Oligomeric Architecture of Mouse Activating Nkrp1 Receptors on Living Cells



Abstract
1. Introduction
2. Results
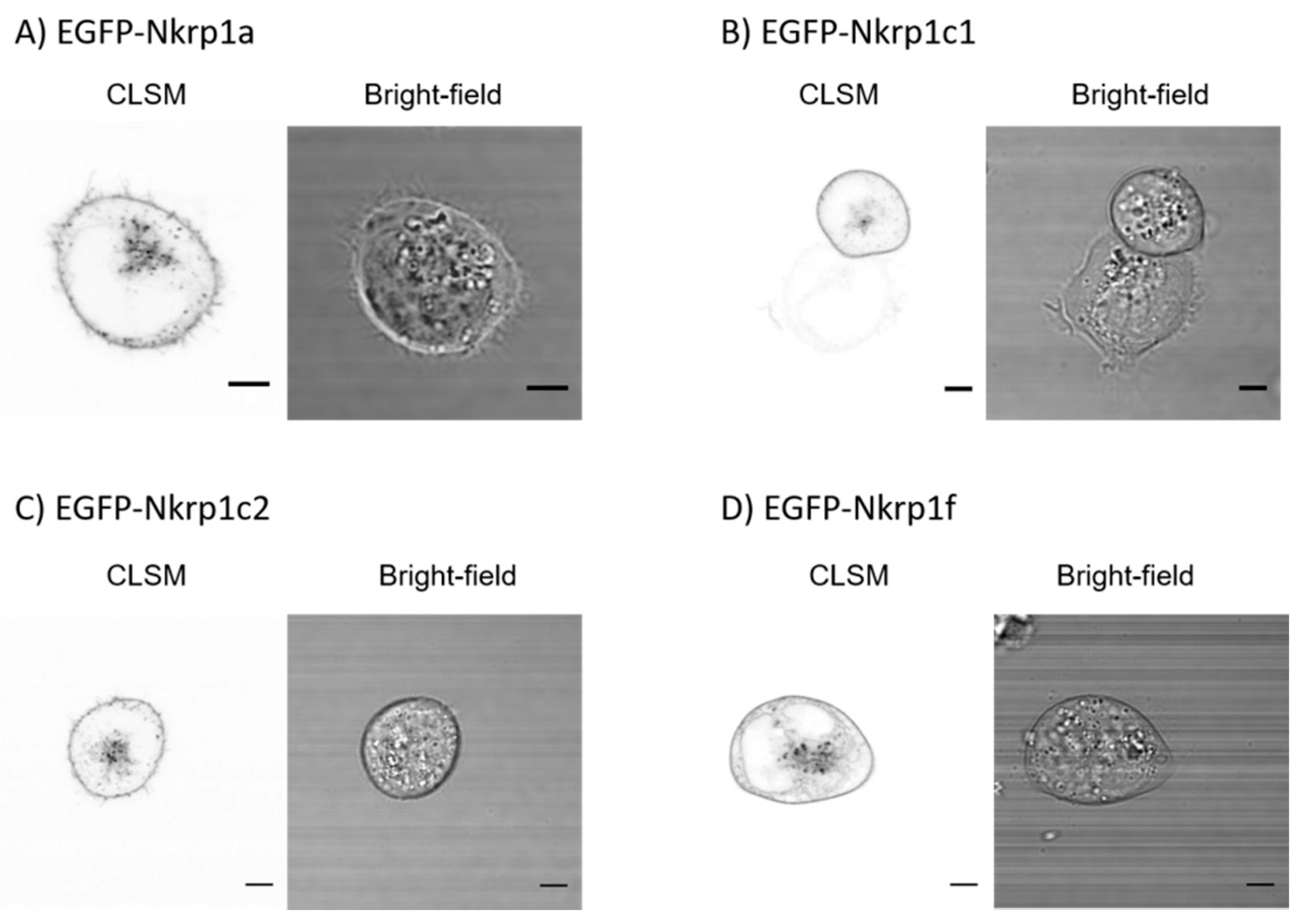
2.1. Surface Localization of Mouse Activating Nkrp1 Proteins in Mammalian Cells
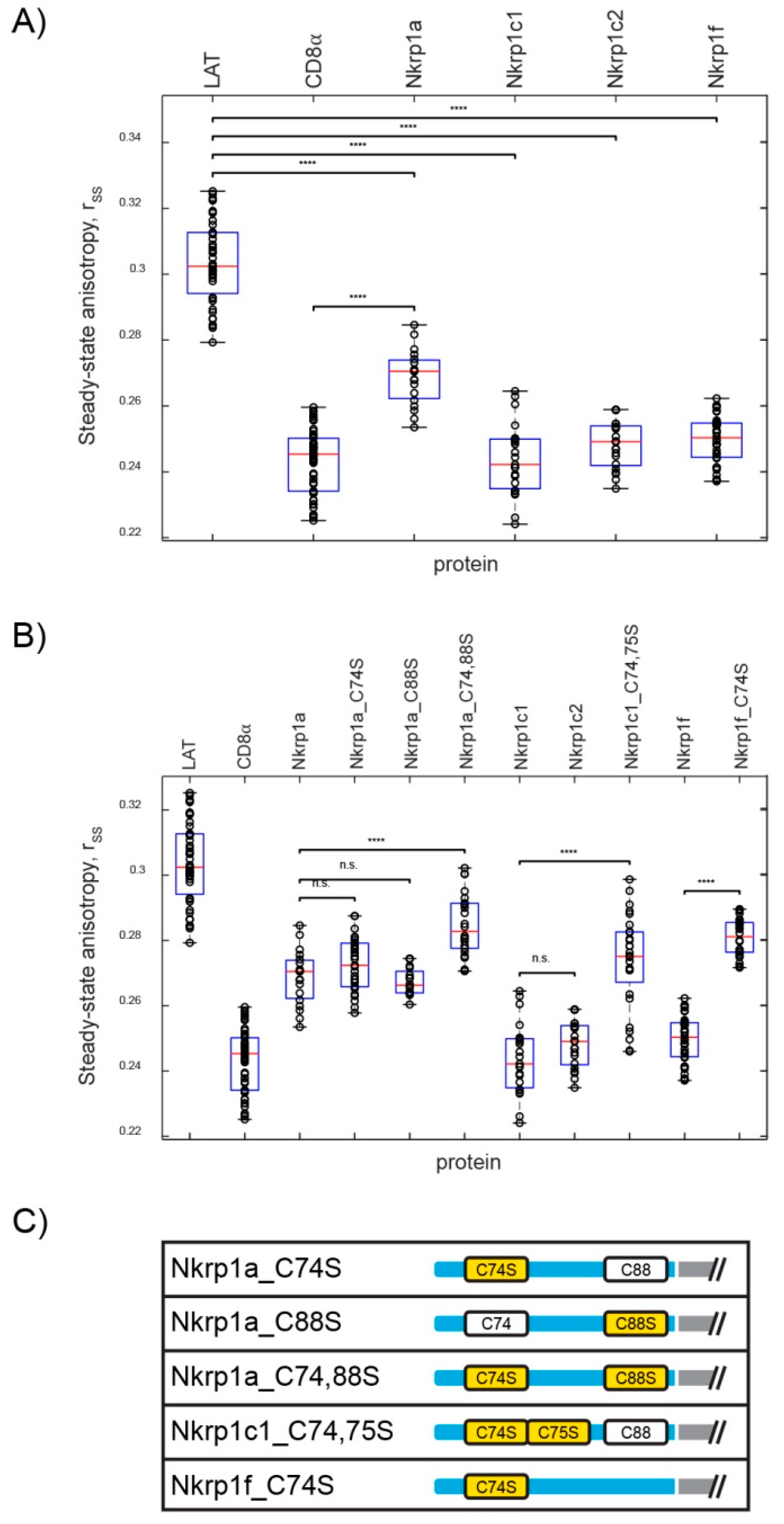
2.2. Stoichiometry of Nkrp1 Proteins in Mammalian Cells
2.2.1. Homo-FRET Analysis of Nkrp1 Proteins in Living Jurkat Cells
2.2.2. Western Blotting of Nkrp1 Protein Variants in COS-7 cell Lysates
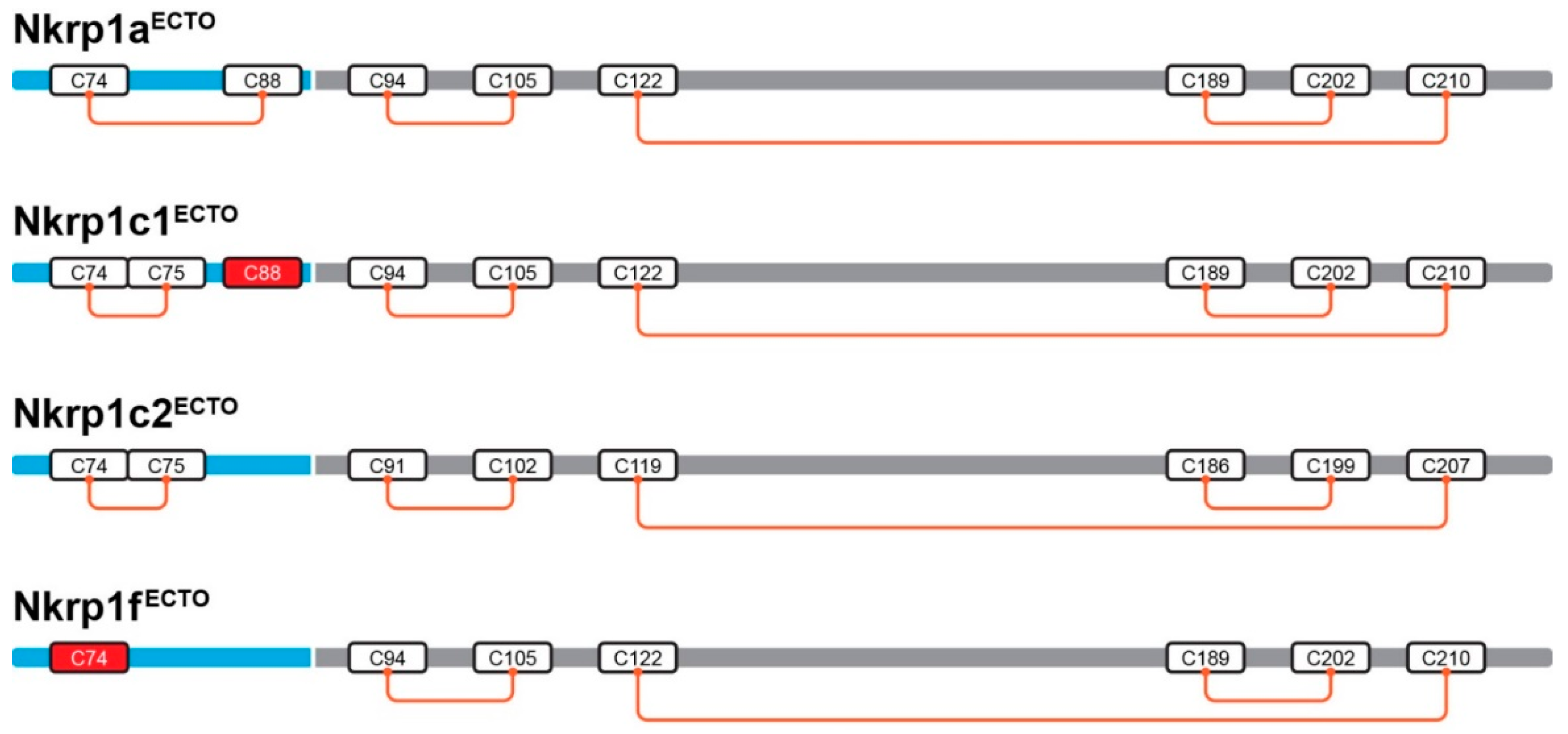
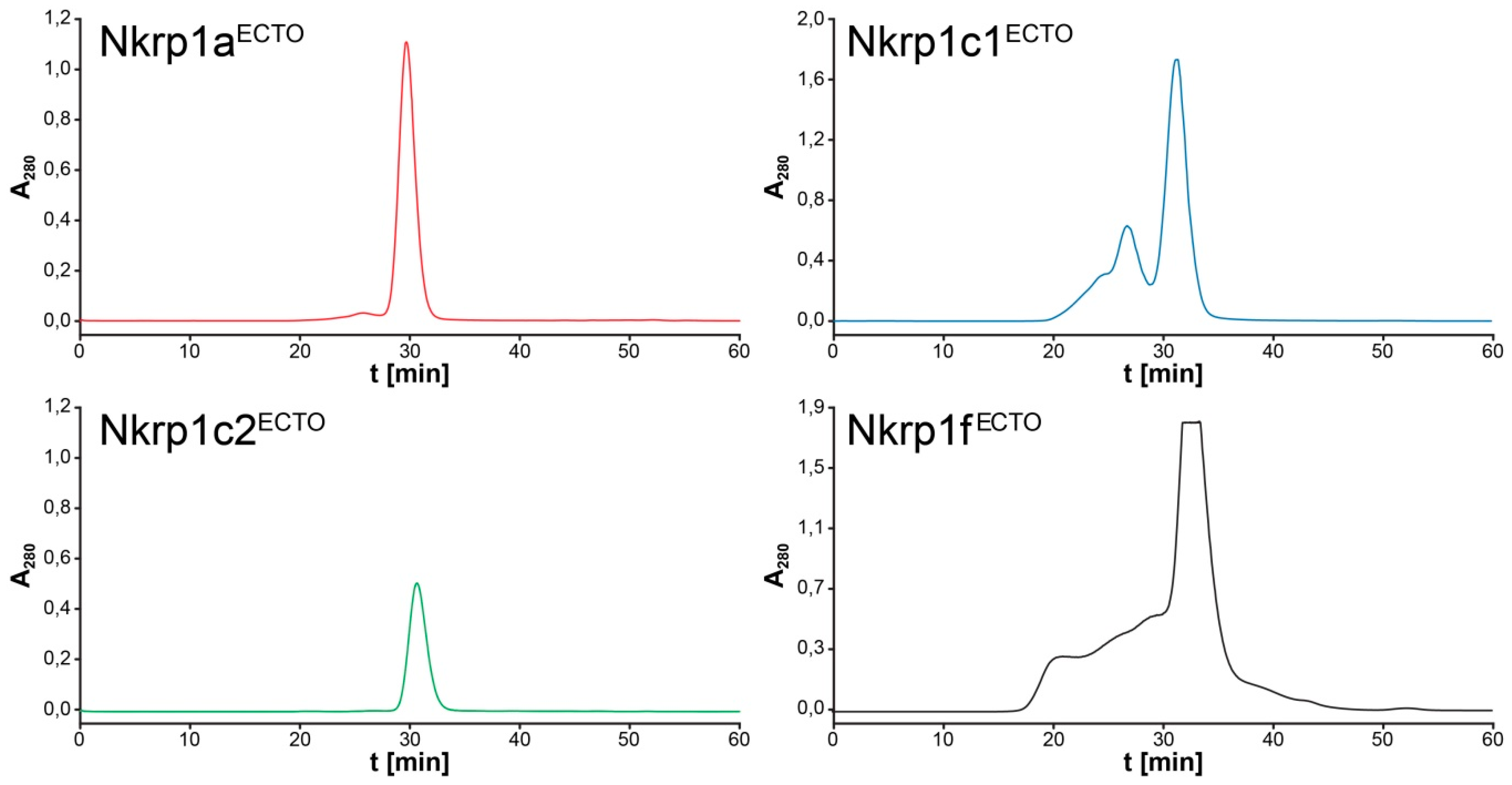
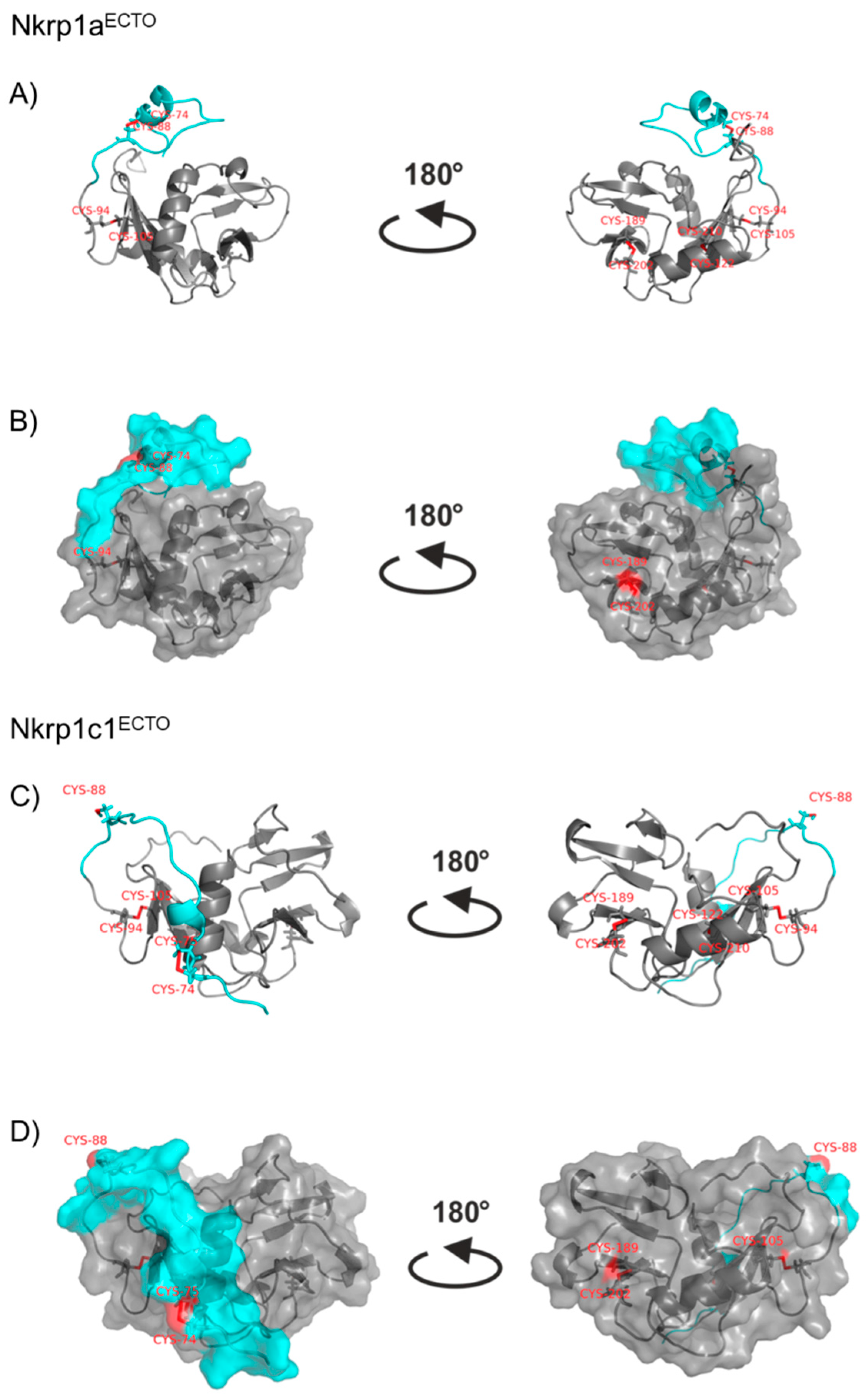
2.3. Refolding, Purification and Evaluation of Disulfide Bonds in Recombinant Murine Activating Nkrp1 Proteins
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. DNA Cloning
4.3. Bacterial Recombinant Expression of Activating Nkrp1 Proteins
4.4. Protein Refolding and Purification
4.5. Evaluation of Disulfide Bonds
4.6. Cell Culture and Transfection
4.7. Live Cell Imaging
4.8. Western Blotting
4.9. Förster Resonance Energy Transfer (Homo-FRET)
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Clr | C-type lectin-related protein |
| CTLD | C-Type Lectin-like Domain |
| CTLR | C-Type Lectin-like Receptor |
| EDTA | Ethylenediaminetetraacetic Acid |
| EGFP | Enhanced Green Fluorescent Protein |
| ESI-FT-ICR MS | Electrospray Ionization Fourier Transform Ion Cyclotron Resonance Mass Spectrometry |
| FRET | Förster Resonance Energy Transfer |
| ILC | Innate Lymphoid Cells |
| LAT | Linker of Activated T cells |
| NK | Natural Killer |
| PVDF | Polyvinylidene Difluoride |
| TCSPC | Time-correlated Single Photon Counting |
| TNFα | Tumor Necrosis Factor α |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Median | Mean | SD | |
|---|---|---|---|
| LAT | 0.3024 | 0.3038 | 0.0125 |
| CD8α | 0.2454 | 0.2435 | 0.0095 |
| Nkrp1a | 0.2705 | 0.2687 | 0.0083 |
| Nkrp1a_C74S | 0.2724 | 0.2726 | 0.0084 |
| Nkrp1a_C88S | 0.2663 | 0.2673 | 0.0042 |
| Nkrp1a_C74,88S | 0.2828 | 0.2849 | 0.0095 |
| Nkrp1c1 | 0.2422 | 0.2437 | 0.0108 |
| Nkrp1c2 | 0.2491 | 0.2483 | 0.0073 |
| Nkrpc1_C74,75S | 0.2751 | 0.2735 | 0.0143 |
| Nkrp1f | 0.2504 | 0.2498 | 0.0072 |
| Nkrp1f_C74S | 0.2812 | 0.2808 | 0.0056 |
| Name | Sequence 5′→ 3′ | Note |
|---|---|---|
| NKRP1A STALK FW | GCCATATGTCAATAGAAAAATGCTATGTG | Nkrp1aECTO for bacterial recombinant expression |
| MNKR1ARE | GCAAGCTTATCAGTGTCCATAACCCACATAGTTGCT | Nkrp1aECTO for bacterial recombinant expression |
| NKRP1C STALK FW | CGCATATGTCAAGAGAAAAATGCTGTGTG | Nkrp1c1ECTO for bacterial recombinant expression |
| MNKR1CRE | GCAAGCTTATCAGGAGTCATTACTCGGGGT | Nkrp1c1ECTO for bacterial recombinant expression |
| Nkrp1f FW | CGCCATATGCAAAAACCACCAATAGAAAAATG | Nkrp1fECTO for bacterial recombinant expression |
| Nkrp1 REV | GCAAGCTTTCAGACATGTATCAGGGTC | Nkrp1fECTO for bacterial recombinant expression |
| mNKRC2Fw2 | TGAATAAAACAACAGTTAATTTAGAGTGCCCAC | DCS sequence deletion in Nkrp1c1 to generate Nkrp1c2 |
| mNKRC2Rev1 | GTGGGCACTCTAAATTAACTGTTGTTTTATTCAGGTTC | DCS sequence deletion in Nkrp1c1 to generate Nkrp1c2 |
| Nkrp1c1_Bam+Xho_Fw | GGCGGATCCATGGACACAGCAAG | Nkrp1c1 and -c2 constructs |
