Control Mechanisms of the Tumor Suppressor PDCD4: Expression and Functions


Abstract
1. Introduction
2. The Function of PDCD4
2.1. PDCD4 Inhibits Neoplastic Transformation
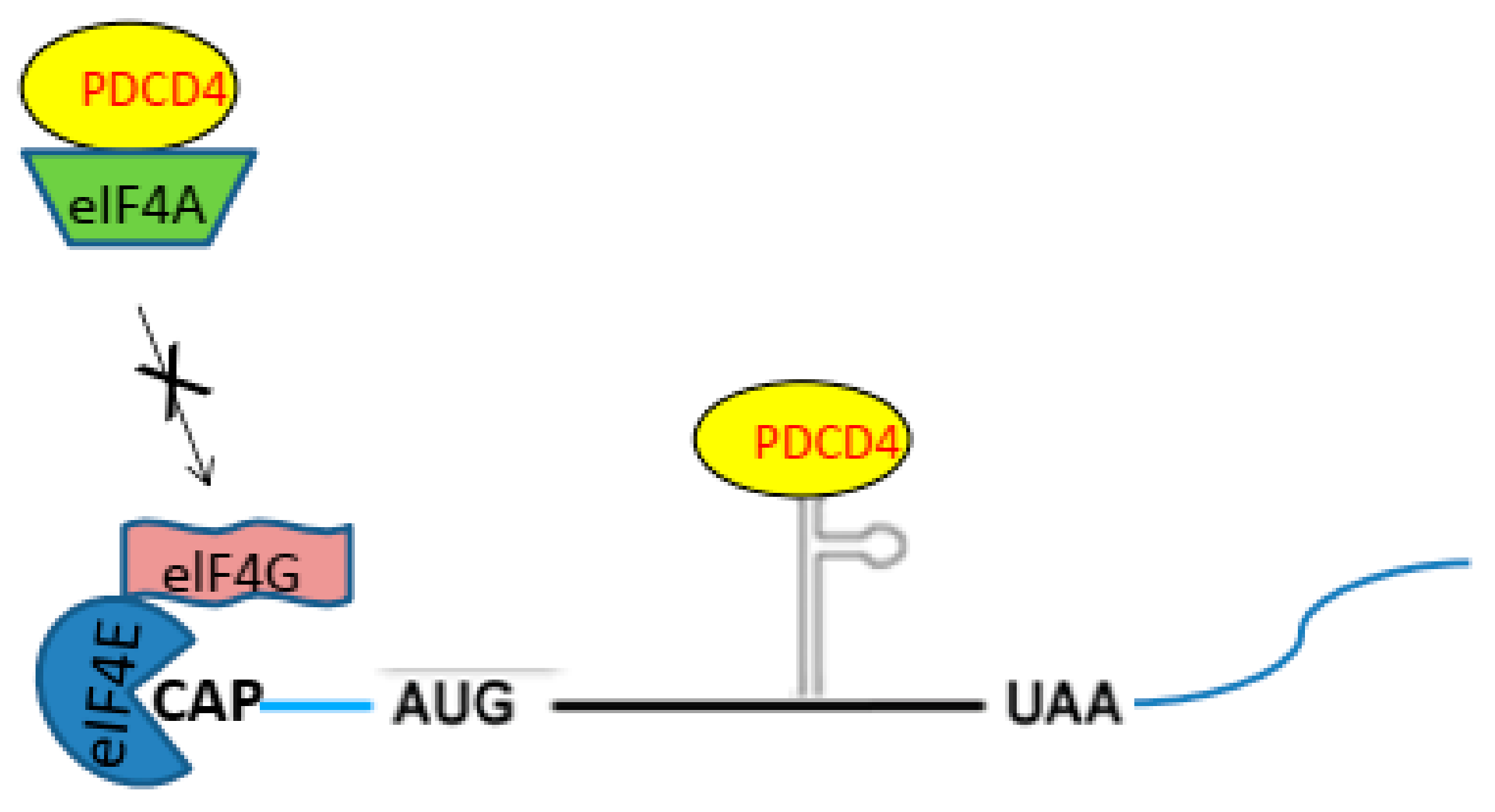
2.2. PDCD4 Controls Translation
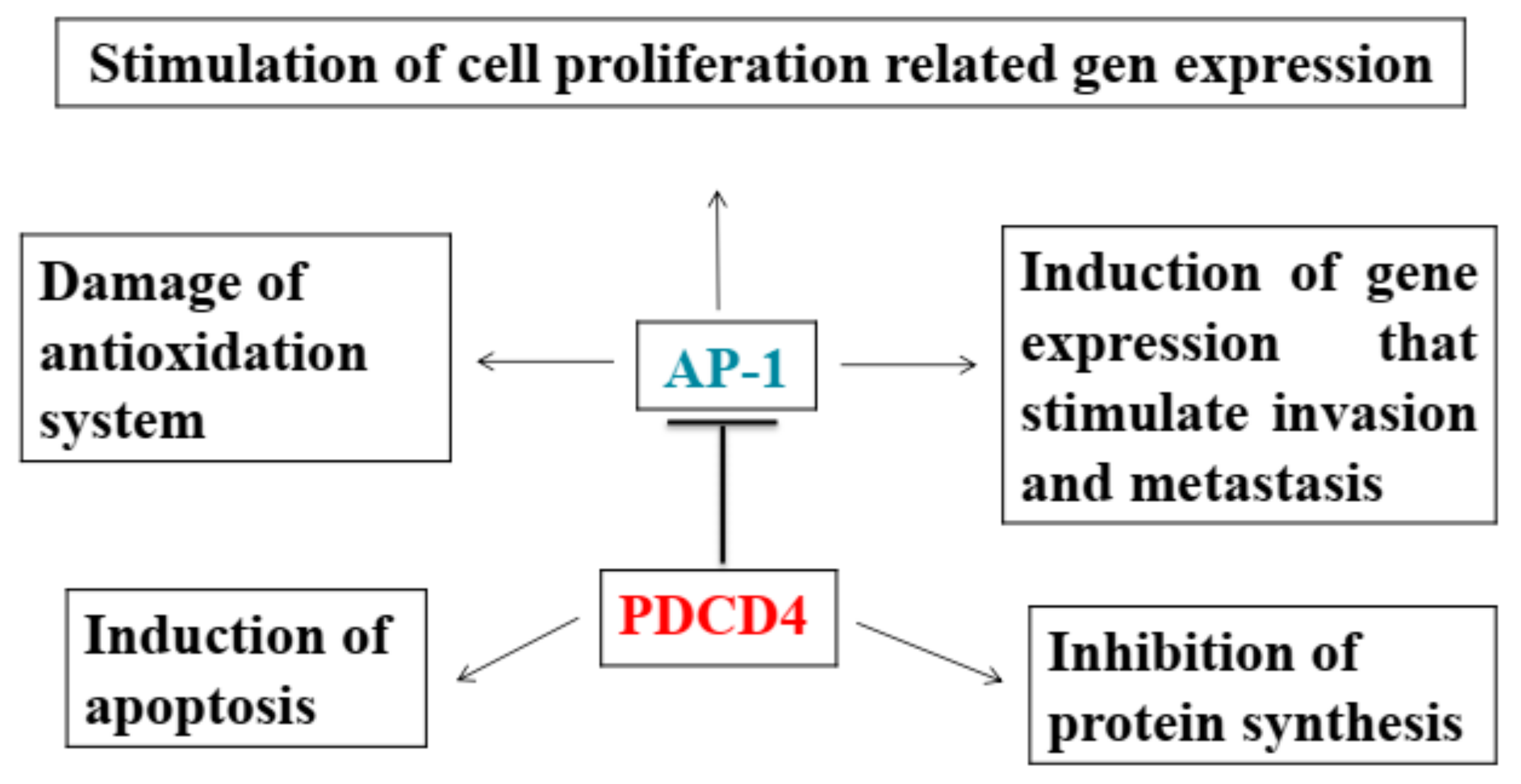
2.3. PDCD4 Knockdown Activates AP-1 and β-catenin/Tcf-dependent Transcriptions
2.4. Inhibition of Transcriptions
2.5. Interaction with Cytoplasmic Factors
2.6. Induction of Apoptosis
3. Regulation of PDCD4 Expression
3.1. Controls at the Transcription
3.2. The Inhibition of PDCD4 Translation by miRNAs
3.3. Controls at Protein Degradation
3.4. Arginine Methylation of PDCD4 Protein
4. The Tumor Promotors EGF and TPA Induce PDCD4 Degradation in Huh7 Hepatoma Cells
5. Ser71 and Ser76 Are Phosphorylated by Different Enzymes
6. EGF Down-Regulates PDCD4 mRNA Levels but TPA Does Not
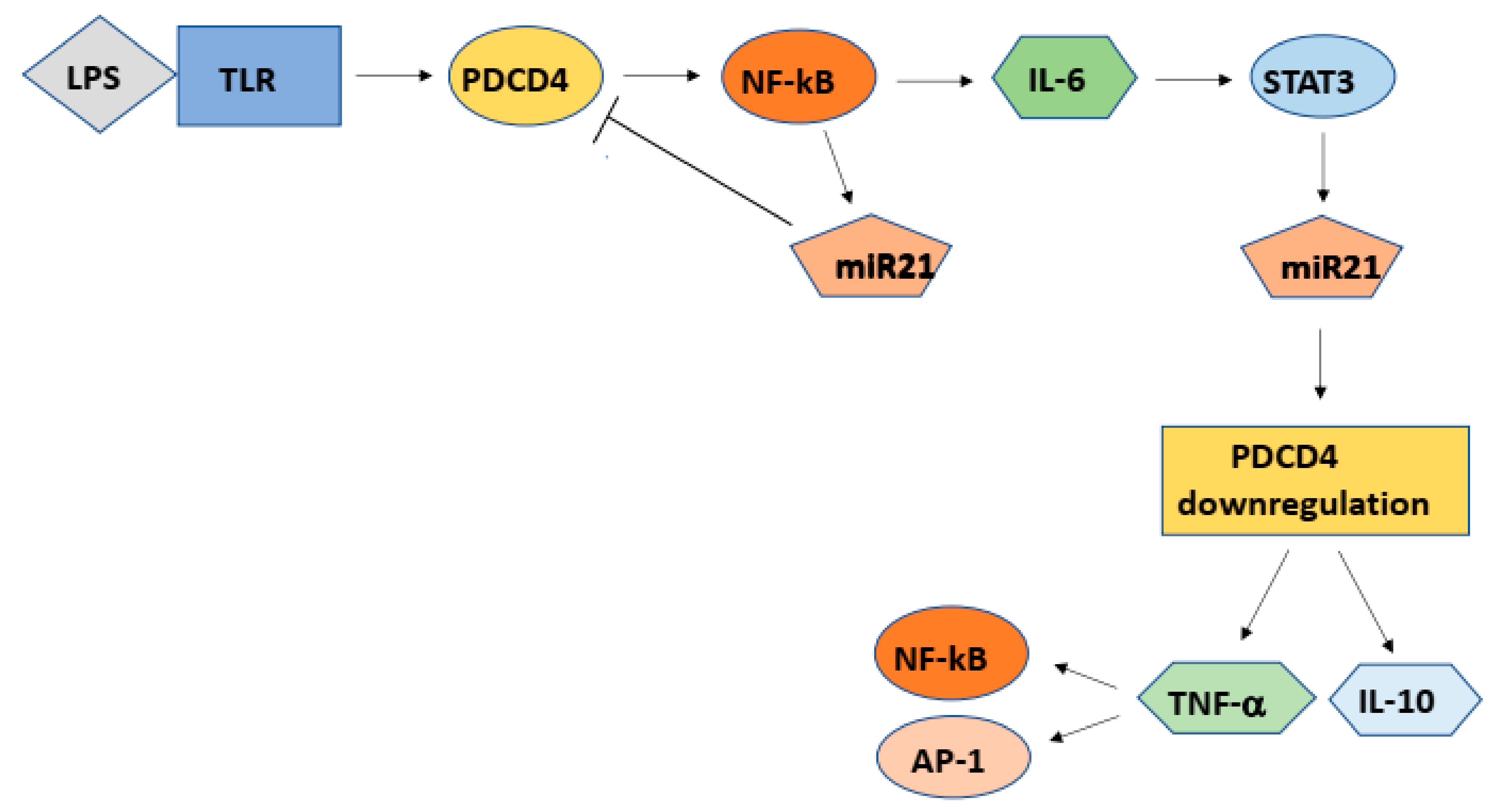
7. Inflammation and Carcinogenesis
8. Clinical Aspects
9. Future Perspectives and Open Questions
Author Contributions
Funding
Conflicts of Interest
References
- Fürstenberger, G.; Berry, D.L.; Sorg, B.; Marks, F. Skin tumor promotion by phorbol esters is a two-stage process. Proc. Natl. Acad Sci. USA 1981, 78, 7722–7726. [Google Scholar]
- Fabregat, I.; Sanchez, A.; Alvarez, A.M.; Nakamura, T.; Benito, M. Epidermal growth factor, but not hepatocyte growth factor, suppresses the apoptosis induced by transforming growth factor-beta in fetal hepatocytes in primary culture. FEBS Lett. 1996, 384, 14–18. [Google Scholar] [CrossRef][Green Version]
- Colburn, N.H.; Former, B.F.; Nelson, K.A.; Yuspa, S.H. Tumour promoter induces anchorage independence irreversibly. Nature 1979, 281, 589–591. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Birrer, M.J.; Watts, R.G.; Matrisian, L.M.; Colburn, N.H. Blocking of tumor promoter-induced AP-1 activity inhibits induced transformation in JB6 mouse epidermal cells. Proc. Natl. Acad Sci. USA 1994, 91, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, L.; Colburn, N. AP1/jun function is differentially induced in promotion-sensitive and resistant JB6 cells. Science 1989, 244, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Cmarik, J.L.; Min, H.; Hegamyer, G.; Zhan, S.; Kulesz-Martin, M.; Yoshinaga, H.; Matsuhashi, S.; Colburn, N.H. Differentially expressed protein Pdcd4 inhibits tumor promoter-induced neoplastic transformation. Proc. Natl. Acad Sci. USA 1999, 96, 14037–14042. [Google Scholar] [CrossRef] [PubMed]
- Matsuhashi, S.; Ozaki, I. Programmed Cell Death 4. In Encyclopedia of Cancer; Schwab, M., Ed.; Springer-Verlag Berlin Heidelberg: Berlin, Germany, 2017; p. 3454. [Google Scholar]
- Azzoni, L.; Zatsepina, O.; Abebe, B.; Bennett, I.M.; Kanakaraj, P.; Perussia, B. Differential Transcriptional Regulation of CD161 and a Novel Gene, 197/15a, by IL-2, IL-15, and IL-12 in NK and T Cells. J. Immunol. 1998, 161, 3493–3500. [Google Scholar] [PubMed]
- Shibahara, K.; Asano, M.; Ishida, Y.; Aoki, T.; Koike, T.; Honjo, T. Isolation of a novel mouse gene MA-3 that is induced upon programmed cell death. Gene 1995, 166, 297–301. [Google Scholar] [CrossRef]
- Onishi, Y.; Hashimoto, S.; Kizaki, H. Cloning of the TIS gene suppressed by topoisomerase inhibitors. Gene 1998, 215, 453–459. [Google Scholar] [CrossRef]
- Göke, A.; Göke, R.; Knolle, A.; Trusheim, H.; Schmidt, H.; Wilmen, A.; Carmody, R.; Göke, B.; Chen, Y.H. DUG is a novel homologue of translation initiation factor 4G that binds eIF4A. Biochem. Biophys. Res. Commun. 2002, 297, 78–82. [Google Scholar] [CrossRef]
- Schlichter, U.; Kattmann, D.; Appl, H.; Miethe, J.; Brehmer-Fastnacht, A.; Klempnauer, K.-H. Identification of the myb-inducible promoter of the chicken Pdcd4 gene1The GeneBank accession number for the Pdcd4 promotor sequence is AF382032.1. Biochim. Biophys. Acta 2001, 1520, 99–104. [Google Scholar] [CrossRef]
- Klein, S.L.; Strausberg, R.L.; Wagner, L.; Pontius, J.; Clifton, S.W.; Richardson, P. Genetic and genomic tools for Xenopus research: The NIH Xenopus initiative. Dev. Dyn. 2002, 225, 384–391. [Google Scholar] [CrossRef]
