Crosstalk between p38 and Erk 1/2 in Downregulation of FGF1-Induced Signaling

 , and
, and
Abstract
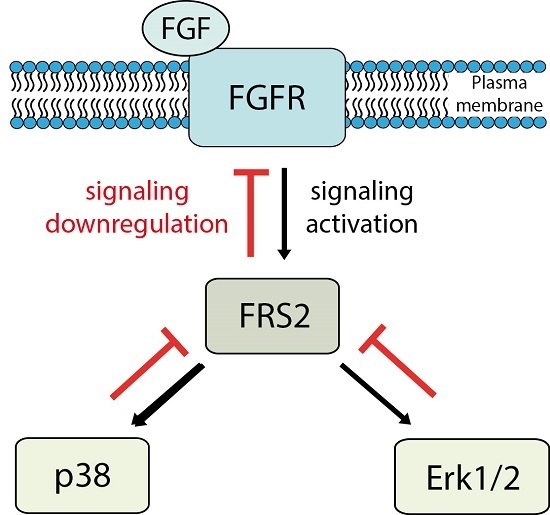
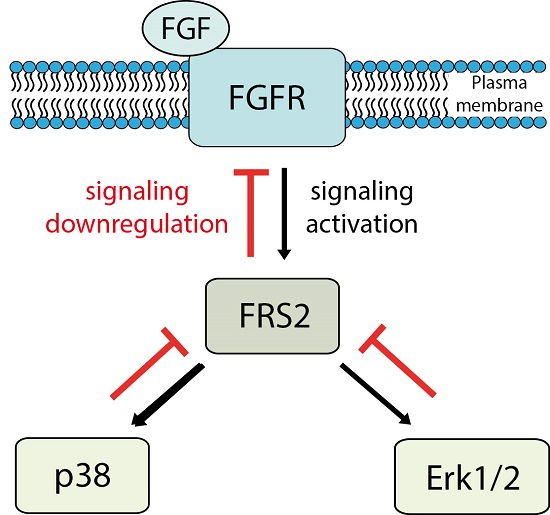{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
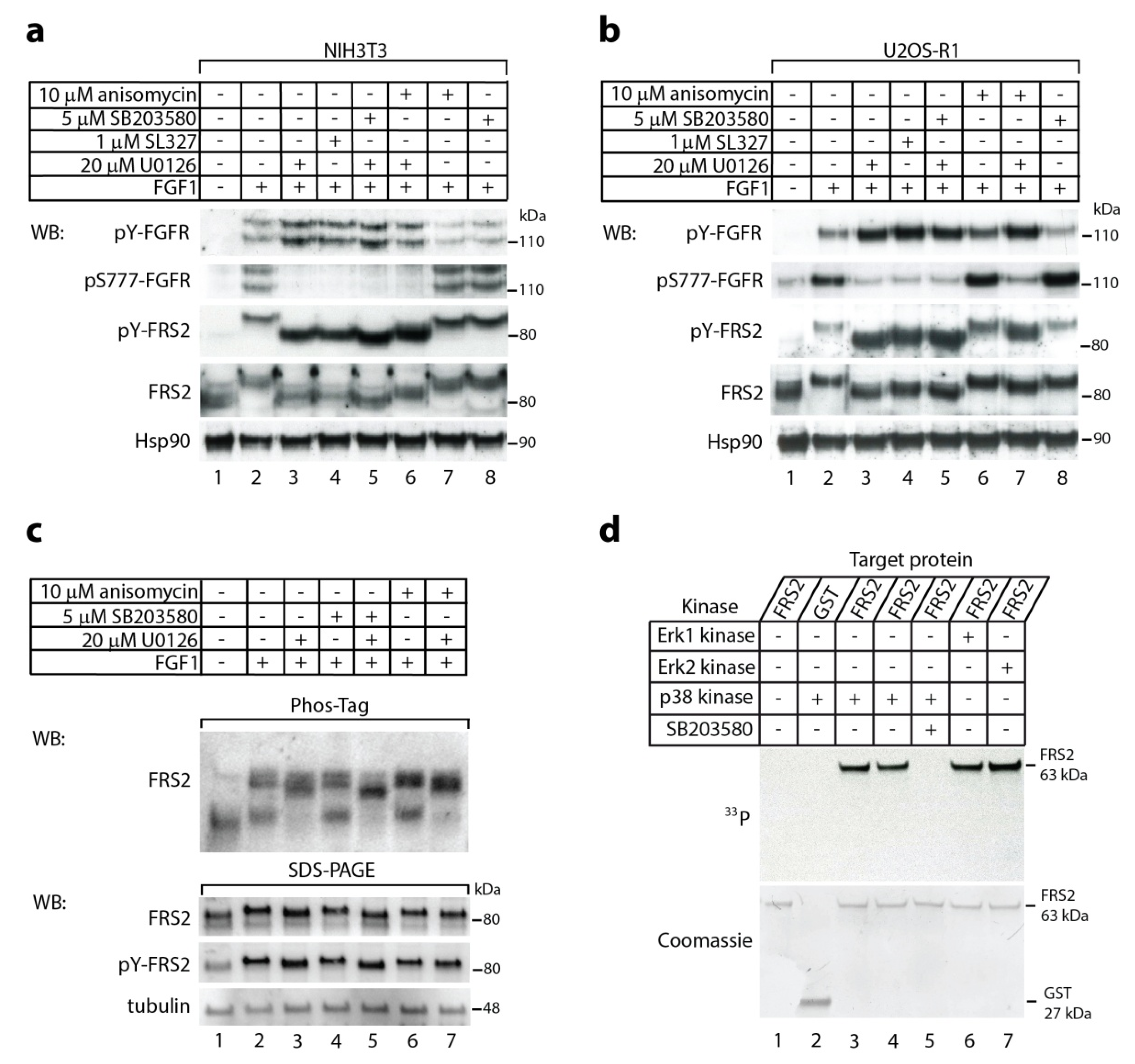
2.1. The p38 Activity Influences Electrophoretic Mobility Shift of FRS2
2.2. In Vitro Phosphorylation of FRS2 by p38α Kinase
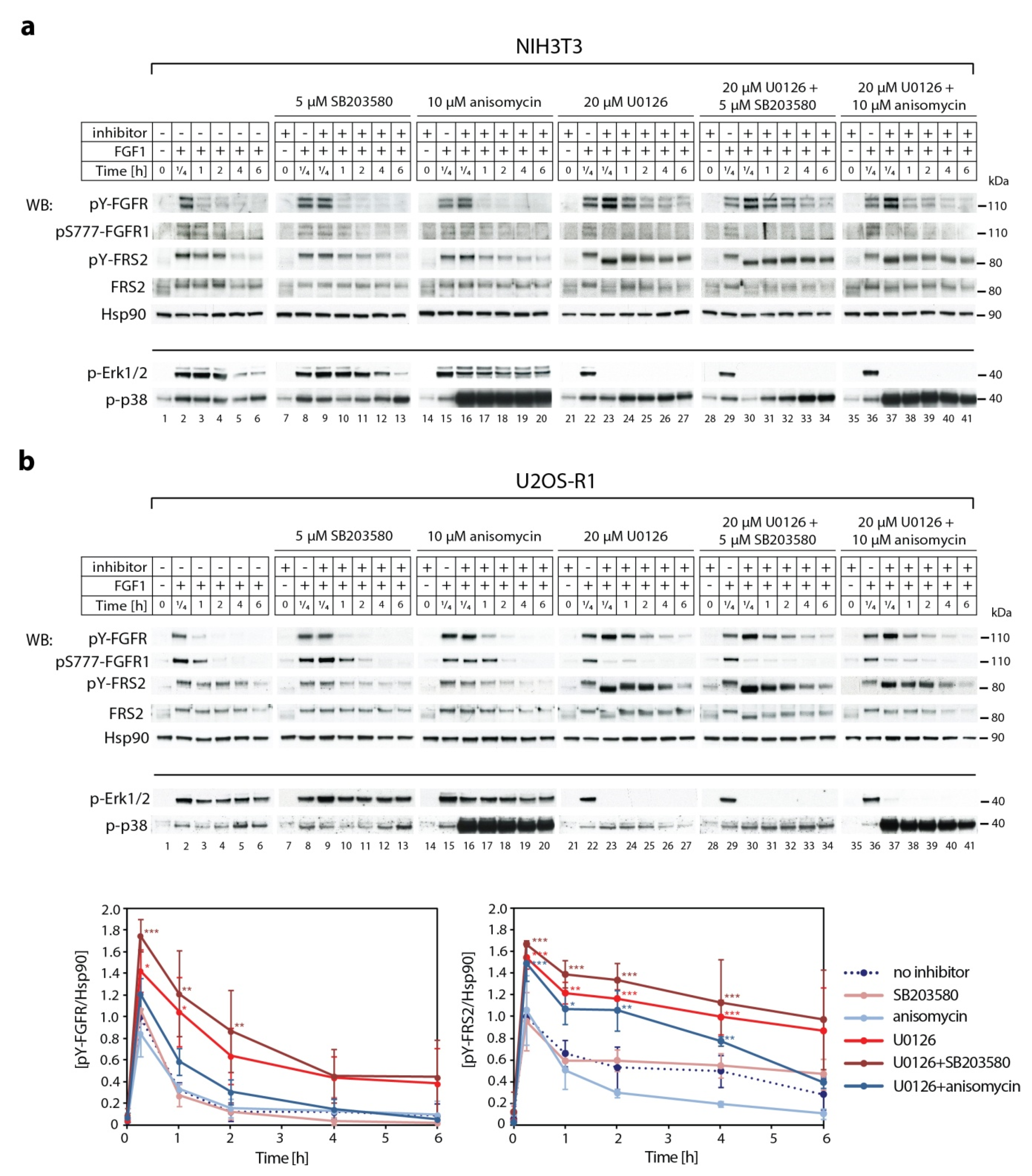
2.3. Synergistic Effect of MEK1/2 and p38 Inhibitors on Kinetics of FGF1-Induced Signaling
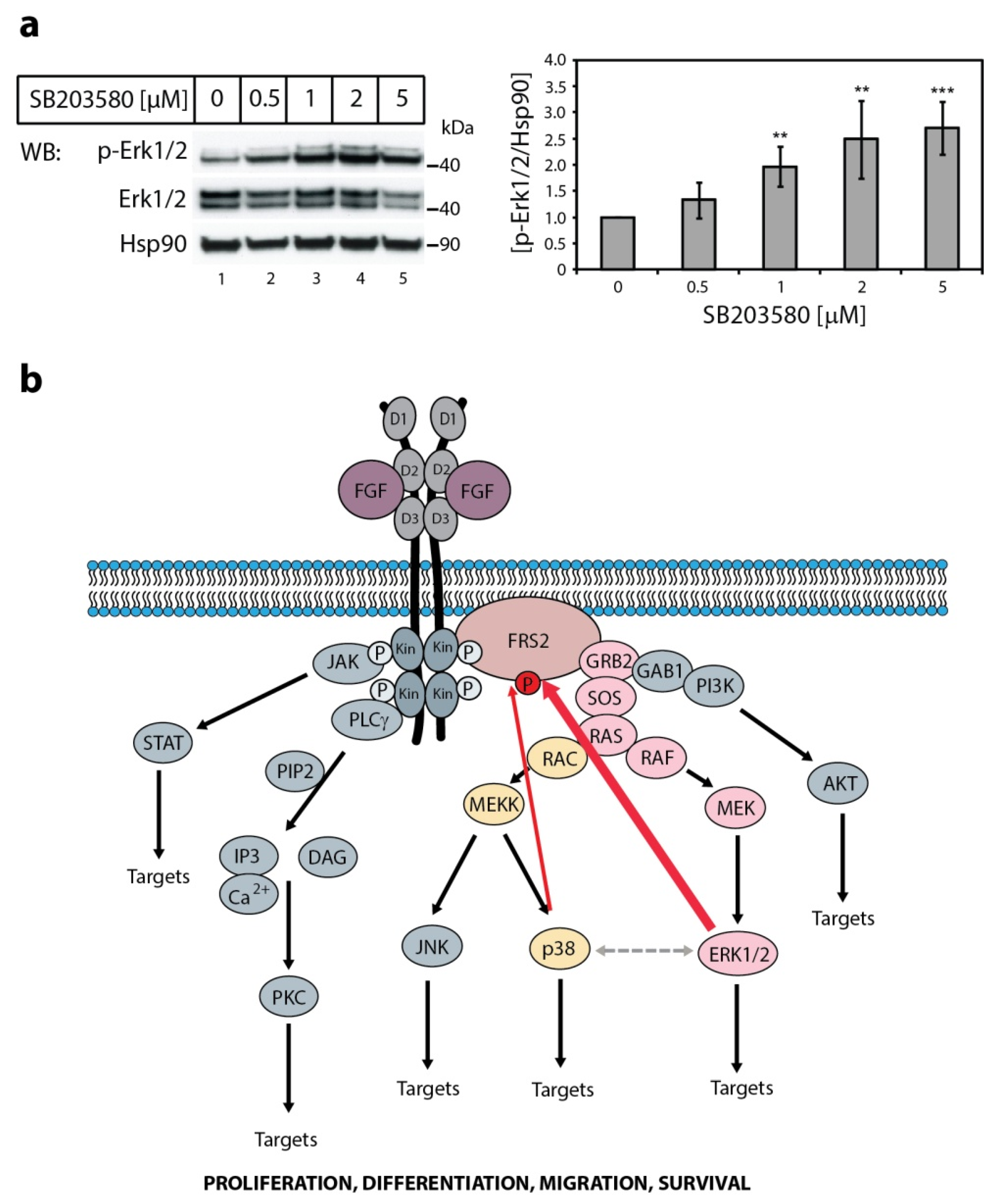
2.4. The Effect of p38 Kinase on Erk1/2 Activity
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Bacterial Strain
4.2. Recombinant Proteins
4.3. Analysis of Signaling Cascades
4.4. In Vitro Phosphorylation of Recombinant FRS2
4.5. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| FGF | Fibroblast Growth Factor |
| FGFR | Fibroblast Growth Factor Receptor |
| FRS2 | FGF receptor substrate 2 |
| MAPK | mitogen-activated protein kinase |
References
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.; Lamothe, B.; Lee, A.; Schlessinger, J.; Lax, I. FRS2 alpha attenuates FGF receptor signaling by Grb2-mediated recruitment of the ubiquitin ligase Cbl. Proc. Natl. Acad. Sci. USA 2002, 99, 6684–6689. [Google Scholar] [CrossRef]
- Cho, J.Y.; Guo, C.; Torello, M.; Lunstrum, G.P.; Iwata, T.; Deng, C.; Horton, W.A. Defective lysosomal targeting of activated fibroblast growth factor receptor 3 in achondroplasia. Proc. Natl. Acad. Sci. USA 2004, 101, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Haugsten, E.M.; Sorensen, V.; Brech, A.; Olsnes, S.; Wesche, J. Different intracellular trafficking of FGF1 endocytosed by the four homologous FGF receptors. J. Cell Sci. 2005, 118, 3869–3881. [Google Scholar] [CrossRef] [PubMed]