| Nkrp1c1_Bam+Xho_Rev | CGTCTAGAAGCCTCGAGTTAGGAGTCATTACTCG | Nkrp1c1 and -c2 constructs |
| Fw_Nkrp1a_Stop | AAGGGATCCATGGACACAGCAAGGGTC | Nkrp1a construct |
| Rev_Nkrp1a_Stop | GCCCTCGAGTCAACCGGTGTGTCCATAACCCACATAGTTG | Nkrp1a construct |
| Fw_Nkrp1f_Stop | AAGGGATCCATGGACACATCAAAGGTCC | Nkrp1f construct |
| Rev_Nkrp1f_Stop | GTTCTCGAGTCAACCGGTGACATGTATCAGGGTCTTTTG | Nkrp1f construct |
| A1_C220S_Fw1 | CCATCAATAGAAAAATCTTATGTGCTTATTCAAGAG | Missense mutation (TGC→TCT) in Nkrp1a construct, generation of Nkrp1a_C74S mutant |
| A1_C220S_Rev1 | CTCTTGAATAAGCACATAAGATTTTTCTATTGATGG | Missense mutation (TGC→TCT) in Nkrp1a construct, generation of Nkrp1a_C74S mutant |
| A1_C262S_Fw2 | GAATAAAACAACAGACAGTTCAGCTAAGCTAGAG | Missense mutation (GATTGT→GACAGT) in Nkrp1a construct, generation of Nkrp1a_C88S mutant |
| A1_C262S_Rev2 | CTCTAGCTTAGCTGAACTGTCTGTTGTTTTATTC | Missense mutation (GATTGT→GACAGT) in Nkrp1a construct, generation of Nkrp1a_C88S mutant |
| C1_CC220SS_Fw | CATCAAGAGAAAAATCATCCGTGTTTATTCAAGAG | Missense mutation (TGCTGT→TCATCC) in Nkrp1c1 construct, generation of Nkrp1c1_C74,75S mutant |
| C1_CC220SS_Rev | CTCTTGAATAAACACGGATGATTTTTCTCTTGATG | Missense mutation (TGCTGT→TCATCC) in Nkrp1c1 construct, generation of Nkrp1c1_C74,75S mutant |
| F_C220S_Fw | CACCAATAGAAAAGTCAAGTGTGGCTGCTC | Missense mutation (AAATGC→AAGTCA) in Nkrp1f construct, generation of Nkrp1f_C74S mutant |
| F_C220S_Rev | GAGCAGCCACACTTGACTTTTCTATTGGTG | Missense mutation (TGCTGT→TCATCC) in Nkrp1c1 construct, generation of Nkrp1c1_C74,75S mutant |
| CD8a_Fw 1 | TTCGAATTCGCCACCATGGCCTCACCGTTG | CD8α construct |
| CD8a_REV | TCCGGATCCCACAATTTTCTCTGAAGGTCTG | CD8α construct |
| CD8a_288_For | CGAGAAGCTCAATTCGTCGAAACTG | Silent mutation (CTG→CTC) in CD8α construct |
| CD8a_288_Rev | CAGTTTCGACGAATTGAGCTTCTCG | Silent mutation (CTG→CTC) in CD8α construct |
| EGFP_Eco+Bam_FW | TTCGAATTCGCGACCATGGTGAGCAAGGGC | Fluorescent protein (EGFP) construct on the N-terminus of Nkrp1 protein |
| EGFP_Eco+Bam_REV | TAGGATCCGCCACCAGAGCCCTTGTACAGCTCGTC | Fluorescent protein (EGFP) construct on the N-terminus of Nkrp1 protein, GSGGGS linker |
| T198 TMD GFP FWD | AACGGATCCGGAGGTGGATCTGTGAGCAAGGGCGAGGAGCTG | EGFP construct on the C-terminus of the target protein, GSGGGS linker |
| T199 TMD GFP REV | gagaattctcgagTTACTTGTACAGCTCGTCCATGCCGAG | EGFP construct on the C-terminus of the target protein, GSGGGS linker |

| Protein 1 | Protein 2 | p-value |
|---|---|---|
| LAT | Nkrp1a | 1.1e-17 (****) |
| LAT | Nkrp1c1 | 3.3e-26 (****) |
| LAT | Nkrp1c2 | 8.4e-31 (****) |
| LAT | Nkrp1f | 6.1e-35 (****) |
| CD8α | Nkrp1c1 | 9.6e-1 (n.s.) |
| CD8α | Nkrp1c2 | 2.5e-2 (*) |
| Protein 1 | Protein 2 | p-value |
| Nkrp1a | Nkrp1a_C74S | 1.2e-1 (n.s.) |
| Nkrp1a | Nkrp1a_C88S | 5.2e-1 (n.s.) |
| Nkrp1a | Nkrp1a_C74,88S | 9.7e-7 (****) |
| Nkrp1c1 | Nkrp1c2 | 1.0e-1 (n.s.) |
| Nkrp1c1 | Nkrp1c1_C74,75S | 3.6e-11 (****) |
| Nkrp1f | Nkrp1f_C74S | 2.4e-26 (****) |


| Protease | C-C Cross-Link | Disulfide-Linked Peptide(s) | Sequence(s) of disulfide-Linked Peptides | Experimental [M + H]+ | Error [ppm] |
