitylated-Rpn10 provides insight into its autoregulation mechanism. Nat. Commun. 2016, 7, 12960. [Google Scholar] [CrossRef]
- Lu, X.; Nowicka, U.; Sridharan, V.; Liu, F.; Randles, L.; Hymel, D.; Dyba, M.; Tarasov, S.G.; Tarasova, N.I.; Zhao, X.Z.; et al. Structure of the RPN13-RPN2 complex provides insights for Rpn13 and Uch37 as anticancer targets. Nat. Commun. 2017, 8, 15540. [Google Scholar] [CrossRef] [PubMed]
- VanderLinden, R.T.; Hemmis, C.W.; Yao, T.; Robinson, H.; Hill, C.P. Structure and energetics of pairwise interactions between proteasome subunits RPN2, RPN13, and ubiquitin clarify a substrate recruitment mechanism. J. Biol. Chem. 2017, 292, 9493–9504. [Google Scholar] [CrossRef]
- Mueller, T.D.; Kamionka, M.; Feigon, J. Specificity of the Interaction between Ubiquitin-associated Domains and Ubiquitin. J. Biol. Chem. 2004, 279, 11926–11936. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Vossler, R.A.; Diaz-Martinez, L.A.; Winter, N.S.; Clarke, D.J.; Walters, K.J. UBL/UBA Ubiquitin Receptor Proteins Bind a Common Tetraubiquitin Chain. J. Mol. Biol. 2006, 356, 1027–1035. [Google Scholar] [CrossRef]
- Husnjak, K.; Dikic, I. Ubiquitin-Binding Proteins: Decoders of Ubiquitin-Mediated Cellular Functions. Annu. Rev. Biochem. 2012, 81, 291–322. [Google Scholar] [CrossRef] [PubMed]
- Buchberger, A. From UBA to UBX: New words in the ubiquitin vocabulary. Trends Cell Biol. 2002, 12, 216–221. [Google Scholar] [CrossRef]
- Ciechanover, A.; Schwartz, A.L. The ubiquitin system: Pathogenesis of human diseases and drug targeting. Biochim. Biophys. Acta Mol. Cell Res. 2004, 1695, 3–17. [Google Scholar] [CrossRef]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019. [CrossRef]
- Kurtishi, A.; Rosen, B.; Patil, K.S.; Alves, G.W.; Møller, S.G. Cellular Proteostasis in Neurodegeneration. Mol. Neurobiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Pace, M.C.; Xu, G.; Fromholt, S.; Howard, J.; Crosby, K.; Giasson, B.I.; Lewis, J.; Borchelt, D.R. Changes in proteome solubility indicate widespread proteostatic disruption in mouse models of neurodegenerative disease. Acta Neuropathol. 2018, 136, 919–938. [Google Scholar] [CrossRef] [PubMed]
- Jantrapirom, S.; Lo Piccolo, L.; Yoshida, H.; Yamaguchi, M. Depletion of Ubiquilin induces an augmentation in soluble ubiquitinated Drosophila TDP-43 to drive neurotoxicity in the fly. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3038–3049. [Google Scholar] [CrossRef]
- Le, N.T.T.; Chang, L.; Kovlyagina, I.; Georgiou, P.; Safren, N.; Braunstein, K.E.; Kvarta, M.D.; Van Dyke, A.M.; LeGates, T.A.; Philips, T.; et al. Motor neuron disease, TDP-43 pathology, and memory deficits in mice expressing ALS–FTD-linked UBQLN2 mutations. Proc. Natl. Acad. Sci. USA 2016, 113, E7580–E7589. [Google Scholar] [CrossRef]
- Liu, Y.; Dai, H.; Xiao, W. UAS MAG1, a yeast cis-acting element that regulates the expression of MAG1, is located within the protein coding region of DDI1. Mol. Gen. Genet. MGG 1997, 255, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Xiao, W. Two alternative cell cycle checkpoint pathways differentially control DNA damage-dependent induction of MAG1 and DDI1 expression in yeast. Mol. Genet. Genom. 2001, 266, 436–444. [Google Scholar]
- Jelinsky, S.A.; Samson, L.D. Global response of Saccharomyces cerevisiae to an alkylating agent. Proc. Natl. Acad. Sci. USA 1999, 96, 1486–1491. [Google Scholar] [CrossRef]
- Gabriely, G.; Kama, R.; Gelin-Licht, R.; Gerst, J.E. Different domains of the UBL-UBA ubiquitin receptor, Ddi1/Vsm1, are involved in its multiple cellular roles. Mol. Biol. Cell 2008, 19, 3625–3637. [Google Scholar] [CrossRef]
- Nowicka, U.; Zhang, D.; Walker, O.; Krutauz, D.; Castañeda, C.A.; Chaturvedi, A.; Chen, T.Y.; Reis, N.; Glickman, M.H.; Fushman, D. DNA-damage-inducible 1 protein (Ddi1) contains an uncharacteristic ubiquitin-like domain that binds ubiquitin. Structure 2015, 23, 542–557. [Google Scholar] [CrossRef]
- Bertolaet, B.L.; Clarke, D.J.; Wolff, M.; Watson, M.H.; Henze, M.; Divita, G.; Reed, S.I. UBA domains of DNA damage-inducible proteins interact with ubiquitin. Nat. Struct. Biol. 2001, 8, 417–422. [Google Scholar] [CrossRef]
- Trempe, J.-F.; Brown, N.R.; Lowe, E.D.; Gordon, C.; Campbell, I.D.; Noble, M.E.M.; Endicott, J.A. Mechanism of Lys48-linked polyubiquitin chain recognition by the Mud1 UBA domain. EMBO J. 2005, 24, 3178–3189. [Google Scholar] [CrossRef] [PubMed]