- Venkatesh, B.; Lee, A.P.; Ravi, V.; Maurya, A.K.; Lian, M.M.; Swann, J.B.; Ohta, Y.; Flajnik, M.F.; Sutoh, Y.; Kasahara, M.; et al. Elephant shark genome provides unique insights into gnathostome evolution. Nature 2014, 505, 174. [Google Scholar] [CrossRef] [PubMed]
- Cash, A.C.; Andrews, J. Fine scale analysis of gene expression in Drosophila melanogaster gonads reveals Programmed cell death 4 promotes the differentiation of female germline stem cells. BMC Dev. Biol. 2012, 12, 4. [Google Scholar] [CrossRef]
- Cheng, S.; Liu, R.; Gallie, D.R. The unique evolution of the programmed cell death 4 protein in plants. BMC Evol. Biol. 2013, 13, 199. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Knösel, T.; Kristiansen, G.; Pietas, A.; Garber, M.E.; Matsuhashi, S.; Ozaki, I.; Petersen, I. Loss of PDCD4 expression in human lung cancer correlates with tumour progression and prognosis. J. Pathol. 2003, 200, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ozaki, I.; Mizuta, T.; Hamajima, H.; Yasutake, T.; Eguchi, Y.; Ideguchi, H.; Yamamoto, K.; Matsuhashi, S. Involvement of programmed cell death 4 in transforming growth factor-β1-induced apoptosis in human hepatocellular carcinoma. Oncogene 2006, 25, 6101. [Google Scholar] [CrossRef]
- Gao, F.; Wang, X.; Zhu, F.; Wang, Q.; Zhang, X.; Guo, C.; Zhou, C.; Ma, C.; Sun, W.; Zhang, Y.; et al. PDCD4 gene silencing in gliomas is associated with 5′CpG island methylation and unfavourable prognosis. J. Cell. Mol. Med. 2009, 13, 4257–4267. [Google Scholar] [CrossRef]
- Kakimoto, T.; Shiraishi, R.; Iwakiri, R.; Fujimoto, K.; Takahashi, H.; Hamajima, H.; Mizuta, T.; Ideguchi, H.; Toda, S.; Kitajima, Y.; et al. Expression patterns of the tumor suppressor PDCD4 and correlation with β-catenin expression in gastric cancers. Oncol. Rep. 2011, 26, 1385–1392. [Google Scholar]
- Hilliard, A.; Hilliard, B.; Zheng, S.-J.; Sun, H.; Miwa, T.; Song, W.; Göke, R.; Chen, Y.H. Translational Regulation of Autoimmune Inflammation and Lymphoma Genesis by Programmed Cell Death 4. J. Immunol. 2006, 177, 8095–8102. [Google Scholar] [CrossRef]
- Jansen, A.P.; Camalier, C.E.; Colburn, N.H. Epidermal Expression of the Translation Inhibitor Programmed Cell Death 4 Suppresses Tumorigenesis. Cancer Res. 2005, 65, 6034–6041. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Koonin, E.V. Eukaryote-specific domains in translation initiation factors: Implications for translation regulation and evolution of the translation system. Genome Res. 2000, 10, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yang, H.-S. The role of Pdcd4 in tumour suppression and protein translation. Biol. Cell 2018, 110, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Wedeken, L.; Singh, P.; Klempnauer, K.-H. Tumor Suppressor Protein Pdcd4 Inhibits Translation of p53 mRNA. J. Biol. Chem. 2011, 286, 42855–42862. [Google Scholar] [CrossRef]
- Wang, Q.; Zhu, J.; Wang, Y.W.; Dai, Y.; Wang, Y.L.; Wang, C.; Liu, J.; Baker, A.; Colburn, N.H.; Yang, H.S. Tumor suppressor Pdcd4 attenuates Sin1 translation to inhibit invasion in colon carcinoma. Oncogene 2017, 36, 6225. [Google Scholar] [CrossRef] [PubMed]
- Böhm, M.; Sawicka, K.; Siebrasse, J.P.; Brehmer-Fastnacht, A.; Peters, R.; Klempnauer, K.-H. The transformation suppressor protein Pdcd4 shuttles between nucleus and cytoplasm and binds RNA. Oncogene 2003, 22, 4905. [Google Scholar] [CrossRef]
- Singh, P.; Wedeken, L.; Waters, L.C.; Carr, M.D.; Klempnauer, K.H. Pdcd4 directly binds the coding region of c-myb mRNA and suppresses its translation. Oncogene 2011, 30, 4864. [Google Scholar] [CrossRef]
- Biyanee, A.; Ohnheiser, J.; Singh, P.; Klempnauer, K.H. A novel mechanism for the control of translation of specific mRNAs by tumor suppressor protein Pdcd4: Inhibition of translation elongation. Oncogene 2014, 34, 1384. [Google Scholar] [CrossRef]
- Liwak, U.; Thakor, N.; Jordan, L.E.; Roy, R.; Lewis, S.M.; Pardo, O.E.; Seckl, M.; Holcik, M. Tumor Suppressor PDCD4 Represses Internal Ribosome Entry Site-Mediated Translation of Antiapoptotic Proteins and Is Regulated by S6 Kinase 2. Mol. Cell. Biol. 2012, 32, 1818–1829. [Google Scholar] [CrossRef]