|---|---|---|---|---|---|
| Nkrp1aECTO | |||||
| Trypsin | C74-C88 | 74-84; 85-91 | CYVLIQENLNK - TTDCSAK | 2058.9835 | 1.56 |
| Trypsin | (C94-C105; C122-C210); (C94-C122; C105-C210) | 92-102; 105-125; 208-212 | LECPQDWLSHR -CFQVSHVSNTWEEGLVDCDGK - WICQK | 4407.9662 | 0.34 |
| Trypsin | C189-C202 | 180-196; 197-207 | ITGDTENDSCAAISGDK - VTFESCNSDNR | 2965.2422 | 1.73 |
| AspN | (C74-C88; C94-C105); (C74-C94; C88-C105) | 70-79; 87-96; 103-120 | SIEKCYVLIQ - DCSAKLECPQ - DKCFQVSHVSNTWEEGLV | 4361.0233 | 0.25 |
| AspN | C122-C210 | 122-133; 205-227 | DCDGKGATLMLIQ - DNRWICQKELYHETLSNYVGYGH | 4186.9419 | 0.72 |
| AspN | C189-C202 | 187-199; 200-204 | DSCAAISG - DKVTFESCNS | 1849.7579 | 1.75 |
| GluC | C74-C88 | 73-80; 81-93 | KCYVLIQE - NLNKTTDCSAKLE | 2429.2051 | 1.04 |
| GluC | C94-C105 | 94-116 | CPQDWLSHRDKCFQVSHVSNTWE | 2800.2355 | 0.65 |
| Nkrp1c1ECTO monomer | |||||
| Trypsin | C74-C75 | 74-84 | CCVFIQENLNK | 1308.6075 | 2.18 |
| Trypsin | (C88-cysteamine; C94-C105; C122-C210); (C88-cysteamine; C94-C122; C105-C210); (C94-cysteamine; C88-C105; C122-C210);(C94-cysteamine; C88-C122; C105-C210); (C105-cysteamine;C88-C122; C94-C210); (C105-cysteamine;C88-C210; C94-C122); (C122-cysteamine; C88-C105; C94-C210); (C122-cysteamine; C88-C210; C94-C105) | 85-102; 105-124; 208-212; cysteamine | TTDCSVNLECPQDWLLHR - CFHVSQVSNTWEEGQADCGR - WICQK | 5129.2091 | 4.34 |
| Trypsin | C189-C202 | 180-196; 197-207 | ITGVTENGSCASILGDK - VTPESCASDNR | 2840.3037 | 2.43 |
| AspN | C74-C75 | 70-86 | SREKCCVFIQENLNKTT | 2010.9736 | 2.10 |
| AspN | (C88-cysteamine; C94-C105); (C94-cysteamine; C88-C105) | 87-96; 103-115; cysteamine | DCSVNLECPQ - DKCFHVSQVSNTW | 2730.1415 | 3.07 |
| AspN | C122-C210 | 121-133; 205-221 | DCGRKGATLLLIQ - DNRWICQKELNHETPSN | 3468.7111 | 2.16 |
| AspN | C189-C202 | 176-194; 195-204 | DVLKITGVTENGSCASILG - DKVTPESCAS | 2910.4071 | 2.52 |
| GluC | C74-C75 | 73-80 | KCCVFIQE | 967.4376 | 2.25 |
| GluC | C88-cysteamine | 81-93; cysteamine | NLNKTTDCSVNLE | 1525.6985 | 2.99 |
| GluC | C94-C105 | 94-116 | CPQDWLSHRDKCFQVSHVSNTWE | 2826.2875 | 2.00 |
| GluC | (C122-C202; C189-C210); (C122-C210; C189-C202) | 118-137; 186-200; 201-213 | GQADCGRKGATLLLIQDQEE -NGSCASILGDKVTPE - SCASDNRWICQKE | 5169.3684 | 4.58 |
| Nkrp1c1ECTO dimer | |||||
| Trypsin | C74-C75 | 74-84 | CCVFIQENLNK | 1308.6075 | 2.18 |
| Trypsin | C189-C202 | 180-196; 197-207 | ITGVTENGSCASILGDK - VTPESCASDNR | 2840.3037 | 2.39 |
| AspN | C74-C75 | 70-86 | SREKCCVFIQENLNKTT | 2010.9736 | 2.10 |
| AspN | C88-C88 | 87-92; 87-92 | DCSVNL - DCSVNL | 1297.5399 | 2.63 |
| AspN | C122-C210 | 121-133; 205-221 | DCGRKGATLLLIQ - DNRWICQKELNHETPSN | 3468.7111 | 2.51 |
| AspN | C189-C202 | 176-194; 195-204 | DVLKITGVTENGSCASILG - DKVTPESCAS | 2910.4071 | 0.12 |
| GluC | C74-C75 | 73-80 | KCCVFIQE | 967.4376 | 2.25 |
| GluC | C94-C105 | 94-116 | CPQDWLSHRDKCFQVSHVSNTWE | 2826.2875 | 2.02 |
| GluC | (C122-C202; C189-C210); (C122-C210; C189-C202) | 118-137; 186-200; 201-213 | GQADCGRKGATLLLIQDQEE -NGSCASILGDKVTPE - SCASDNRWICQKE | 5169.3710 | 4.20 |
| Nkrp1c2ECTO | |||||
| Trypsin | C74-C75 | 74-84 | CCVFIQENLNK | 1308.6075 | 2.65 |