- Sivá, M.; Svoboda, M.; Veverka, V.; Trempe, J.-F.; Hofmann, K.; Kožíšek, M.; Hexnerová, R.; Sedlák, F.; Belza, J.; Brynda, J.; et al. Human DNA-Damage-Inducible 2 Protein Is Structurally and Functionally Distinct from Its Yeast Ortholog. Sci. Rep. 2016, 6, 30443. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Martínez, L.A.; Kang, Y.; Walters, K.J.; Clarke, D.J. Yeast UBL-UBA proteins have partially redundant functions in cell cycle control. Cell Div. 2006, 1, 28. [Google Scholar] [CrossRef] [PubMed]
- Sirkis, R.; Gerst, J.E.; Fass, D. Ddi1, a Eukaryotic Protein With the Retroviral Protease Fold. J. Mol. Biol. 2006, 364, 376–387. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Bronner, V.; Zhang, D.; Fushman, D.; Glickman, M.H. Rpn1 and Rpn2 coordinate ubiquitin processing factors at proteasome. J. Biol. Chem. 2012, 287, 14659–14671. [Google Scholar] [CrossRef]
- Gomez, T.A.; Kolawa, N.; Gee, M.; Sweredoski, M.J.; Deshaies, R.J. Identification of a functional docking site in the Rpn1 LRR domain for the UBA-UBL domain protein Ddi1. BMC Biol. 2011, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Lehrbach, N.J.; Ruvkun, G. Proteasome dysfunction triggers activation of SKN-1A/Nrf1 by the aspartic protease DDI-1. Elife 2016, 5, e17721. [Google Scholar] [CrossRef]
- Arlt, A.; Bauer, I.; Schafmayer, C.; Tepel, J.; Müerköster, S.S.; Brosch, M.; Röder, C.; Kalthoff, H.; Hampe, J.; Moyer, M.P.; et al. Increased proteasome subunit protein expression and proteasome activity in colon cancer relate to an enhanced activation of nuclear factor E2-related factor 2 (Nrf2). Oncogene 2009, 28, 3983–3996. [Google Scholar] [CrossRef]
- Radhakrishnan, S.K.; Lee, C.S.; Young, P.; Beskow, A.; Chan, J.Y.; Deshaies, R.J. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell 2010, 38, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Steffen, J.; Seeger, M.; Koch, A.; Krüger, E. Proteasomal Degradation Is Transcriptionally Controlled by TCF11 via an ERAD-Dependent Feedback Loop. Mol. Cell 2010, 40, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.; Kalev, O.; Mehrabian, S.; Traykov, L.; Raycheva, M.; Kanakis, D.; Drineas, P.; Lutz, M.I.; Ströbel, T.; Penz, T.; et al. Familial early-onset dementia with complex neuropathologic phenotype and genomic background. Neurobiol. Aging 2016, 42, 199–204. [Google Scholar] [CrossRef]
- Morawe, T.; Honemann-Capito, M.; von Stein, W.; Wodarz, A. Loss of the extraproteasomal ubiquitin receptor Rings lost impairs ring canal growth in Drosophila oogenesis. J. Cell Biol. 2011, 193, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.; Lectez, B.; Osinalde, N.; Sivá, M.; Elu, N.; Aloria, K.; Procházková, M.; Perez, C.; Martínez-Hernández, J.; Barrio, R.; et al. Quantitative proteomics reveals neuronal ubiquitination of Rngo/Ddi1 and several proteasomal subunits by Ube3a, accounting for the complexity of Angelman syndrome. Hum. Mol. Genet. 2018, 27, 1955–1971. [Google Scholar] [CrossRef] [PubMed]
- Buiting, K.; Williams, C.; Horsthemke, B. Angelman syndrome—Insights into a rare neurogenetic disorder. Nat. Rev. Neurol. 2016, 12, 584–593. [Google Scholar] [CrossRef]
- Kito, K.; Yeh, E.T.H.; Kamitani, T. NUB1, a NEDD8-interacting Protein, Is Induced by Interferon and Down-regulates the NEDD8 Expression. J. Biol. Chem. 2001, 276, 20603–20609. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, G.; Kalveram, B.; Groettrup, M. Degradation of FAT10 by the 26S proteasome is independent of ubiquitylation but relies on NUB1L. FEBS Lett. 2009, 583, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Raasi, S.; Groettrup, M.; Schmidtke, G. NEDD8 ultimate buster-1L interacts with the ubiquitin-like protein FAT10 and accelerates its degradation. J. Biol. Chem. 2004, 279, 16503–16510. [Google Scholar] [CrossRef] [PubMed]
- Tanji, K.; Tanaka, T.; Kamitani, T. Interaction of NUB1 with the proteasome subunit S5a. Biochem. Biophys. Res. Commun. 2005, 337, 116–120. [Google Scholar] [CrossRef]
- Rani, N.; Aichem, A.; Schmidtke, G.; Kreft, S.G.; Groettrup, M. FAT10 and NUB1L bind to the VWA domain of Rpn10 and Rpn1 to enable proteasome-mediated proteolysis. Nat. Commun. 2012, 3, 749. [Google Scholar] [CrossRef]
- Kamitani, T.; Kito, K.; Fukuda-Kamitani, T.; Yeh, E.T.H. Targeting of NEDD8 and Its Conjugates for Proteasomal Degradation by NUB1. J. Biol. Chem. 2001, 276, 46655–46660. [Google Scholar] [CrossRef]