- Haas, A.; Ulrich, D.; Müller, J.P.; Ohnheiser, J.; Fehler, O.; Singh, P.; Klempnauer, K.-H. An evolutionarily conserved interaction of tumor suppressor protein Pdcd4 with the poly(A)-binding protein contributes to translation suppression by Pdcd4. Nucleic Acids Res. 2014, 42, 11107–11118. [Google Scholar]
- Ozanne, B.W.; McGarry, L.; Spence, H.J.; Johnston, I.; Winnie, J.; Meagher, L.; Stapleton, G. Transcriptional regulation of cell invasion: AP-1 regulation of a multigenic invasion programme. Eur. J. Cancer 2000, 36, 1640–1648. [Google Scholar] [CrossRef]
- Yang, H.-S.; Matthews, C.P.; Clair, T.; Wang, Q.; Baker, A.R.; Li, C.-C.H.; Tan, T.-H.; Colburn, N.H. Tumorigenesis Suppressor Pdcd4 Down-Regulates Mitogen-Activated Protein Kinase Kinase Kinase Kinase 1 Expression to Suppress Colon Carcinoma Cell Invasion. Mol. Cell. Biol. 2006, 26, 1297–1306. [Google Scholar] [CrossRef]
- Bitomsky, N.; Böhm, M.; Klempnauer, K.-H. Transformation suppressor protein Pdcd4 interferes with JNK-mediated phosphorylation of c-Jun and recruitment of the coactivator p300 by c-Jun. Oncogene 2004, 23, 7484. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Yang, H.-S. Pdcd4 knockdown up-regulates MAP4K1 expression and activation of AP-1 dependent transcription through c-Myc. Biochim. Biophys. Acta 2012, 1823, 1807–1814. [Google Scholar] [CrossRef]
- Wang, Q.; Sun, Z.X.; Allgayer, H.; Yang, H.S. Downregulation of E-cadherin is an essential event in activating β-catenin/Tcf-dependent transcription and expression of its target genes in Pdcd4 knockdown cells. Oncogene 2009, 29, 128. [Google Scholar] [CrossRef]
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; García de Herreros, A. The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Izumi, H.; Tanimoto, A.; Takahashi, M.; Miyamoto, N.; Kashiwagi, E.; Kidani, A.; Hirano, G.; Masubuchi, D.; Fukunaka, Y.; et al. Programmed Cell Death Protein 4 Down-regulates Y-Box Binding Protein-1 Expression via a Direct Interaction with Twist1 to Suppress Cancer Cell Growth. Cancer Res. 2009, 69, 3148–3156. [Google Scholar] [CrossRef]
- Leupold, J.H.; Yang, H.S.; Colburn, N.H.; Asangani, I.; Post, S.; Allgayer, H. Tumor suppressor Pdcd4 inhibits invasion/intravasation and regulates urokinase receptor (u-PAR) gene expression via Sp-transcription factors. Oncogene 2007, 26, 4550. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.R.; Young, M.R.; Colburn, N.H.; Hwang, S.-K. Tumor suppressor PDCD4 inhibits NF-κB-dependent transcription in human glioblastoma cells by direct interaction with p65. Carcinogenesis 2014, 35, 1469–1480. [Google Scholar]
- Jo, S.-H.; Kim, D.E.; Clocchiatti, A.; Dotto, G.P. PDCD4 is a CSL associated protein with a transcription repressive function in cancer associated fibroblast activation. Oncotarget 2016, 7, 58717–58727. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kumar, N.; Wethkamp, N.; Waters, L.C.; Carr, M.D.; Klempnauer, K.H. Tumor suppressor protein Pdcd4 interacts with Daxx and modulates the stability of Daxx and the Hipk2-dependent phosphorylation of p53 at serine 46. Oncogenesis 2013, 2, e37. [Google Scholar] [CrossRef]
- Eto, K.; Goto, S.; Nakashima, W.; Ura, Y.; Abe, S.I. Loss of programmed cell death 4 induces apoptosis by promoting the translation of procaspase-3 mRNA. Cell Death Differ. 2011, 19, 573. [Google Scholar] [CrossRef]
- Guo, J.; Ozaki, I.; Xia, J.; Kuwashiro, T.; Kojima, M.; Takahashi, H.; Ashida, K.; Anzai, K.; Matsuhashi, S. PDCD4 Knockdown Induces Senescence in Hepatoma Cells by Up-Regulating the p21 Expression. Front. Oncol 2019, 8, 661. [Google Scholar] [CrossRef]
- Boward, B.; Wu, T.; Dalton, S. Concise Review: Control of Cell Fate Through Cell Cycle and Pluripotency Networks. Stem Cells 2016, 34, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Alessio, N.; Capasso, S.; Ferone, A.; Di Bernardo, G.; Cipollaro, M.; Casale, F.; Peluso, G.; Giordano, A.; Galderisi, U. Misidentified Human Gene Functions with Mouse Models: The Case of the Retinoblastoma Gene Family in Senescence. Neoplasia (New York, N.Y.) 2017, 19, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.-J.; Ahn, H.-S.; Lee, J.-Y.; Matsuhashi, S.; Park, W.-Y. Up-regulation of PDCD4 in senescent human diploid fibroblasts. Biochem. Biophys. Res. Commun. 2002, 293, 617–621. [Google Scholar] [CrossRef]