| Trypsin | (C91-C102; C119-C207); (C91-C119; C102-C207) | 85-99; 102-121; 205-209 | TTVNLECPQDWLLHR - CFHVSQVSNTWEEGQADCGR - WICQK | 4749.1429 | 1.26 |
| Trypsin | C186-C199 | 177-193; 194-204 | ITGVTENGSCASILGDK - VTPESCASDNR | 2840.3037 | 2.28 |
| AspN | (C74-C75; C91-C102); (C74-C91; C75-C102); (C74-C102; C75-C91) | 70-93; 100-117 | SREKCCVFIQENLNKTTVNLECPQ - DKCFHVSQVSNTWEEGQA | 4856.2171 | 0.77 |
| AspN | C119-C207 | 118-130; 202-218 | DCGRKGATLLLIQ - DNRWICQKELNHETPSN | 3468.7111 | 2.02 |
| AspN | C186-C199 | 173-191; 192-201 | DVLKITGVTENGSCASILG - DKVTPESCAS | 2910.4071 | 2.03 |
| GluC | C74-C75 | 73-80 | KCCVFIQE | 967.4376 | 2.94 |
| GluC | C91-C102 | 91-113 | CPQDWLLHRDKCFHVSQVSNTWE | 2826.2875 | 3.70 |
| GluC | (C119-C199; C189-C207); (C119-C207; C119-C189) | 115-134; 183-197; 198-210 | GQADCGRKGATLLLIQDQEE - NGSCASILGDKVTPE - SCASDNRWICQKE | 5169.3759 | 4.26 |
| Nkrp1fECTO | |||||
| Trypsin | C74-C74 | 74-82; 74-82 | CSVAAQENR - CSVAAQENR | 1951.8709 | 0.31 |
| Trypsin | C94-C105 | 86-96; 105-113 | SAILECPR – CLFVSQISR | 1937.9928 | 0.6 |
| AspN | C122-C210 | 120-125; 205-212 | DACSME – DNHWICQK | 1695.6535 | 1.2 |
| AspN | C189-C202 | 186-204 | ENSCAIISHTEVFSDSCSS | 2031.8295 | 0.8 |

References
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L. NK cell receptors. Annu. Rev. Immunol. 1998, 16, 359–393. [Google Scholar] [CrossRef] [PubMed]
- Giorda, R.; Trucco, M. Mouse NKR-P1. A family of genes selectively coexpressed in adherent lymphokine-activated killer cells. J. Immunol. 1991, 147, 1701–1708. [Google Scholar]
- Giorda, R.; Rudert, W.A.; Vavassori, C.; Chambers, W.H.; Hiserodt, J.C.; Trucco, M. NKR-P1, a signal transduction molecule on natural killer cells. Science 1990, 249, 1298–1300. [Google Scholar] [CrossRef] [PubMed]
- Chambers, W.H.; Vujanovic, N.L.; DeLeo, A.B.; Olszowy, M.W.; Herberman, R.B.; Hiserodt, J.C. Monoclonal antibody to a triggering structure expressed on rat natural killer cells and adherent lymphokine-activated killer cells. J. Exp. Med. 1989, 169, 1373–1389. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.C.; Niemi, E.C.; Goldfien, R.D.; Hiserodt, J.C.; Seaman, W.E. NKR-P1, an activating molecule on rat natural killer cells, stimulates phosphoinositide turnover and a rise in intracellular calcium. J. Immunol. 1991, 147, 3244–3250. [Google Scholar]
- Yokoyama, W.M.; Ryan, J.C.; Hunter, J.J.; Smith, H.R.; Stark, M.; Seaman, W.E. cDNA cloning of mouse NKR-P1 and genetic linkage with LY-49. Identification of a natural killer cell gene complex on mouse chromosome 6. J. Immunol. 1991, 147, 3229–3236. [Google Scholar] [PubMed]
- Kveberg, L.; Dai, K.-Z.; Inngjerdingen, M.; Brooks, C.G.; Fossum, S.; Vaage, J.T. Phylogenetic and functional conservation of the NKR-P1F and NKR-P1G receptors in rat and mouse. Immunogenetics 2011, 63, 429–436. [Google Scholar] [CrossRef]
- Kveberg, L.; Dai, K.-Z.; Westgaard, I.H.; Daws, M.R.; Fossum, S.; Naper, C.; Vaage, J.T. Two major groups of rat NKR-P1 receptors can be distinguished based on chromosomal localization, phylogenetic analysis and Clr ligand binding. Eur. J. Immunol. 2009, 39, 541–551. [Google Scholar] [CrossRef]