- Bonacci, T.; Audebert, S.; Camoin, L.; Baudelet, E.; Iovanna, J.-L.; Soubeyran, P. Regulation of NUB1 Activity through Non-Proteolytic Mdm2-Mediated Ubiquitination. PLoS ONE 2017, 12, e0169988. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Kawashima, H.; Yeh, E.T.H.; Kamitani, T. Regulation of the NEDD8 Conjugation System by a Splicing Variant, NUB1L. J. Biol. Chem. 2003, 278, 32905–32913. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yang, H.; Zhao, J.; Zhang, Y.-H.; Song, A.-X.; Hu, H.-Y. NEDD8 ultimate buster-1 long (NUB1L) protein promotes transfer of NEDD8 to proteasome for degradation through the P97UFD1/NPL4 complex. J. Biol. Chem. 2013, 288, 31339–31349. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Rabouille, C.; Newman, R.; Levine, T.P.; Pappin, D.; Freemont, P.; Warren, G. p47 is a cofactor for p97-mediated membrane fusion. Nature 1997, 388, 75–78. [Google Scholar] [CrossRef]
- Meyer, H.H.; Shorter, J.G.; Seemann, J.; Pappin, D.; Warren, G. A complex of mammalian Ufd1 and Npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J. 2000, 19, 2181–2192. [Google Scholar] [CrossRef]
- Li, J.; Ma, W.; Li, H.; Hou, N.; Wang, X.; Kim, I.; Li, F.; Su, H. NEDD8 Ultimate Buster 1 Long (NUB1L) Protein Suppresses Atypical Neddylation and Promotes the Proteasomal Degradation of Misfolded Proteins. J. Biol. Chem. 2015, 290, 23850–23862. [Google Scholar] [CrossRef]
- Liu, G.; Xirodimas, D.P. NUB1 promotes cytoplasmic localization of p53 through cooperation of the NEDD8 and ubiquitin pathways. Oncogene 2010, 29, 2252–2261. [Google Scholar] [CrossRef]
- Mori, F.; Tanji, K.; Odagiri, S.; Hattori, M.; Hoshikawa, Y.; Kono, C.; Yasui, K.; Yokoi, S.; Hasegawa, Y.; Kamitani, T.; et al. Ubiquitin-related proteins in neuronal and glial intranuclear inclusions in intranuclear inclusion body disease. Pathol. Int. 2012, 62, 407–411. [Google Scholar] [CrossRef]
- Tanji, K.; Mori, F.; Kakita, A.; Zhang, H.; Kito, K.; Kamitani, T.; Takahashi, H.; Wakabayashi, K. Immunohistochemical localization of NUB1, a synphilin-1-binding protein, in neurodegenerative disorders. Acta Neuropathol. 2007, 114, 365–371. [Google Scholar] [CrossRef]
- Tanji, K.; Mori, F.; Kito, K.; Kakita, A.; Mimura, J.; Itoh, K.; Takahashi, H.; Kamitani, T.; Wakabayashi, K. Synphilin-1-Binding Protein NUB1 is Colocalized With Nonfibrillar, Proteinase K-Resistant α-Synuclein in Presynapses in Lewy Body Disease. J. Neuropathol. Exp. Neurol. 2011, 70, 879–889. [Google Scholar] [CrossRef]
- Tanji, K.; Tanaka, T.; Mori, F.; Kito, K.; Takahashi, H.; Wakabayashi, K.; Kamitani, T. NUB1 Suppresses the Formation of Lewy Body-Like Inclusions by Proteasomal Degradation of Synphilin-1. Am. J. Pathol. 2006, 169, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.J.; Hernández, F.; Gómez-Ramos, P.; Morán, M.A.; Hen, R.; Avila, J. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 2001, 20, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.V.W.; Stoothoff, W.H. Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci. 2004, 117, 5721–5729. [Google Scholar] [CrossRef] [PubMed]
- Rankin, C.A.; Sun, Q.; Gamblin, T.C. Pre-assembled tau filaments phosphorylated by GSK-3b form large tangle-like structures. Neurobiol. Dis. 2008, 31, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Richet, E.; Pooler, A.M.; Rodriguez, T.; Novoselov, S.S.; Schmidtke, G.; Groettrup, M.; Hanger, D.P.; Cheetham, M.E.; van der Spuy, J. NUB1 modulation of GSK3β reduces tau aggregation. Hum. Mol. Genet. 2012, 21, 5254–5267. [Google Scholar] [CrossRef][Green Version]
- Lu, B.; Al-Ramahi, I.; Valencia, A.; Wang, Q.; Berenshteyn, F.; Yang, H.; Gallego-Flores, T.; Ichcho, S.; Lacoste, A.; Hild, M.; et al. Identification of NUB1 as a suppressor of mutant Huntingtin toxicity via enhanced protein clearance. Nat. Neurosci. 2013, 16, 562–570. [Google Scholar] [CrossRef]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Yokoi, M.; Hanaoka, F. Two mammalian homologs of yeast Rad23, HR23A and HR23B, as multifunctional proteins. Gene 2017, 597, 1–9. [Google Scholar] [CrossRef]
- Dantuma, N.P.; Heinen, C.; Hoogstraten, D. The ubiquitin receptor Rad23: At the crossroads of nucleotide excision repair and proteasomal degradation. DNA Repair 2009, 8, 449–460. [Google Scholar] [CrossRef]
- Hiyama, H.; Yokoi, M.; Masutani, C.; Sugasawa, K.; Maekawa, T.; Tanaka, K.; Hoeijmakers, J.H.; Hanaoka, F. Interaction of hHR23 with S5a. The ubiquitin-like domain of hHR23 mediates interaction with S5a subunit of 26 S proteasome. J. Biol. Chem. 1999, 274, 28019–28025. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Madura, K. Rad23 promotes the targeting of proteolytic substrates to the proteasome. Mol. Cell. Biol. 2002, 22, 4902–4913. [Google Scholar] [CrossRef] [PubMed]