- Guo, X.; Li, W.; Wang, Q.; Yang, H.-S. AKT Activation by Pdcd4 Knockdown Up-Regulates Cyclin D1 Expression and Promotes Cell Proliferation. Genes Cancer 2011, 2, 818. [Google Scholar] [CrossRef]
- Leupold, J.H.; Asangani, I.A.; Mudduluru, G.; Allgayer, H. Promoter cloning and characterization of the human programmed cell death protein 4 (pdcd4) gene: Evidence for ZBP-89 and Sp-binding motifs as essential Pdcd4 regulators. Biosci. Rep. 2012, 32, 281–297. [Google Scholar] [CrossRef]
- Appl, H.; Klempnauer, K.-H. Targeted disruption of c-myb in the chicken pre B-cell line DT40. Oncogene 2002, 21, 3076. [Google Scholar] [CrossRef][Green Version]
- Jansson, M.D.; Lund, A.H. MicroRNA and cancer. Mol. Oncol. 2012, 6, 590–610. [Google Scholar] [CrossRef]
- Melnik, B.C. MiR-21: An environmental driver of malignant melanoma? J. Transl. Med. 2015, 13, 202. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, Y.B.; Zhang, X.H.; Yu, X.L.; Wang, Z.B.; Cheng, X.C. MicroRNA-21 gene and cancer. Med. Oncol. 2013, 30, 376. [Google Scholar] [CrossRef] [PubMed]
- Selcuklu, S.D.; Donoghue, M.T.; Spillane, C. miR-21 as a key regulator of oncogenic processes. Biochem. Soc. Trans. 2009, 37, 918–925. [Google Scholar] [CrossRef]
- Medina, P.P.; Nolde, M.; Slack, F.J. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature 2010, 467, 86. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.A.; Rasheed, S.A.K.; Nikolova, D.A.; Leupold, J.H.; Colburn, N.H.; Post, S.; Allgayer, H. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 2007, 27, 2128. [Google Scholar] [CrossRef] [PubMed]
- Frankel, L.B.; Christoffersen, N.R.; Jacobsen, A.; Lindow, M.; Krogh, A.; Lund, A.H. Programmed Cell Death 4 (PDCD4) Is an Important Functional Target of the MicroRNA miR-21 in Breast Cancer Cells. J. Biol. Chem. 2008, 283, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Ning, F.-L.; Wang, F.; Li, M.-L.; Yu, Z.-S.; Hao, Y.-Z.; Chen, S.-S. MicroRNA-182 modulates chemosensitivity of human non-small cell lung cancer to cisplatin by targeting PDCD4. Diagn. Pathol. 2014, 9, 143. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Xu, Z.; Yuan, M.; Zhang, Y.; Zhao, B.; Wang, J.; Zhang, A.; Li, G. MicroRNA-16 suppresses the activation of inflammatory macrophages in atherosclerosis by targeting PDCD4. Int. J. Mol. Med. 2016, 37, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, J.; Li, J.; Wang, X.; Song, W. MicroRNA-150 promotes cell proliferation, migration, and invasion of cervical cancer through targeting PDCD4. Biomed. Pharmacother. 2018, 97, 511–517. [Google Scholar] [CrossRef]
- Zhang, X.; Gee, H.; Rose, B.; Lee, C.S.; Clark, J.; Elliott, M.; Gamble, J.R.; Cairns, M.J.; Harris, A.; Khoury, S.; et al. Regulation of the tumour suppressor PDCD4 by miR-499 and miR-21 in oropharyngeal cancers. BMC Cancer 2016, 16, 86. [Google Scholar] [CrossRef]
- Dorrello, N.V.; Peschiaroli, A.; Guardavaccaro, D.; Colburn, N.H.; Sherman, N.E.; Pagano, M. S6K1- and ßTRCP-Mediated Degradation of PDCD4 Promotes Protein Translation and Cell Growth. Science 2006, 314, 467–471. [Google Scholar] [CrossRef]
- Schmid, T.; Jansen, A.P.; Baker, A.R.; Hegamyer, G.; Hagan, J.P.; Colburn, N.H. Translation Inhibitor Pdcd4 Is Targeted for Degradation during Tumor Promotion. Cancer Res. 2008, 68, 1254–1260. [Google Scholar] [CrossRef]
- Fay, M.M.; Clegg, J.M.; Uchida, K.A.; Powers, M.A.; Ullman, K.S. Enhanced Arginine Methylation of Programmed Cell Death 4 Protein during Nutrient Deprivation Promotes Tumor Cell Viability. J. Biol. Chem. 2014, 289, 17541–17552. [Google Scholar] [CrossRef]
- Shima, Y.; Nakao, K.; Nakashima, T.; Kawakami, A.; Nakata, K.; Hamasaki, K.; Kato, Y.; Eguchi, K.; Ishii, N. Activation of caspase-8 in transforming growth factor-β–induced apoptosis of human hepatoma cells. Hepatology 1999, 30, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Matsuhashi, S.; Hamajima, H.; Xia, J.; Zhang, H.; Mizuta, T.; Anzai, K.; Ozaki, I. Control of a tumor suppressor PDCD4: Degradation mechanisms of the protein in hepatocellular carcinoma cells. Cell. Signal. 2014, 26, 603–610. [Google Scholar] [CrossRef]