- Dufour, C.; Guenou, H.; Kaabeche, K.; Bouvard, D.; Sanjay, A.; Marie, P.J. FGFR2-Cbl interaction in lipid rafts triggers attenuation of PI3K/Akt signaling and osteoblast survival. Bone 2008, 42, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Zakrzewska, M.; Haugsten, E.M.; Nadratowska-Wesolowska, B.; Oppelt, A.; Hausott, B.; Jin, Y.; Otlewski, J.; Wesche, J.; Wiedlocha, A. ERK-mediated phosphorylation of fibroblast growth factor receptor 1 on Ser777 inhibits signaling. Sci. Signal. 2013, 6, ra11. [Google Scholar] [CrossRef]
- Lonic, A.; Barry, E.F.; Quach, C.; Kobe, B.; Saunders, N.; Guthridge, M.A. Fibroblast growth factor receptor 2 phosphorylation on serine 779 couples to 14-3-3 and regulates cell survival and proliferation. Mol. Cell. Biol. 2008, 28, 3372–3385. [Google Scholar] [CrossRef]
- Lonic, A.; Powell, J.A.; Kong, Y.; Thomas, D.; Holien, J.K.; Truong, N.; Parker, M.W.; Guthridge, M.A. Phosphorylation of serine 779 in fibroblast growth factor receptor 1 and 2 by protein kinase C(epsilon) regulates Ras/mitogen-activated protein kinase signaling and neuronal differentiation. J. Biol. Chem. 2013, 288, 14874–14885. [Google Scholar] [CrossRef]