- Elsasser, S.; Chandler-Militello, D.; Müller, B.; Hanna, J.; Finley, D. Rad23 and Rpn10 Serve as Alternative Ubiquitin Receptors for the Proteasome. J. Biol. Chem. 2004, 279, 26817–26822. [Google Scholar] [CrossRef]
- Kim, I.; Mi, K.; Rao, H. Multiple interactions of rad23 suggest a mechanism for ubiquitylated substrate delivery important in proteolysis. Mol. Biol. Cell 2004, 15, 3357–3365. [Google Scholar] [CrossRef] [PubMed]
- Elsasser, S.; Gali, R.R.; Schwickart, M.; Larsen, C.N.; Leggett, D.S.; Müller, B.; Feng, M.T.; Tübing, F.; Dittmar, G.A.G.; Finley, D. Proteasome subunit Rpn1 binds ubiquitin-like protein domains. Nat. Cell Biol. 2002, 4, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Shinde, U.; Ortolan, T.G.; Madura, K. Ubiquitin-associated (UBA) domains in Rad23 bind ubiquitin and promote inhibition of multi-ubiquitin chain assembly. EMBO Rep. 2001, 2, 933–938. [Google Scholar] [CrossRef]
- Raasi, S.; Pickart, C.M. Rad23 ubiquitin-associated domains (UBA) inhibit 26 S proteasome-catalyzed proteolysis by sequestering lysine 48-linked polyubiquitin chains. J. Biol. Chem. 2003, 278, 8951–8959. [Google Scholar] [CrossRef]
- Raasi, S.; Orlov, I.; Fleming, K.G.; Pickart, C.M. Binding of polyubiquitin chains to ubiquitin-associated (UBA) domains of HHR23A. J. Mol. Biol. 2004, 341, 1367–1379. [Google Scholar] [CrossRef]
- Kang, Y.; Zhang, N.; Koepp, D.M.; Walters, K.J. Ubiquitin receptor proteins hHR23a and hPLIC2 interact. J. Mol. Biol. 2007, 365, 1093–1101. [Google Scholar] [CrossRef]
- Heinen, C.; Acs, K.; Hoogstraten, D.; Dantuma, N.P. C-terminal UBA domains protect ubiquitin receptors by preventing initiation of protein degradation. Nat. Commun. 2011, 2, 191. [Google Scholar] [CrossRef]
- Liang, R.-Y.; Chen, L.; Ko, B.-T.; Shen, Y.-H.; Li, Y.-T.; Chen, B.-R.; Lin, K.-T.; Madura, K.; Chuang, S.-M. Rad23 Interaction with the Proteasome Is Regulated by Phosphorylation of Its Ubiquitin-Like (UbL) Domain. J. Mol. Biol. 2014, 426, 4049–4060. [Google Scholar] [CrossRef]
- Medicherla, B.; Kostova, Z.; Schaefer, A.; Wolf, D.H. A genomic screen identifies Dsk2p and Rad23p as essential components of ER-associated degradation. EMBO Rep. 2004, 5, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Gödderz, D.; Heinen, C.; Marchese, F.P.; Kurz, T.; Acs, K.; Dantuma, N.P. Cdc48-independent proteasomal degradation coincides with a reduced need for ubiquitylation. Sci. Rep. 2015, 5, 7615. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Ahn, J.; Liu, C.; Tanabe, K.; Apodaca, J.; Suzuki, T.; Rao, H. The Png1–Rad23 complex regulates glycoprotein turnover. J. Cell Biol. 2006, 172, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Katiyar, S.; Lennarz, W.J. Misfolding of glycoproteins is a prerequisite for peptide: N-glycanase mediated deglycosylation. FEBS Lett. 2005, 579, 823–826. [Google Scholar] [CrossRef]
- Hwang, G.-W.; Sasaki, D.; Naganuma, A. Overexpression of Rad23 confers resistance to methylmercury in saccharomyces cerevisiae via inhibition of the degradation of ubiquitinated proteins. Mol. Pharmacol. 2005, 68, 1074–1078. [Google Scholar] [CrossRef] [PubMed]
- Glockzin, S.; Ogi, F.-X.; Hengstermann, A.; Scheffner, M.; Blattner, C. Involvement of the DNA repair protein hHR23 in p53 degradation. Mol. Cell. Biol. 2003, 23, 8960–8969. [Google Scholar] [CrossRef] [PubMed]
- Brignone, C.; Bradley, K.E.; Kisselev, A.F.; Grossman, S.R. A post-ubiquitination role for MDM2 and hHR23A in the p53 degradation pathway. Oncogene 2004, 23, 4121–4129. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Blount, J.R.; Tsou, W.-L.; Ristic, G.; Burr, A.A.; Ouyang, M.; Galante, H.; Scaglione, K.M.; Todi, S.V. Ubiquitin-binding site 2 of ataxin-3 prevents its proteasomal degradation by interacting with Rad23. Nat. Commun. 2014, 5, 4638. [Google Scholar] [CrossRef]
- Paulson, H.L.; Shakkottai, V.G.; Clark, H.B.; Orr, H.T. Polyglutamine spinocerebellar ataxias—From genes to potential treatments. Nat. Rev. Neurosci. 2017, 18, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Lundgren, J.; Masson, P.; Mirzaei, Z.; Young, P. Identification and characterization of a Drosophila proteasome regulatory network. Mol. Cell. Biol. 2005, 25, 4662–4675. [Google Scholar] [CrossRef] [PubMed]