- Wang, Y.; Mei, H.; Shao, Q.; Wang, J.; Lin, Z. Association of ribosomal protein S6 kinase 1 with cellular radiosensitivity of non-small lung cancer. Int. J. Radiat. Biol. 2017, 93, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, M.; Hamajima, H.; Xia, J.; Iwane, S.; Kwaguchi, Y.; Eguchi, Y.; Mizuta, T.; Fujimoto, K.; Ozaki, I.; Matsuhashi, S. Regulation of tumor suppressor PDCD4 by novel protein kinase C isoforms. Biochim. Biophys. Acta 2010, 1803, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Cardozo, T.; Pagano, M. The SCF ubiquitin ligase: Insights into a molecular machine. Nat. Rev. Mol. Cell. Biol. 2004, 5, 739. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Hu, K.; Zhang, S.; Dong, X.; Yin, Z.; Meng, R.; Zhao, Y.; Dai, X.; Zhang, T.; Yang, K.; et al. S6K1 phosphorylation-dependent degradation of Mxi1 by β-Trcp ubiquitin ligase promotes Myc activation and radioresistance in lung cancer. Theranostics 2018, 8, 1286–1300. [Google Scholar] [CrossRef]
- Galan, J.A.; Geraghty, K.M.; Lavoie, G.; Kanshin, E.; Tcherkezian, J.; Calabrese, V.; Jeschke, G.R.; Turk, B.E.; Ballif, B.A.; Blenis, J.; et al. Phosphoproteomic analysis identifies the tumor suppressor PDCD4 as a RSK substrate negatively regulated by 14-3-3. Proc. Natl. Acad Sci. USA 2014, 111, E2918–E2927. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, R.; Holz, M.K. RSK-mediated down-regulation of PDCD4 is required for proliferation, survival, and migration in a model of triple-negative breast cancer. Oncotarget 2016, 7, 27567–27583. [Google Scholar] [CrossRef]
- Vikhreva, P.N.; Shepelev, M.V.; Korobko, I.V. mTOR-dependent transcriptional repression of Pdcd4 tumor suppressor in lung cancer cells. Biochim. Biophys. Acta 2014, 1839, 43–49. [Google Scholar] [CrossRef]
- Weigert, A.; Brüne, B.; Rübsamen, D.; Blees, J.S.; Schulz, K.; Milke, L.; Eifler, L.K.; Bajer, M.M.; Schmid, T.; Baker, A.R.; et al. Inflammation-induced loss of Pdcd4 is mediated by phosphorylation-dependent degradation. Carcinogenesis 2011, 32, 1427–1433. [Google Scholar]
- Yasuda, M.; Schmid, T.; Rübsamen, D.; Colburn, N.H.; Irie, K.; Murakami, A. Downregulation of programmed cell death 4 by inflammatory conditions contributes to the generation of the tumor promoting microenvironment. Mol. Carcinog. 2010, 49, 837–848. [Google Scholar] [CrossRef]
- Sheedy, F.J.; Palsson-McDermott, E.; Hennessy, E.J.; Martin, C.; O’Leary, J.J.; Ruan, Q.; Johnson, D.S.; Chen, Y.; O’Neill, L.A.J. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat. Immunol. 2009, 11, 141. [Google Scholar] [CrossRef]
- Barker, J.R.; Becker Buscaglia, L.E.; Ma, X.; Li, Y. MicroRNAs in NF-κB signaling. J. Mol. Cell. Biol. 2011, 3, 159–166. [Google Scholar]
- Ng, E.K.O.; Shin, V.Y.; Chu, K.-M.; Jin, H.; Cheng, A.S.L.; Wong, C.Y.P.; Sung, J.J.Y.; Leung, W.K.; Chong, W.W.S. NF-κB targets miR-16 and miR-21 in gastric cancer: Involvement of prostaglandin E receptors. Carcinogenesis 2010, 32, 240–245. [Google Scholar]
- Li, J.; Wang, K.; Chen, X.; Meng, H.; Song, M.; Wang, Y.; Xu, X.; Bai, Y. Transcriptional activation of microRNA-34a by NF-kappa B in human esophageal cancer cells. BMC Mol. Biol. 2012, 13, 4. [Google Scholar] [CrossRef]
- Matsusaka, T.; Fujikawa, K.; Nishio, Y.; Mukaida, N.; Matsushima, K.; Kishimoto, T.; Akira, S. Transcription factors NF-IL6 and NF-kappa B synergistically activate transcription of the inflammatory cytokines, interleukin 6 and interleukin 8. Proc. Natl. Acad Sci. USA 1993, 90, 10193–10197. [Google Scholar] [CrossRef]
- Kitamura, H.; Ohno, Y.; Toyoshima, Y.; Ohtake, J.; Homma, S.; Kawamura, H.; Takahashi, N.; Taketomi, A. Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci. 2017, 108, 1947–1952. [Google Scholar] [CrossRef]