- Plougastel, B.; Matsumoto, K.; Dubbelde, C.; Yokoyama, W.M. Analysis of a 1-Mb BAC contig overlapping the mouse Nkrp1 cluster of genes: Cloning of three new Nkrp1 members, Nkrp1d, Nkrp1e, and Nkrp1f. Immunogenetics 2001, 53, 592–598. [Google Scholar] [CrossRef]
- Kung, S.K.; Su, R.C.; Shannon, J.; Miller, R.G. The NKR-P1B gene product is an inhibitory receptor on SJL/J NK cells. J. Immunol. 1999, 162, 5876–5887. [Google Scholar]
- Glimcher, L.; Shen, F.W.; Cantor, H. Identification of a cell-surface antigen selectively expressed on the natural killer cell. J. Exp. Med. 1977, 145, 1–9. [Google Scholar] [CrossRef]
- Sentman, C.L.; Hacjett, J.; Moore, T.A.; Tutt, M.M.; Bennett, M.; Kumar, V. Pan Natural Killer Cell Monoclonal Antibodies and Their Relationship to the NK1.1 Antigen. Hybridoma 1989, 8, 605–614. [Google Scholar] [CrossRef]
- Karlhofer, F.M.; Yokoyama, W.M. Stimulation of murine natural killer (NK) cells by a monoclonal antibody specific for the NK1.1 antigen. IL-2-activated NK cells possess additional specific stimulation pathways. J. Immunol. 1991, 146, 3662–3673. [Google Scholar]
- Burton, R.C.; Smart, Y.C.; Koo, G.C.; Winn, H.J. Studies on murine natural killer (NK) cells. V. Genetic analysis of NK cell markers. Cell. Immunol. 1991, 135, 445–453. [Google Scholar] [CrossRef]
- Ryan, J.C.; Turck, J.; Niemi, E.C.; Yokoyama, W.M.; Seaman, W.E. Molecular cloning of the NK1.1 antigen, a member of the NKR-P1 family of natural killer cell activation molecules. J. Immunol. 1992, 149, 1631–1635. [Google Scholar]
- Iizuka, K.; Naidenko, O.V.; Plougastel, B.F.M.; Fremont, D.H.; Yokoyama, W.M. Genetically linked C-type lectin-related ligands for the NKRP1 family of natural killer cell receptors. Nat. Immunol. 2003, 4, 801–807. [Google Scholar] [CrossRef]
- Carlyle, J.R.; Jamieson, A.M.; Gasser, S.; Clingan, C.S.; Arase, H.; Raulet, D.H. Missing self-recognition of Ocil/Clr-b by inhibitory NKR-P1 natural killer cell receptors. Proc. Natl. Acad. Sci. USA 2004, 101, 3527–3532. [Google Scholar] [CrossRef]
- Plougastel, B.; Dubbelde, C.; Yokoyama, W.M. Cloning of Clr, a new family of lectin-like genes localized between mouse Nkrp1a and Cd69. Immunogenetics 2001, 53, 209–214. [Google Scholar] [CrossRef]
- Aguilar, O.A.; Berry, R.; Rahim, M.M.A.; Reichel, J.J.; Popović, B.; Tanaka, M.; Fu, Z.; Balaji, G.R.; Lau, T.N.H.; Tu, M.M.; et al. A Viral Immunoevasin Controls Innate Immunity by Targeting the Prototypical Natural Killer Cell Receptor Family. Cell 2017, 169, 58–71.e14. [Google Scholar] [CrossRef]
- Zelensky, A.N.; Gready, J.E. The C-type lectin-like domain superfamily. FEBS J. 2005, 272, 6179–6217. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Guidotti, G. Identification of the cysteine residues involved in the class I disulfide bonds of the human insulin receptor: properties of insulin receptor monomers. Mol. Biol. Cell 1996, 7, 679–691. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zal, T.; Gascoigne, N.R. Using live FRET imaging to reveal early protein–protein interactions during T cell activation. Curr. Opin. Immunol. 2004, 16, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Bader, A.N.; Hofman, E.G.; Voortman, J.; van Bergen en Henegouwen, P.M.P.; Gerritsen, H.C. Homo-FRET imaging enables quantification of protein cluster sizes with subcellular resolution. Biophys. J. 2009, 97, 2613–2622. [Google Scholar] [CrossRef] [PubMed]