- Tsou, W.-L.; Ouyang, M.; Hosking, R.R.; Sutton, J.R.; Blount, J.R.; Burr, A.A.; Todi, S.V. The deubiquitinase ataxin-3 requires Rad23 and DnaJ-1 for its neuroprotective role in Drosophila melanogaster. Neurobiol. Dis. 2015, 82, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Sutton, J.R.; Blount, J.R.; Libohova, K.; Tsou, W.-L.; Joshi, G.S.; Paulson, H.L.; Costa, M.d.C.; Scaglione, K.M.; Todi, S.V. Interaction of the polyglutamine protein ataxin-3 with Rad23 regulates toxicity in Drosophila models of Spinocerebellar Ataxia Type 3. Hum. Mol. Genet. 2017, 26, 1419–1431. [Google Scholar] [CrossRef]
- Kamura, T.; Hara, T.; Matsumoto, M.; Ishida, N.; Okumura, F.; Hatakeyama, S.; Yoshida, M.; Nakayama, K.; Nakayama, K.I. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27 Kip1 at G1 phase. Nat. Cell Biol. 2004, 6, 1229–1235. [Google Scholar] [CrossRef]
- Kotoshiba, S.; Kamura, T.; Hara, T.; Ishida, N.; Nakayama, K.I. Molecular Dissection of the Interaction between p27 and Kip1 Ubiquitylation-promoting Complex, the Ubiquitin Ligase That Regulates Proteolysis of p27 in G1 Phase. J. Biol. Chem. 2005, 280, 17694–17700. [Google Scholar] [CrossRef]
- Hara, T.; Kamura, T.; Kotoshiba, S.; Takahashi, H.; Fujiwara, K.; Onoyama, I.; Shirakawa, M.; Mizushima, N.; Nakayama, K.I. Role of the UBL-UBA Protein KPC2 in Degradation of p27 at G1 Phase of the Cell Cycle. Mol. Cell. Biol. 2005, 25, 9292–9303. [Google Scholar] [CrossRef]
- Bridoux, L.; Bergiers, I.; Draime, A.; Halbout, M.; Deneyer, N.; Twizere, J.-C.; Rezsohazy, R. KPC2 relocalizes HOXA2 to the cytoplasm and decreases its transcriptional activity. Biochim. Biophys. Acta Gene Regul. Mech. 2015, 1849, 1298–1311. [Google Scholar] [CrossRef]
- Mah, A.L.; Perry, G.; Smith, M.A.; Monteiro, M.J. Identification of ubiquilin, a novel presenilin interactor that increases presenilin protein accumulation. J. Cell Biol. 2000, 151, 847–862. [Google Scholar] [CrossRef]
- Conklin, D.; Holderman, S.; Whitmore, T.E.; Maurer, M.; Feldhaus, A.L. Molecular cloning, chromosome mapping and characterization of UBQLN3 a testis-specific gene that contains an ubiquitin-like domain. Gene 2000, 249, 91–98. [Google Scholar] [CrossRef]
- Marín, I. The ubiquilin gene family: Evolutionary patterns and functional insights. BMC Evol. Biol. 2014, 14, 63. [Google Scholar] [CrossRef]
- Deng, H.-X.; Chen, W.; Hong, S.-T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Teyssou, E.; Chartier, L.; Amador, M.-D.-M.; Lam, R.; Lautrette, G.; Nicol, M.; Machat, S.; Da Barroca, S.; Moigneu, C.; Mairey, M.; et al. Novel UBQLN2 mutations linked to amyotrophic lateral sclerosis and atypical hereditary spastic paraplegia phenotype through defective HSP70-mediated proteolysis. Neurobiol. Aging 2017, 58, 239-e11. [Google Scholar] [CrossRef] [PubMed]
- Sharkey, L.M.; Safren, N.; Pithadia, A.S.; Gerson, J.E.; Dulchavsky, M.; Fischer, S.; Patel, R.; Lantis, G.; Ashraf, N.; Kim, J.H.; et al. Mutant UBQLN2 promotes toxicity by modulating intrinsic self-assembly. Proc. Natl. Acad. Sci. USA 2018, 115, E10495–E10504. [Google Scholar] [CrossRef] [PubMed]
- Hjerpe, R.; Bett, J.S.; Keuss, M.J.; Solovyova, A.; McWilliams, T.G.; Johnson, C.; Sahu, I.; Varghese, J.; Wood, N.; Wightman, M.; et al. UBQLN2 Mediates Autophagy-Independent Protein Aggregate Clearance by the Proteasome. Cell 2016, 166, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Kleijnen, M.F.; Alarcon, R.M.; Howley, P.M. The ubiquitin-associated domain of hPLIC-2 interacts with the proteasome. Mol. Biol. Cell 2003, 14, 3868–3875. [Google Scholar] [CrossRef] [PubMed]
- Seok Ko, H.; Uehara, T.; Tsuruma, K.; Nomura, Y. Ubiquilin interacts with ubiquitylated proteins and proteasome through its ubiquitin-associated and ubiquitin-like domains. FEBS Lett. 2004, 566, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Walters, K.J.; Kleijnen, M.F.; Goh, A.M.; Wagner, G.; Howley, P.M. Structural studies of the interaction between ubiquitin family proteins and proteasome subunit S5a. Biochemistry 2002, 41, 1767–1777. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Yan, L.H.; Huang, B.; Liu, M.; Liu, X.; Huang, C. Pathogenic mutation of UBQLN2 impairs its interaction with UBXD8 and disrupts endoplasmic reticulum-associated protein degradation. J. Neurochem. 2014, 129, 99–106. [Google Scholar] [CrossRef]
- Lim, P.J.; Danner, R.; Liang, J.; Doong, H.; Harman, C.; Srinivasan, D.; Rothenberg, C.; Wang, H.; Ye, Y.; Fang, S.; et al. Ubiquilin and p97/VCP bind erasin, forming a complex involved in ERAD. J. Cell Biol. 2009, 187, 201–217. [Google Scholar] [CrossRef]
- Kim, T.-Y.; Kim, E.; Yoon, S.K.; Yoon, J.-B. Herp enhances ER-associated protein degradation by recruiting ubiquilins. Biochem. Biophys. Res. Commun. 2008, 369, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef]