- Nadratowska-Wesolowska, B.; Haugsten, E.M.; Zakrzewska, M.; Jakimowicz, P.; Zhen, Y.; Pajdzik, D.; Wesche, J.; Wiedlocha, A. RSK2 regulates endocytosis of FGF receptor 1 by phosphorylation on serine 789. Oncogene 2014, 33, 4823–4836. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, Z.Y. The mechanism of dephosphorylation of extracellular signal-regulated kinase 2 by mitogen-activated protein kinase phosphatase 3. J. Biol. Chem. 2001, 276, 32382–32391. [Google Scholar] [CrossRef]
- Cabrita, M.A.; Christofori, G. Sprouty proteins, masterminds of receptor tyrosine kinase signaling. Angiogenesis 2008, 11, 53–62. [Google Scholar] [CrossRef]
- Martinez, N.; Garcia-Dominguez, C.A.; Domingo, B.; Oliva, J.L.; Zarich, N.; Sanchez, A.; Gutierrez-Eisman, S.; Llopis, J.; Rojas, J.M. Sprouty2 binds Grb2 at two different proline-rich regions, and the mechanism of ERK inhibition is independent of this interaction. Cell Signal. 2007, 19, 2277–2285. [Google Scholar] [CrossRef]
- Kovalenko, D.; Yang, X.; Nadeau, R.J.; Harkins, L.K.; Friesel, R. Sef inhibits fibroblast growth factor signaling by inhibiting FGFR1 tyrosine phosphorylation and subsequent ERK activation. J. Biol. Chem. 2003, 278, 14087–14091. [Google Scholar] [CrossRef]
- Tsang, M.; Dawid, I.B. Promotion and attenuation of FGF signaling through the Ras-MAPK pathway. Sci. STKE 2004, 2004, pe17. [Google Scholar] [CrossRef]
- Kostas, M.; Haugsten, E.M.; Zhen, Y.; Sorensen, V.; Szybowska, P.; Fiorito, E.; Lorenz, S.; Jones, N.; de Souza, G.A.; Wiedlocha, A.; et al. Protein Tyrosine Phosphatase Receptor Type G (PTPRG) Controls Fibroblast Growth Factor Receptor (FGFR) 1 Activity and Influences Sensitivity to FGFR Kinase Inhibitors. Mol. Cell. Proteom. 2018, 17, 850–870. [Google Scholar] [CrossRef]