- Rozovski, U.; Calin, G.A.; Setoyama, T.; D’Abundo, L.; Harris, D.M.; Li, P.; Liu, Z.; Grgurevic, S.; Ferrajoli, A.; Faderl, S.; et al. Signal transducer and activator of transcription (STAT)-3 regulates microRNA gene expression in chronic lymphocytic leukemia cells. Mol. Cancer 2013, 12, 50. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Jaeger, S.A.; Hirsch, H.A.; Bulyk, M.L.; Struhl, K. STAT3 Activation of miR-21 and miR-181b-1 via PTEN and CYLD Are Part of the Epigenetic Switch Linking Inflammation to Cancer. Mol. Cell 2010, 39, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.Y.W.; Earle, C.; Wong, G.; Spevak, C.C.; Krueger, K. Stem cell marker (Nanog) and Stat-3 signaling promote MicroRNA-21 expression and chemoresistance in hyaluronan/CD44-activated head and neck squamous cell carcinoma cells. Oncogene 2011, 31, 149. [Google Scholar] [CrossRef] [PubMed]
- Merline, R.; Moreth, K.; Beckmann, J.; Nastase, M.V.; Zeng-Brouwers, J.; Tralhão, J.G.; Lemarchand, P.; Pfeilschifter, J.; Schaefer, R.M.; Iozzo, R.V.; et al. Signaling by the Matrix Proteoglycan Decorin Controls Inflammation and Cancer Through PDCD4 and MicroRNA-21. Sci. Signal. 2011, 4, ra75. [Google Scholar] [CrossRef] [PubMed]
- Binia, A.; Van Stiphout, N.; Liang, L.; Michel, S.; Bhavsar, P.K.; Fan Chung, K.; Brightling, C.E.; Barnes, P.J.; Kabesch, M.; Bush, A.; et al. A Polymorphism Affecting MYB Binding within the Promoter of the PDCD4 Gene is Associated with Severe Asthma in Children. Hum. Mutat. 2013, 34, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Gao, Q.; Wang, L.; Guo, C.; Zhu, F.; Wang, B.; Wang, Q.; Gao, F.; Chen, Y.; Zhang, L. Deficiency of programmed cell death 4 results in increased IL-10 expression by macrophages and thereby attenuates atherosclerosis in hyperlipidemic mice. Cell. Mol. Immunol. 2015, 13, 524. [Google Scholar] [CrossRef]
- Ye, J.; Guo, R.; Shi, Y.; Qi, F.; Guo, C.; Yang, L. miR-155 Regulated Inflammation Response by the SOCS1-STAT3-PDCD4 Axis in Atherogenesis. Mediat. Inflamm. 2016, 2016, 14. [Google Scholar] [CrossRef]
- Chen, Z.; Yuan, Y.-C.; Wang, Y.; Liu, Z.; Chan, H.J.; Chen, S. Down-regulation of programmed cell death 4 (PDCD4) is associated with aromatase inhibitor resistance and a poor prognosis in estrogen receptor-positive breast cancer. Breast Cancer Res. Treat. 2015, 152, 29–39. [Google Scholar] [CrossRef]
- Mudduluru, G.; Medved, F.; Grobholz, R.; Jost, C.; Gruber, A.; Leupold, J.H.; Post, S.; Jansen, A.; Colburn, N.H.; Allgayer, H. Loss of programmed cell death 4 expression marks adenoma-carcinoma transition, correlates inversely with phosphorylated protein kinase B, and is an independent prognostic factor in resected colorectal cancer. Cancer 2007, 110, 1697–1707. [Google Scholar] [CrossRef]
- Wei, N.; Liu, S.S.; Leung, T.H.; Tam, K.F.; Liao, X.Y.; Cheung, A.N.; Chan, K.K.; Ngan, H.Y. Loss of Programmed cell death 4 (Pdcd4) associates with the progression of ovarian cancer. Mol. Cancer 2009, 8, 70. [Google Scholar] [CrossRef]
- Li, X.; Xin, S.; Yang, D.; Li, X.; He, Z.; Che, X.; Wang, J.; Chen, F.; Wang, X.; Song, X. Down-regulation of PDCD4 expression is an independent predictor of poor prognosis in human renal cell carcinoma patients. J. Cancer Res. Clin. Oncol. 2012, 138, 529–535. [Google Scholar] [CrossRef]
- Mudduluru, G.; George-William, J.N.; Muppala, S.; Asangani, I.A.; Kumarswamy, R.; Nelson, L.D.; Allgayer, H. Curcumin regulates miR-21 expression and inhibits invasion and metastasis in colorectal cancer. Biosci. Rep. 2011, 31, 185–197. [Google Scholar] [CrossRef]
- Mirzaei, H.; Masoudifar, A.; Sahebkar, A.; Zare, N.; Sadri Nahand, J.; Rashidi, B.; Mehrabian, E.; Mohammadi, M.; Mirzaei, H.R.; Jaafari, M.R. MicroRNA: A novel target of curcumin in cancer therapy. J. Cell. Physiol. 2018, 233, 3004–3015. [Google Scholar] [CrossRef]
- Tili, E.; Michaille, J.-J.; Alder, H.; Volinia, S.; Delmas, D.; Latruffe, N.; Croce, C.M. Resveratrol modulates the levels of microRNAs targeting genes encoding tumor-suppressors and effectors of TGFβ signaling pathway in SW480 cells. Biochem. Pharmacol. 2010, 80, 2057–2065. [Google Scholar] [CrossRef]
- Sheth, S.; Jajoo, S.; Kaur, T.; Mukherjea, D.; Sheehan, K.; Rybak, L.P.; Ramkumar, V. Resveratrol reduces prostate cancer growth and metastasis by inhibiting the Akt/MicroRNA-21 pathway. PLoS ONE 2012, 7, e51655. [Google Scholar] [CrossRef]