- N’Diaye, E.N.; Kajihara, K.K.; Hsieh, I.; Morisaki, H.; Debnath, J.; Brown, E.J. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep. 2009, 10, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Li, Y.; Yuan, X.; Zhao, M.; Wang, J.; Li, Y.; Li, Y.; Lin, H.; Zhang, Q.; Wang, W.; et al. The UbL-UBA Ubiquilin4 protein functions as a tumor suppressor in gastric cancer by p53-dependent and p53-independent regulation of p21. Cell Death Differ. 2019, 26, 516–530. [Google Scholar] [CrossRef]
- Kessler, R.; Tisserand, J.; Font-Burgada, J.; Reina, O.; Coch, L.; Attolini, C.S.; Garcia-Bassets, I.; Azorín, F. dDsk2 regulates H2Bub1 and RNA polymerase II pausing at dHP1c complex target genes. Nat. Commun. 2015, 6, 7049. [Google Scholar] [CrossRef] [PubMed]
- Hartmann-Petersen, R.; Wallace, M.; Hofmann, K.; Koch, G.; Johnsen, A.H.; Hendil, K.B.; Gordon, C. The Ubx2 and Ubx3 cofactors direct Cdc48 activity to proteolytic and nonproteolytic ubiquitin-dependent processes. Curr. Biol. 2004, 14, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Regan-Klapisz, E.; Sorokina, I.; Voortman, J.; de Keizer, P.; Roovers, R.C.; Verheesen, P.; Urbé, S.; Fallon, L.; Fon, E.A.; Verkleij, A.; et al. Ubiquilin recruits Eps15 into ubiquitin-rich cytoplasmic aggregates via a UIM-UBL interaction. J. Cell Sci. 2005, 118, 4437–4450. [Google Scholar] [CrossRef] [PubMed]
- Kleijnen, M.F.; Shih, A.H.; Zhou, P.; Kumar, S.; Soccio, R.E.; Kedersha, N.L.; Gill, G.; Howley, P.M. The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol. Cell 2000, 6, 409–419. [Google Scholar] [CrossRef]
- Massey, L.K.; Mah, A.L.; Ford, D.L.; Miller, J.; Liang, J.; Doong, H.; Monteiro, M.J. Overexpression of ubiquilin decreases ubiquitination and degradation of presenilin proteins. J. Alzheimers Dis. 2004, 6, 79–92. [Google Scholar] [CrossRef]
- Bedford, F.K.; Kittler, J.T.; Muller, E.; Thomas, P.; Uren, J.M.; Merlo, D.; Wisden, W.; Triller, A.; Smart, T.G.; Moss, S.J. GABAA receptor cell surface number and subunit stability are regulated by the ubiquitin-like protein Plic-1. Nat. Neurosci. 2001, 4, 908–916. [Google Scholar] [CrossRef]
- Zhang, D.; Raasi, S.; Fushman, D. Affinity Makes the Difference: Nonselective Interaction of the UBA Domain of Ubiquilin-1 with Monomeric Ubiquitin and Polyubiquitin Chains. J. Mol. Biol. 2008, 377, 162–180. [Google Scholar] [CrossRef]
- Kaye, F.J.; Modi, S.; Ivanovska, I.; Koonin, E.V.; Thress, K.; Kubo, A.; Kornbluth, S.; Rose, M.D. A family of ubiquitin-like proteins binds the ATPase domain of Hsp70-like Stch. FEBS Lett. 2000, 467, 348–355. [Google Scholar] [CrossRef]
- Haapasalo, A.; Viswanathan, J.; Bertram, L.; Soininen, H.; Tanzi, R.E.; Hiltunen, M. Emerging role of Alzheimer’s disease-associated ubiquilin-1 in protein aggregation: Figure 1. Biochem. Soc. Trans. 2010, 38, 150–155. [Google Scholar] [CrossRef] [PubMed]
- El Ayadi, A.; Stieren, E.S.; Barral, J.M.; Boehning, D. Ubiquilin-1 and protein quality control in Alzheimer disease. Prion 2013, 7, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.Y.; Yang, S.; Warraich, S.T.; Blair, I.P. Ubiquilin 2: A component of the ubiquitin-proteasome system with an emerging role in neurodegeneration. Int. J. Biochem. Cell Biol. 2014, 50, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.-C.; Polymenidou, M.; Cleveland, D.W. Converging Mechanisms in ALS and FTD: Disrupted RNA and Protein Homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef]
- Viswanathan, J.; Haapasalo, A.; Böttcher, C.; Miettinen, R.; Kurkinen, K.M.A.; Lu, A.; Thomas, A.; Maynard, C.J.; Romano, D.; Hyman, B.T.; et al. Alzheimer’s Disease-Associated Ubiquilin-1 Regulates Presenilin-1 Accumulation and Aggresome Formation. Traffic 2011, 12, 330–348. [Google Scholar] [CrossRef] [PubMed]
- Stieren, E.S.; El Ayadi, A.; Xiao, Y.; Siller, E.; Landsverk, M.L.; Oberhauser, A.F.; Barral, J.M.; Boehning, D. Ubiquilin-1 is a molecular chaperone for the amyloid precursor protein. J. Biol. Chem. 2011, 286, 35689–35698. [Google Scholar] [CrossRef]
- Daoud, H.; Suhail, H.; Szuto, A.; Camu, W.; Salachas, F.; Meininger, V.; Bouchard, J.-P.; Dupré, N.; Dion, P.A.; Rouleau, G.A. UBQLN2 mutations are rare in French and French–Canadian amyotrophic lateral sclerosis. Neurobiol. Aging 2012, 33, 2230.e1. [Google Scholar] [CrossRef]
- Synofzik, M.; Maetzler, W.; Grehl, T.; Prudlo, J.; vom Hagen, J.M.; Haack, T.; Rebassoo, P.; Munz, M.; Schöls, L.; Biskup, S. Screening in ALS and FTD patients reveals 3 novel UBQLN2 mutations outside the PXX domain and a pure FTD phenotype. Neurobiol. Aging 2012, 33, 2949.e13. [Google Scholar] [CrossRef]