- Pompach, P.; Man, P.; Kavan, D.; Hofbauerová, K.; Kumar, V.; Bezouška, K.; Havlíček, V.; Novák, P. Modified electrophoretic and digestion conditions allow a simplified mass spectrometric evaluation of disulfide bonds. J. Mass Spectrom. 2009, 44, 1571–1578. [Google Scholar] [CrossRef]
- Angers, S.; Salahpour, A.; Bouvier, M. Dimerization: an emerging concept for G protein-coupled receptor ontogeny and function. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 409–435. [Google Scholar] [CrossRef]
- Gomes, I.; Jordan, B.A.; Gupta, A.; Rios, C.; Trapaidze, N.; Devi, L.A. G protein coupled receptor dimerization: implications in modulating receptor function. J. Mol. Med. 2001, 79, 226–242. [Google Scholar] [CrossRef]
- Middleton, R.E.; Pheasant, D.J.; Miller, C. Homodimeric architecture of a CIC-type chloride ion channel. Nature 1996, 383, 337–340. [Google Scholar] [CrossRef]
- Ward, C.W.; Menting, J.G.; Lawrence, M.C. The insulin receptor changes conformation in unforeseen ways on ligand binding: Sharpening the picture of insulin receptor activation. BioEssays 2013, 35, 945–954. [Google Scholar] [CrossRef]
- Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef]
- Postel-Vinay, M.C.; Finidori, J. Growth hormone receptor: Structure and signal transduction. Eur. J. Endocrinol. 1995, 133, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Ward, L.D.; Hammacher, A.; Howlett, G.J.; Matthews, J.M.; Fabri, L.; Moritz, R.L.; Nice, E.C.; Weinstock, J.; Simpson, R.J. Influence of interleukin-6 (IL-6) dimerization on formation of the high affinity hexameric IL-6.receptor complex. J. Biol. Chem. 1996, 271, 20138–20144. [Google Scholar] [CrossRef]
- Bochtler, M.; Ditzel, L.; Groll, M.; Hartmann, C.; Huber, R. The proteasome. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 295–317. [Google Scholar] [CrossRef]
- Bentley, G.A.; Lewit-Bentley, A.; Finch, J.T.; Podjarny, A.D.; Roth, M. Crystal structure of the nucleosome core particle at 16 Å resolution. J. Mol. Biol. 1984, 176, 55–75. [Google Scholar] [CrossRef]
- Matadeen, R.; Patwardhan, A.; Gowen, B.; Orlova, E.V.; Pape, T.; Cuff, M.; Mueller, F.; Brimacombe, R.; van Heel, M. The Escherichia coli large ribosomal subunit at 7.5 Å resolution. Structure 1999, 7, 1575–1583. [Google Scholar] [CrossRef]
- Adámková, L.; Institute of Microbiology, Prague, Czech Republic. Life cell imaging of Nkrp1 proteins at the surface of COS-7, RAW264.7 and HeLa cells. Unpublished work. 2015. [Google Scholar]
- Rozbesky, D.; Man, P.; Kavan, D.; Chmelik, J.; Cerny, J.; Bezouska, K.; Novak, P. Chemical cross-linking and H/D exchange for fast refinement of protein crystal structure. Anal. Chem. 2012, 84, 867–870. [Google Scholar] [CrossRef]