- Lax, I.; Wong, A.; Lamothe, B.; Lee, A.; Frost, A.; Hawes, J.; Schlessinger, J. The docking protein FRS2alpha controls a MAP kinase-mediated negative feedback mechanism for signaling by FGF receptors. Mol. Cell 2002, 10, 709–719. [Google Scholar] [CrossRef]
- Ong, S.H.; Guy, G.R.; Hadari, Y.R.; Laks, S.; Gotoh, N.; Schlessinger, J.; Lax, I. FRS2 proteins recruit intracellular signaling pathways by binding to diverse targets on fibroblast growth factor and nerve growth factor receptors. Mol. Cell. Biol. 2000, 20, 979–989. [Google Scholar] [CrossRef]
- Kouhara, H.; Hadari, Y.R.; Spivak-Kroizman, T.; Schilling, J.; Bar-Sagi, D.; Lax, I.; Schlessinger, J. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell 1997, 89, 693–702. [Google Scholar] [CrossRef]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef]
- Stramucci, L.; Pranteda, A.; Bossi, G. Insights of Crosstalk between p53 Protein and the MKK3/MKK6/p38 MAPK Signaling Pathway in Cancer. Cancers (Basel) 2018, 10, 131. [Google Scholar] [CrossRef]
- Sheridan, D.L.; Kong, Y.; Parker, S.A.; Dalby, K.N.; Turk, B.E. Substrate discrimination among mitogen-activated protein kinases through distinct docking sequence motifs. J. Biol. Chem. 2008, 283, 19511–19520. [Google Scholar] [CrossRef]
- Sorensen, V.; Zhen, Y.; Zakrzewska, M.; Haugsten, E.M.; Walchli, S.; Nilsen, T.; Olsnes, S.; Wiedlocha, A. Phosphorylation of fibroblast growth factor (FGF) receptor 1 at Ser777 by p38 mitogen-activated protein kinase regulates translocation of exogenous FGF1 to the cytosol and nucleus. Mol. Cell. Biol. 2008, 28, 4129–4141. [Google Scholar] [CrossRef]