- Williams, K.L.; Warraich, S.T.; Yang, S.; Solski, J.A.; Fernando, R.; Rouleau, G.A.; Nicholson, G.A.; Blair, I.P. UBQLN2/ubiquilin 2 mutation and pathology in familial amyotrophic lateral sclerosis. Neurobiol. Aging 2012, 33, 2527.e3. [Google Scholar] [CrossRef]
- Brettschneider, J.; Van Deerlin, V.M.; Robinson, J.L.; Kwong, L.; Lee, E.B.; Ali, Y.O.; Safren, N.; Monteiro, M.J.; Toledo, J.B.; Elman, L.; et al. Pattern of ubiquilin pathology in ALS and FTLD indicates presence of C9ORF72 hexanucleotide expansion. Acta Neuropathol. 2012, 123, 825–839. [Google Scholar] [CrossRef] [PubMed]
- Riley, B.E.; Xu, Y.; Zoghbi, H.Y.; Orr, H.T. The effects of the polyglutamine repeat protein ataxin-1 on the UbL-UBA protein A1Up. J. Biol. Chem. 2004, 279, 42290–42301. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yue, H.-W.; He, W.-T.; Hong, J.-Y.; Jiang, L.-L.; Hu, H.-Y. PolyQ-expanded huntingtin and ataxin-3 sequester ubiquitin adaptors hHR23B and UBQLN2 into aggregates via conjugated ubiquitin. FASEB J. 2018, 32, 2923–2933. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, N.J.; Lewis, J.; Clippinger, A.K.; Thomas, M.A.; Adamson, J.; Cruz, P.E.; Cannon, A.; Xu, G.; Golde, T.E.; Shaw, G.; et al. Unbiased screen reveals ubiquilin-1 and -2 highly associated with huntingtin inclusions. Brain Res. 2013, 1524, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Stiles, S.G.; Feichtmeier, J.M.; Ramesh, N.; Zhan, L.; Scalf, M.A.; Smith, L.M.; Bhan Pandey, U.; Tibbetts, R.S. Mutation-dependent aggregation and toxicity in a Drosophila model for UBQLN2-associated ALS. Hum. Mol. Genet. 2018, 27, 322–337. [Google Scholar] [CrossRef]
- Hanson, K.A.; Kim, S.H.; Wassarman, D.A.; Tibbetts, R.S. Ubiquilin Modifies TDP-43 Toxicity in a Drosophila Model of Amyotrophic Lateral Sclerosis (ALS). J. Biol. Chem. 2010, 285, 11068–11072. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Shi, Y.; Hanson, K.A.; Williams, L.M.; Sakasai, R.; Bowler, M.J.; Tibbetts, R.S. Potentiation of Amyotrophic Lateral Sclerosis (ALS)-associated TDP-43 Aggregation by the Proteasome-targeting Factor, Ubiquilin 1. J. Biol. Chem. 2009, 284, 8083–8092. [Google Scholar] [CrossRef]
- Picher-Martel, V.; Dutta, K.; Phaneuf, D.; Sobue, G.; Julien, J.P. Ubiquilin-2 drives NF-κB activity and cytosolic TDP-43 aggregation in neuronal cells. Mol. Brain 2015, 8, 71. [Google Scholar] [CrossRef]
- Gkazi, S.A.; Troakes, C.; Topp, S.; Miller, J.W.; Vance, C.A.; Sreedharan, J.; Al-Chalabi, A.; Kirby, J.; Shaw, P.J.; Al-Sarraj, S.; et al. Striking phenotypic variation in a family with the P506S UBQLN2 mutation including amyotrophic lateral sclerosis, spastic paraplegia, and frontotemporal dementia. Neurobiol. Aging 2019, 73, 229.e5. [Google Scholar] [CrossRef]
- Osaka, M.; Ito, D.; Suzuki, N. Disturbance of proteasomal and autophagic protein degradation pathways by amyotrophic lateral sclerosis-linked mutations in ubiquilin 2. Biochem. Biophys. Res. Commun. 2016, 472, 324–331. [Google Scholar] [CrossRef]
- Li, A.; Xie, Z.; Dong, Y.; McKay, K.M.; McKee, M.L.; Tanzi, R.E. Isolation and characterization of the Drosophila ubiquilin ortholog dUbqln: In vivo interaction with early-onset Alzheimer disease genes. Hum. Mol. Genet. 2007, 16, 2626–2639. [Google Scholar] [CrossRef]
- Jantrapirom, S.; Lo Piccolo, L.; Yoshida, H.; Yamaguchi, M. A new Drosophila model of Ubiquilin knockdown shows the effect of impaired proteostasis on locomotive and learning abilities. Exp. Cell Res. 2017, 362, 461–471. [Google Scholar] [CrossRef]
- Atkin, G.; Paulson, H. Ubiquitin pathways in neurodegenerative disease. Front. Mol. Neurosci. 2014, 7, 63. [Google Scholar] [CrossRef]
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018, 9, 1097. [Google Scholar] [CrossRef]
- Venkatraman, P.; Wetzel, R.; Tanaka, M.; Nukina, N.; Goldberg, A.L. Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol. Cell 2004, 14, 95–104. [Google Scholar] [CrossRef]
- Holmberg, C.I.; Staniszewski, K.E.; Mensah, K.N.; Matouschek, A.; Morimoto, R.I. Inefficient degradation of truncated polyglutamine proteins by the proteasome. EMBO J. 2004, 23, 4307–4318. [Google Scholar] [CrossRef]
- Jana, N.R.; Zemskov, E.A.; Wang, G.H.; Nukina, N. Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum. Mol. Genet. 2001, 10, 1049–1059. [Google Scholar] [CrossRef]
- Lee, F.K.M.; Wong, A.K.Y.; Lee, Y.W.; Wan, O.W.; Edwin Chan, H.Y.; Chung, K.K.K. The role of ubiquitin linkages on α-synuclein induced-toxicity in a Drosophila model of Parkinson’s disease. J. Neurochem. 2009, 110, 208–219. [Google Scholar] [CrossRef]