- Rozbeský, D.; Adámek, D.; Pospíšilová, E.; Novák, P.; Chmelík, J. Solution structure of the lymphocyte receptor Nkrp1a reveals a distinct conformation of the long loop region as compared to in the crystal structure. Proteins 2016, 84, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Kolenko, P.; Rozbeský, D.; Vaněk, O.; Kopecký, V.; Hofbauerová, K.; Novák, P.; Pompach, P.; Hašek, J.; Skálová, T.; Bezouška, K.; et al. Molecular architecture of mouse activating NKR-P1 receptors. J. Struct. Biol. 2011, 175, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Spreu, J.; Kuttruff, S.; Stejfova, V.; Dennehy, K.M.; Schittek, B.; Steinle, A. Interaction of C-type lectin-like receptors NKp65 and KACL facilitates dedicated immune recognition of human keratinocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 5100–5105. [Google Scholar] [CrossRef]
- Li, Y.; Mariuzza, R.A. Structural basis for recognition of cellular and viral ligands by NK cell receptors. Front. Immunol. 2014, 5, 1–20. [Google Scholar] [CrossRef]
- Luken, B.M.; Winn, L.Y.N.; Emsley, J.; Lane, D.A.; Crawley, J.T.B. The importance of vicinal cysteines, C1669 and C1670, for von Willebrand factor A2 domain function. Blood 2010, 115, 4910–4913. [Google Scholar] [CrossRef]
- Miller, S.M.; Moore, M.J.; Massey, V.; Williams, C.H.; Distefano, M.D.; Ballou, D.P.; Walsh, C.T. Evidence for the participation of Cys558 and Cys559 at the active site of mercuric reductase. Biochemistry 1989, 28, 1194–1205. [Google Scholar] [CrossRef]
- Blake, C.C.; Ghosh, M.; Harlos, K.; Avezoux, A.; Anthony, C. The active site of methanol dehydrogenase contains a disulphide bridge between adjacent cysteine residues. Nat. Struct. Biol. 1994, 1, 102–105. [Google Scholar] [CrossRef]
- Klco, J.M.; Lassere, T.B.; Baranski, T.J. C5a receptor oligomerization. I. Disulfide trapping reveals oligomers and potential contact surfaces in a G protein-coupled receptor. J. Biol. Chem. 2003, 278, 35345–35353. [Google Scholar] [CrossRef]
- Back, J.; Malchiodi, E.L.; Cho, S.; Scarpellino, L.; Schneider, P.; Kerzic, M.C.; Mariuzza, R.A.; Held, W. Distinct conformations of Ly49 natural killer cell receptors mediate MHC class I recognition in trans and cis. Immunity 2009, 31, 598–608. [Google Scholar] [CrossRef]
- Held, W.; Mariuzza, R.A. Cis interactions of immunoreceptors with MHC and non-MHC ligands. Nat. Rev. Immunol. 2008, 8, 269–278. [Google Scholar] [CrossRef]
- Dai, Y.; Walker, S.A.; de Vet, E.; Cook, S.; Welch, H.C.E.; Lockyer, P.J. Ca2+-dependent monomer and dimer formation switches CAPRI Protein between Ras GTPase-activating protein (GAP) and RapGAP activities. J. Biol. Chem. 2011, 286, 19905–19916. [Google Scholar] [CrossRef]
- Carlyle, J.R.; Martin, A.; Mehra, A.; Attisano, L.; Tsui, F.W.; Zúñiga-Pflücker, J.C. Mouse NKR-P1B, a novel NK1.1 antigen with inhibitory function. J. Immunol. 1999, 162, 5917–5923. [Google Scholar]
- Balaji, G.R.; Aguilar, O.A.; Tanaka, M.; Shingu-Vazquez, M.A.; Fu, Z.; Gully, B.S.; Lanier, L.L.; Carlyle, J.R.; Rossjohn, J.; Berry, R. Recognition of host Clr-b by the inhibitory NKR-P1B receptor provides a basis for missing-self recognition. Nat. Commun. 2018, 9, 4623. [Google Scholar] [CrossRef]