- Zhou, X.; Ren, Y.; Liu, A.; Jin, R.; Jiang, Q.; Huang, Y.; Kong, L.; Wang, X.; Zhang, L. WP1066 sensitizes oral squamous cell carcinoma cells to cisplatin by targeting STAT3/miR-21 axis. Sci. Rep. 2014, 4, 7461. [Google Scholar] [CrossRef]
- Schmid, T.; Blees, J.S.; Bajer, M.M.; Wild, J.; Pescatori, L.; Crucitti, G.C.; Scipione, L.; Costi, R.; Henrich, C.J.; Brüne, B. Diaryl disulfides as novel stabilizers of tumor suppressor Pdcd4. PLoS ONE 2016, 11, e0151643. [Google Scholar] [CrossRef]
- Grkovic, T.; Blees, J.; Bayer, M.; Colburn, N.; Thomas, C.; Henrich, C.; Peach, M.; McMahon, J.; Schmid, T.; Gustafson, K. Tricyclic guanidine alkaloids from the marine sponge Acanthella cavernosa that stabilize the tumor suppressor PDCD4. Mar. Drugs 2014, 12, 4593–4601. [Google Scholar] [CrossRef]
- Nedaeinia, R.; Sharifi, M.; Avan, A.; Kazemi, M.; Nabinejad, A.; Ferns, G.A.; Ghayour-Mobarhan, M.; Salehi, R. Inhibition of microRNA-21 via locked nucleic acid-anti-miR suppressed metastatic features of colorectal cancer cells through modulation of programmed cell death 4. Tumor Biol. 2017, 39, 1010428317692261. [Google Scholar] [CrossRef]
- Yheskel, M.; Patel, V. Therapeutic microRNAs in polycystic kidney disease. Curr. Opin. Nephrol. Hypertens. 2017, 26, 282. [Google Scholar] [CrossRef]
- Yu, G.; Jia, B.; Cheng, Y.; Zhou, L.; Qian, B.; Liu, Z.; Wang, Y. MicroRNA-429 sensitizes pancreatic cancer cells to gemcitabine through regulation of PDCD4. Am. J. Transl. Res. 2017, 9, 5048. [Google Scholar]
- Hwang, S.K.; Jin, H.; Kwon, J.T.; Chang, S.H.; Kim, T.H.; Cho, C.S.; Lee, K.H.; Young, M.R.; Colburn, N.H.; Beck Jr, G.R.; et al. Aerosol-delivered programmed cell death 4 enhanced apoptosis, controlled cell cycle and suppressed AP-1 activity in the lungs of AP-1 luciferase reporter mice. Gene Ther. 2007, 14, 1353. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhai, R.; Zhao, J.; Kong, F.; Wang, J.; Jiang, W.; Xin, Q.; Xue, X.; Luan, Y. Programmed cell death 4 overexpression enhances sensitivity to cisplatin via the JNK/c-Jun signaling pathway in bladder cancer. Int. J. Oncol. 2018, 52, 1633–1642. [Google Scholar] [CrossRef] [PubMed]
- Jansen, A.P.; Camalier, C.E.; Stark, C.; Colburn, N.H. Characterization of programmed cell death 4 in multiple human cancers reveals a novel enhancer of drug sensitivity. Mol. Cancer Ther. 2004, 3, 103–110. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Function of PDCD4 | Experimental System or Target | Effects of PDCD4 | Ref. |
|---|---|---|---|
| Neoplastic transformation inhibitor | JB6 mouse epidermal cells | Cells with high Pdcd4 levels resist to TPA induced neoplastic transformation | [6] |
| Pdcd4 knockout mice | Induction of spontaneous B-lymphoma | [21] | |
| transgenic mice with epidermal specific Pdcd4 expression | The epidermis resists to TPA-induced carcinogenesis | [22] | |
| Inhibition of translation | SIN1 cap-dependent translation | Inhibition of colon carcinoma invasion | [26] |
| TP53 cap-dependent translation | Maintenance of TP53 levels | [25] | |
| XIAP and Bcl-X1 IRES-mediated translation | Stimulation of apoptosis | [30] | |
| CMyb and AMyb protein elongation inhibition | A regulatory feedback loop, Myb is a transcription factor to stimulate PDCD4 expression | [28,29] | |
| Inhibition of AP-1 activation | Via inhibition of the upstream kinase MAP4K1 suppression | Suppression of human colon carcinoma cell invasion | [33] |
| Maintenance of E-cadherin levels | PDCD4 knockdown down-regulated E-cadherin expression via SNAIL. | Suppression of invasion via reduction of catenin/Tcf-dependent transcription. | [36] |
| Inhibition of transcription factors | PDCD4 binds to TWIST DNA binding site | Suppression of cell growth downregulating the target gene YB1 expression | [38] |
| Sp family; PDCD4 binds to Sp1/Sp3 and inhibits u-PAR transcription | Suppression of colon carcinoma cell invasion/intravasion | [39] | |
| NF-κB; PDCD4 binds with p65 subunit | Inhibition of glioma cell growth | [40] | |
| PDCD4 is a part of CSL transcription inhibitor complex | Negative control of stromal fibroblast conversion into cancer associated fibroblasts | [41] | |