- Al-Ramahi, I.; Lam, Y.C.; Chen, H.-K.; de Gouyon, B.; Zhang, M.; Pérez, A.M.; Branco, J.; de Haro, M.; Patterson, C.; Zoghbi, H.Y.; et al. CHIP Protects from the Neurotoxicity of Expanded and Wild-type Ataxin-1 and Promotes Their Ubiquitination and Degradation. J. Biol. Chem. 2006, 281, 26714–26724. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Fishman, P.S.; Thakor, N.V.; Oyler, G.A. Parkin Facilitates the Elimination of Expanded Polyglutamine Proteins and Leads to Preservation of Proteasome Function. J. Biol. Chem. 2003, 278, 22044–22055. [Google Scholar] [CrossRef]
- Schmidt, M.; Finley, D. Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 13–25. [Google Scholar] [CrossRef]
- Seo, H.; Sonntag, K.-C.; Kim, W.; Cattaneo, E.; Isacson, O. Proteasome Activator Enhances Survival of Huntington’s Disease Neuronal Model Cells. PLoS ONE 2007, 2, e238. [Google Scholar] [CrossRef]
- Chondrogianni, N.; Gonos, E.S. Overexpression of hUMP1/POMP proteasome accessory protein enhances proteasome-mediated antioxidant defence. Exp. Gerontol. 2007, 42, 899–903. [Google Scholar] [CrossRef]
- Leestemaker, Y.; de Jong, A.; Witting, K.F.; Penning, R.; Schuurman, K.; Rodenko, B.; Zaal, E.A.; van de Kooij, B.; Laufer, S.; Heck, A.J.R.; et al. Proteasome Activation by Small Molecules. Cell Chem. Biol. 2017, 24, 725–736. [Google Scholar] [CrossRef]
- Dantuma, N.P.; Bott, L.C. The ubiquitin-proteasome system in neurodegenerative diseases: Precipitating factor, yet part of the solution. Front. Mol. Neurosci. 2014, 7, 70. [Google Scholar] [CrossRef]
- Lee, B.-H.; Lee, M.J.; Park, S.; Oh, D.-C.; Elsasser, S.; Chen, P.-C.; Gartner, C.; Dimova, N.; Hanna, J.; Gygi, S.P.; et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 2010, 467, 179–184. [Google Scholar] [CrossRef]
- Yuan, N.-N.; Cai, C.-Z.; Wu, M.-Y.; Zhu, Q.; Su, H.; Li, M.; Ren, J.; Tan, J.-Q.; Lu, J.-H. Canthin-6-One Accelerates Alpha-Synuclein Degradation by Enhancing UPS Activity: Drug Target Identification by CRISPR-Cas9 Whole Genome-Wide Screening Technology. Front. Pharmacol. 2019, 10, 16. [Google Scholar] [CrossRef]
- Huang, L.; Ho, P.; Chen, C.-H. Activation and inhibition of the proteasome by betulinic acid and its derivatives. FEBS Lett. 2007, 581, 4955–4959. [Google Scholar] [CrossRef]
- Myeku, N.; Clelland, C.L.; Emrani, S.; Kukushkin, N.V.; Yu, W.H.; Goldberg, A.L.; Duff, K.E. Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat. Med. 2016, 22, 46–53. [Google Scholar] [CrossRef]
- Liu, Y.; Hettinger, C.L.; Zhang, D.; Rezvani, K.; Wang, X.; Wang, H. Sulforaphane enhances proteasomal and autophagic activities in mice and is a potential therapeutic reagent for Huntington’s disease. J. Neurochem. 2014, 129, 539–547. [Google Scholar] [CrossRef]
- Newman, T.; Sinadinos, C.; Johnston, A.; Sealey, M.; Mudher, A. Using Drosophila models of neurodegenerative diseases for drug discovery. Expert Opin. Drug Discov. 2011, 6, 129–140. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Uniprot | Human Orthologs | Protein Identity (%) | Domain Identity (%) |
|---|---|---|---|---|
| rngo | Q9VXF9 | Ddi1 Ddi2 | 49.1 | UbL (39.7) UbL (39.7) |
| NUB1 | Q9VRF3 | NUB1 | 31.9 | UbL (30.3) UbA-1 (50) UBA-2 (26.7) |
| Rad23/1 | Q9V3W9 | Rad23A Rad23A | 36.7 36.7 | Rad23A/UbL (52.2) Rad23A/UbA-1 (100) Rad23A/UbA-2 (84.2) |
| ISOT-3A | Q86LF0 | KPC2 | 31.3 | UbA-1 (37.1) UbA-2 (53.1) |
| Ubqn | Q9VWD9 | UBQLN1 UBQLN2 UBQLN4 | 50.2 47.6 46.5 | UBQLN2/UbL (56.3) UBQLN2/UbA (84.6) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jantrapirom, S.; Lo Piccolo, L.; Yamaguchi, M. Non-Proteasomal UbL-UbA Family of Proteins in Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 1893. https://doi.org/10.3390/ijms20081893
Jantrapirom S, Lo Piccolo L, Yamaguchi M. Non-Proteasomal UbL-UbA Family of Proteins in Neurodegeneration. International Journal of Molecular Sciences. 2019; 20(8):1893. https://doi.org/10.3390/ijms20081893
Chicago/Turabian StyleJantrapirom, Salinee, Luca Lo Piccolo, and Masamitsu Yamaguchi. 2019. "Non-Proteasomal UbL-UbA Family of Proteins in Neurodegeneration" International Journal of Molecular Sciences 20, no. 8: 1893. https://doi.org/10.3390/ijms20081893
APA StyleJantrapirom, S., Lo Piccolo, L., & Yamaguchi, M. (2019). Non-Proteasomal UbL-UbA Family of Proteins in Neurodegeneration. International Journal of Molecular Sciences, 20(8), 1893. https://doi.org/10.3390/ijms20081893