- Schmidt, O.; Harbauer, A.B.; Rao, S.; Eyrich, B.; Zahedi, R.P.; Stojanovski, D.; Schonfisch, B.; Guiard, B.; Sickmann, A.; Pfanner, N.; et al. Regulation of mitochondrial protein import by cytosolic kinases. Cell 2011, 144, 227–239. [Google Scholar] [CrossRef]
- Harbauer, A.B.; Opalinska, M.; Gerbeth, C.; Herman, J.S.; Rao, S.; Schonfisch, B.; Guiard, B.; Schmidt, O.; Pfanner, N.; Meisinger, C. Mitochondria. Cell cycle-dependent regulation of mitochondrial preprotein translocase. Science 2014, 346, 1109–1113. [Google Scholar] [CrossRef]
- Tanoue, T.; Nishida, E. Molecular recognitions in the MAP kinase cascades. Cell Signal. 2003, 15, 455–462. [Google Scholar] [CrossRef]
- Barsyte-Lovejoy, D.; Galanis, A.; Sharrocks, A.D. Specificity determinants in MAPK signaling to transcription factors. J. Biol. Chem. 2002, 277, 9896–9903. [Google Scholar] [CrossRef]
- Akella, R.; Moon, T.M.; Goldsmith, E.J. Unique MAP Kinase binding sites. Biochim. Biophys. Acta 2008, 1784, 48–55. [Google Scholar] [CrossRef][Green Version]
- Cowan, K.J.; Storey, K.B. Mitogen-activated protein kinases: New signaling pathways functioning in cellular responses to environmental stress. J. Exp. Biol. 2003, 206, 1107–1115. [Google Scholar] [CrossRef]
- Yordy, J.S.; Muise-Helmericks, R.C. Signal transduction and the Ets family of transcription factors. Oncogene 2000, 19, 6503–6513. [Google Scholar] [CrossRef]
- Kefaloyianni, E.; Gaitanaki, C.; Beis, I. ERK1/2 and p38-MAPK signalling pathways, through MSK1, are involved in NF-kappaB transactivation during oxidative stress in skeletal myoblasts. Cell Signal. 2006, 18, 2238–2251. [Google Scholar] [CrossRef]