- Xiao, J.H.; Davidson, I.; Matthes, H.; Garnier, J.-M.; Chambon, P. Cloning, expression, and transcriptional properties of the human enhancer factor TEF-1. Cell 1991, 65, 551–568. [Google Scholar] [CrossRef]
- Rozbeský, D.; Kavan, D.; Chmelík, J.; Novák, P.; Vaněk, O.; Bezouška, K. High-level expression of soluble form of mouse natural killer cell receptor NKR-P1C(B6) in Escherichia coli. Protein Expr. Purif. 2011, 77, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Chum, T.; Glatzová, D.; Kvíčalová, Z.; Malínský, J.; Brdička, T.; Cebecauer, M. The role of palmitoylation and transmembrane domain in sorting of transmembrane adaptor proteins. J. Cell Sci. 2016, 129, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Hernychová, L.; Mrázek, H.; Ivanova, L.; Kukačka, Z.; Chmelík, J.; Novák, P. Recombinant expression, in vitro refolding and characterizing disulfide bonds of a mouse inhibitory C-type lectin-like receptor Nkrp1b. Physiol. Res. 2015, 64 (Suppl. 1), S85–S93. [Google Scholar]
- Koshioka, M.; Sasaki, K.; Masuhara, H. Time-Dependent Fluorescence Depolarization Analysis in Three-Dimensional Microspectroscopy. Appl. Spectrosc. 1995, 49, 224–228. [Google Scholar] [CrossRef]
- Digman, M.A.; Caiolfa, V.R.; Zamai, M.; Gratton, E. The phasor approach to fluorescence lifetime imaging analysis. Biophys. J. 2008, 94, L14–L16. [Google Scholar] [CrossRef]





| Protein | Experimental [M + H]+, Daltons(error, ppm) | |
|---|---|---|
| Native Form | Reduced Form | |
| Nkrp1aECTO | 18168.5538 (0.03) | 18176.6566 (2.18) |
| Nkrp1c1ECTO monomer modified by cysteamine binding | 17678.3851 (0.01) | 17611.4335 (0.01) |
| Nkrp1c1ECTO dimer | 35204.4478 (7.91) | 17611.5326 (5.63) |
| Nkrp1c2ECTO | 17298.3286 (1.50) | 17306.4254 (3.47) |
| Nkrp1fECTO monomer modified by cysteamine binding | 17713.9178 (2.21) | 17644.9799 (0.55) |
| Nkrp1fECTO dimer | 35274.7974 (1.84) | 17644.9904 (0.05) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adámková, L.; Kvíčalová, Z.; Rozbeský, D.; Kukačka, Z.; Adámek, D.; Cebecauer, M.; Novák, P. Oligomeric Architecture of Mouse Activating Nkrp1 Receptors on Living Cells. Int. J. Mol. Sci. 2019, 20, 1884. https://doi.org/10.3390/ijms20081884
Adámková L, Kvíčalová Z, Rozbeský D, Kukačka Z, Adámek D, Cebecauer M, Novák P. Oligomeric Architecture of Mouse Activating Nkrp1 Receptors on Living Cells. International Journal of Molecular Sciences. 2019; 20(8):1884. https://doi.org/10.3390/ijms20081884
Chicago/Turabian StyleAdámková, Ljubina, Zuzana Kvíčalová, Daniel Rozbeský, Zdeněk Kukačka, David Adámek, Marek Cebecauer, and Petr Novák. 2019. "Oligomeric Architecture of Mouse Activating Nkrp1 Receptors on Living Cells" International Journal of Molecular Sciences 20, no. 8: 1884. https://doi.org/10.3390/ijms20081884
APA StyleAdámková, L., Kvíčalová, Z., Rozbeský, D., Kukačka, Z., Adámek, D., Cebecauer, M., & Novák, P. (2019). Oligomeric Architecture of Mouse Activating Nkrp1 Receptors on Living Cells. International Journal of Molecular Sciences, 20(8), 1884. https://doi.org/10.3390/ijms20081884