| Binding with cytoplasmic protein | DAXX, a scaffold protein with roles in diverse processes | Controls of the activity of DAXX binding partner proteins | [42] |
| Induction of apoptosis | Overexpression of PDCD4 | Apoptosis induction of tumor cells | [18] |
| PDCD4 knockdown | Induction of senescence and/or apoptosis | [43,44] |
| Tumors | Roles of PDCD4 | Ref. |
|---|---|---|
| Brest cancer | PDCD4 overexpression sensitizes aromatase inhibitor (AI)-resistant cells to AI and PDCD4 downregulation is associated with a lower survival of patient in estrogen receptor positive breast cancer. | [89] |
| Colon carcinoma | PDCD4 overexpression inhibits cancer cell invasion/intravation. | [24,39] |
| Colorectal cancer | PDCD4 expression is downregulated in the tumor tissues and the loss of PDCD4 correlated with patient survival. | [90] |
| Epidermal tumor | Epidermis of transgenic mice with epidermis specific Pdcd4 expression resists to TPA-induced carcinogenesis. | [22] |
| Glioma cells | PDCD4 expression is suppressed by the methylation of CpG islands in the promoter region of tumor tissues. The loss of PDCD4 is associated with poor prognosis. | [19] |
| Hepatoma cells | PDCD4 expression is suppressed in the hepatoma tissues from patients, and PDCD4 overexpression induces apoptosis of hepatoma cells. | [18] |
| Lung cancer | PDCD4 expression is downregulated in the tumor tissues and the loss of PDCD4 correlated with poor prognosis in the patients. | [17] |
| B-lymphoma | Pdcd4 knockout mice induce spontaneous B-lymphoma. | [21] |
| Ovarian cancer | PDCD4 expression in the tumor cells suppresses the malignant phenotype. The loss of PDCD4 is correlated with patient poor survival. | [91] |
| Renal cell carcinoma (RCC) | PDCD4 expression is downregulated in RCC tumor tissues and correlated to RCC stage, grade, metastasis and survival. | [92] |
| Drug | Function and Pre-Clinical and Clinical Trials | Ref. |
|---|---|---|
| Curcumin | Upregulation of PDCD4-protein levels by inhibiting mir21 expression | [93,94] |
| Resveratrol | Upregulation of PDCD4-protein levels via Akt/mir21 inhibition. Resveratrol has used for clinical trials | [95,96] |
| WP1066 (STAT3 inhibitor) | Upregulation of PDCD4-protein levels via inhibition of STAT3-mir21 axis | [97] |
| Diaryl disulfides | Stabilization of PDCD4-protein by inhibiting the degradation in ubiquitin-proteasome system | [98] |
| Tricyclic guanidine alkaloids from the marine sponge Acanthella cavernosa | Stabilization of PDCD4-protein by inhibiting the degradation in ubiquitin-proteasome system | [99] |
| Anti-mir21 nucleotide | Stimulation of PDCD4-protein synthesisAn anti-mir21 drug has used for clinical trial in polycystic kidney disease | [100,101] |
| Mir429 | Ectopic expression increased cellular sensitivity to gemcitabine via PDCD4 upregulation | [102] |
| PDCD4 cDNA | Ectopic expression of PDCD4 induces tumor cell apoptosisAerosol delivery of Pdcd4 cDNA into a mouse lung with tumors inhibits cell proliferation and induces apoptosis of the tumor cells | [18,103] |
| PDCD4 specific siRNA | siRNA-mediated PDCD4 knockdown induces tumor cell death | [43,44] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsuhashi, S.; Manirujjaman, M.; Hamajima, H.; Ozaki, I. Control Mechanisms of the Tumor Suppressor PDCD4: Expression and Functions. Int. J. Mol. Sci. 2019, 20, 2304. https://doi.org/10.3390/ijms20092304
Matsuhashi S, Manirujjaman M, Hamajima H, Ozaki I. Control Mechanisms of the Tumor Suppressor PDCD4: Expression and Functions. International Journal of Molecular Sciences. 2019; 20(9):2304. https://doi.org/10.3390/ijms20092304
Chicago/Turabian StyleMatsuhashi, Sachiko, M. Manirujjaman, Hiroshi Hamajima, and Iwata Ozaki. 2019. "Control Mechanisms of the Tumor Suppressor PDCD4: Expression and Functions" International Journal of Molecular Sciences 20, no. 9: 2304. https://doi.org/10.3390/ijms20092304
APA StyleMatsuhashi, S., Manirujjaman, M., Hamajima, H., & Ozaki, I. (2019). Control Mechanisms of the Tumor Suppressor PDCD4: Expression and Functions. International Journal of Molecular Sciences, 20(9), 2304. https://doi.org/10.3390/ijms20092304