- Todd, D.E.; Densham, R.M.; Molton, S.A.; Balmanno, K.; Newson, C.; Weston, C.R.; Garner, A.P.; Scott, L.; Cook, S.J. ERK1/2 and p38 cooperate to induce a p21CIP1-dependent G1 cell cycle arrest. Oncogene 2004, 23, 3284–3295. [Google Scholar] [CrossRef][Green Version]
- Ng, D.C.; Long, C.S.; Bogoyevitch, M.A. A role for the extracellular signal-regulated kinase and p38 mitogen-activated protein kinases in interleukin-1 beta-stimulated delayed signal tranducer and activator of transcription 3 activation, atrial natriuretic factor expression, and cardiac myocyte morphology. J. Biol. Chem. 2001, 276, 29490–29498. [Google Scholar]
- Sundaramurthy, P.; Gakkhar, S.; Sowdhamini, R. Computational prediction and analysis of impact of the cross-talks between JNK and P38 kinase cascades. Bioinformation 2009, 3, 250–254. [Google Scholar] [CrossRef]
- Burotto, M.; Chiou, V.L.; Lee, J.M.; Kohn, E.C. The MAPK pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Peng, W.; Calvo, E. Rational Approaches for Combination Therapy Strategies Targeting the MAP Kinase Pathway in Solid Tumors. Mol. Cancer Ther. 2018, 17, 3–16. [Google Scholar] [CrossRef]
- Grimaldi, A.M.; Simeone, E.; Festino, L.; Vanella, V.; Palla, M.; Ascierto, P.A. Novel mechanisms and therapeutic approaches in melanoma: Targeting the MAPK pathway. Discov. Med. 2015, 19, 455–461. [Google Scholar]
- Neuzillet, C.; Tijeras-Raballand, A.; de Mestier, L.; Cros, J.; Faivre, S.; Raymond, E. MEK in cancer and cancer therapy. Pharmacol. Ther. 2014, 141, 160–171. [Google Scholar] [CrossRef]
- Haugsten, E.M.; Malecki, J.; Bjorklund, S.M.; Olsnes, S.; Wesche, J. Ubiquitination of fibroblast growth factor receptor 1 is required for its intracellular sorting but not for its endocytosis. Mol. Biol. Cell 2008, 19, 3390–3403. [Google Scholar] [CrossRef]
- Zakrzewska, M.; Krowarsch, D.; Wiedlocha, A.; Otlewski, J. Design of fully active FGF-1 variants with increased stability. Protein Eng. Des. Sel. 2004, 17, 603–611. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zakrzewska, M.; Opalinski, L.; Haugsten, E.M.; Otlewski, J.; Wiedlocha, A. Crosstalk between p38 and Erk 1/2 in Downregulation of FGF1-Induced Signaling. Int. J. Mol. Sci. 2019, 20, 1826. https://doi.org/10.3390/ijms20081826
Zakrzewska M, Opalinski L, Haugsten EM, Otlewski J, Wiedlocha A. Crosstalk between p38 and Erk 1/2 in Downregulation of FGF1-Induced Signaling. International Journal of Molecular Sciences. 2019; 20(8):1826. https://doi.org/10.3390/ijms20081826
Chicago/Turabian StyleZakrzewska, Malgorzata, Lukasz Opalinski, Ellen M. Haugsten, Jacek Otlewski, and Antoni Wiedlocha. 2019. "Crosstalk between p38 and Erk 1/2 in Downregulation of FGF1-Induced Signaling" International Journal of Molecular Sciences 20, no. 8: 1826. https://doi.org/10.3390/ijms20081826
APA StyleZakrzewska, M., Opalinski, L., Haugsten, E. M., Otlewski, J., & Wiedlocha, A. (2019). Crosstalk between p38 and Erk 1/2 in Downregulation of FGF1-Induced Signaling. International Journal of Molecular Sciences, 20(8), 1826. https://doi.org/10.3390/ijms20081826