When CAR Meets Stem Cells

Abstract
:1. Introduction
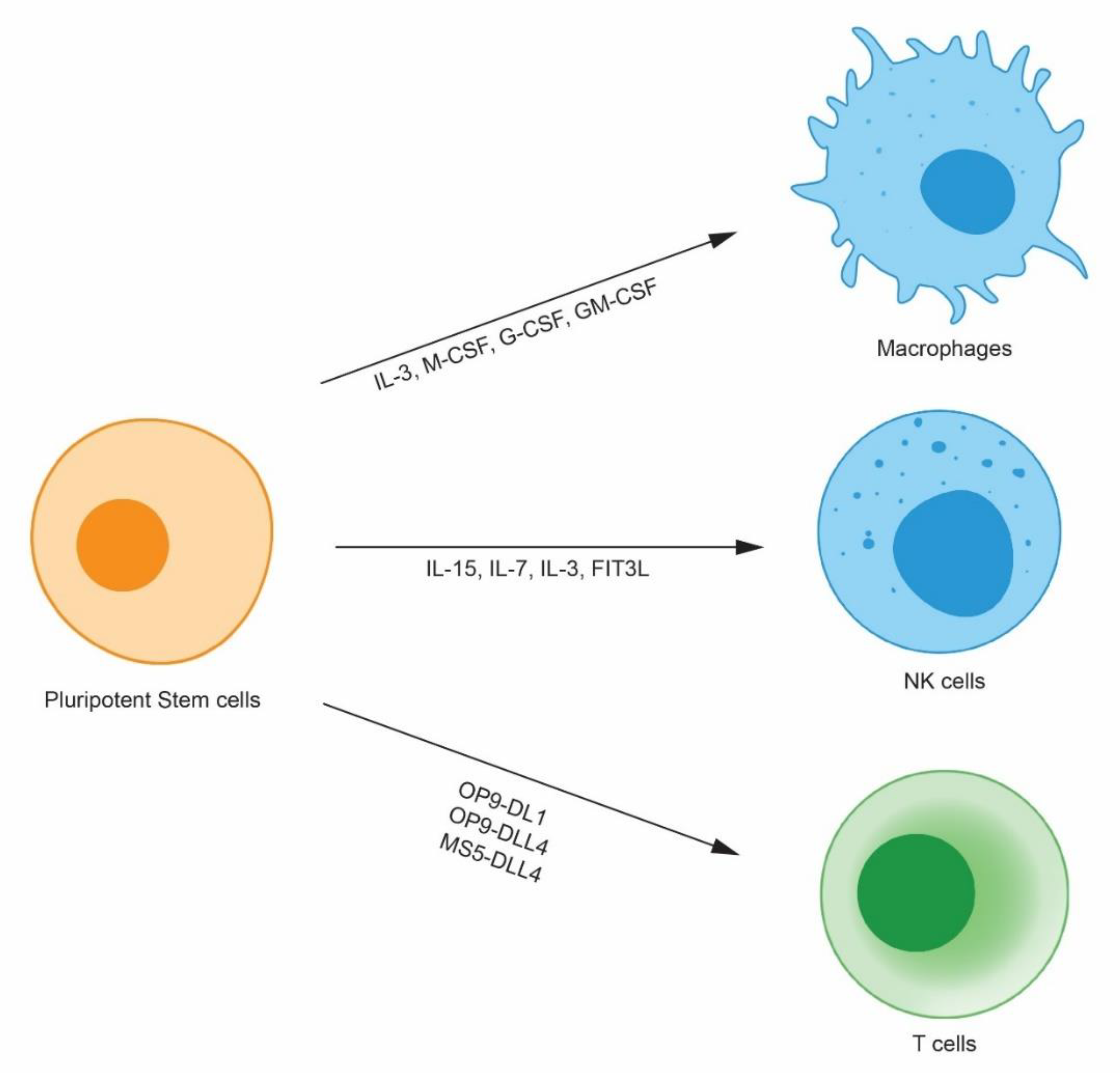
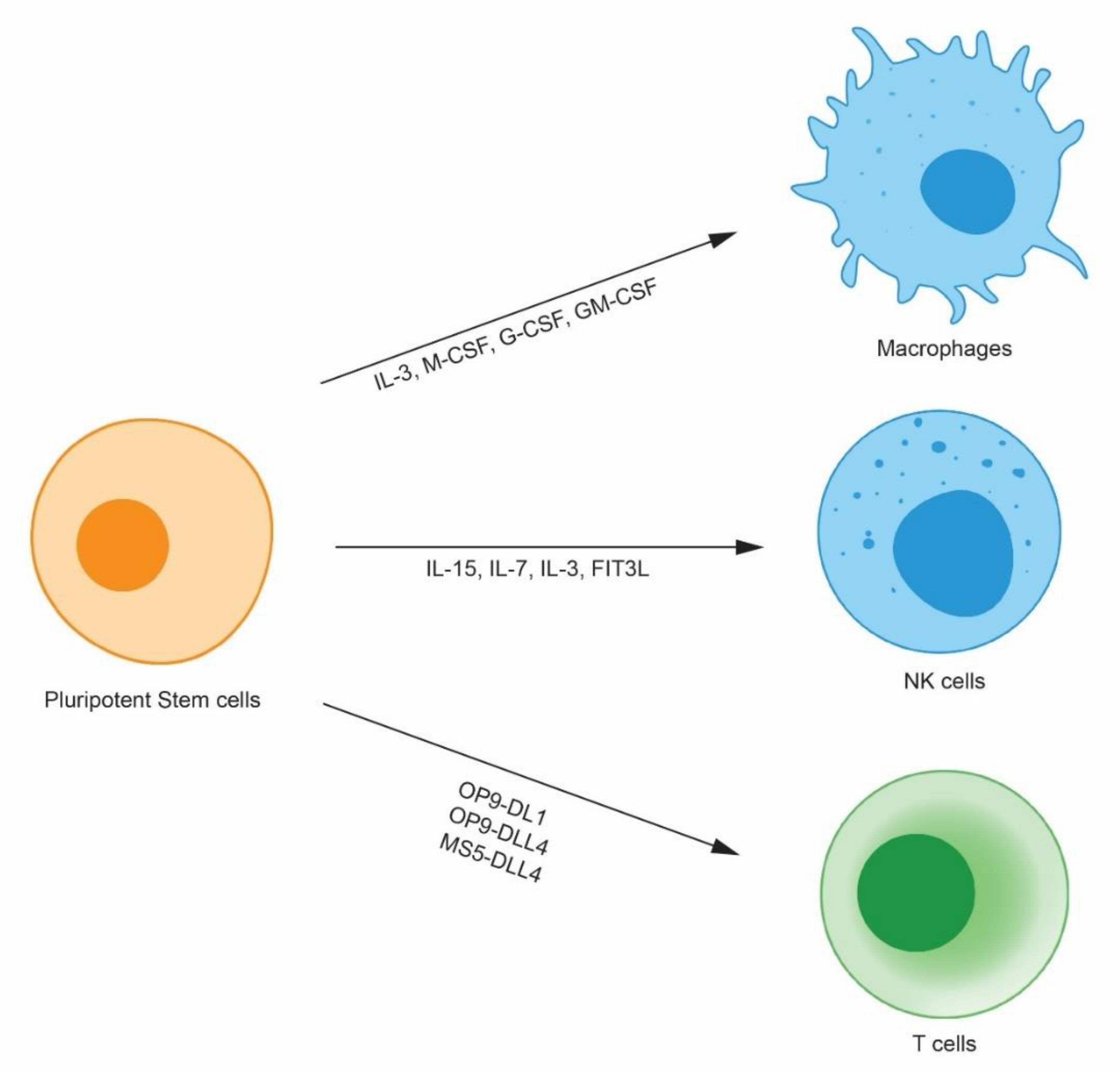
2. Pluripotent Stem-Cell Derived Immune Cells
2.1. Pluripotent Stem Cell-Derived Natural Killer Cells
2.2. Pluripotent Stem Cell-Derived-Macrophages
2.3. Pluripotent Stem Cell-Derived T Cells
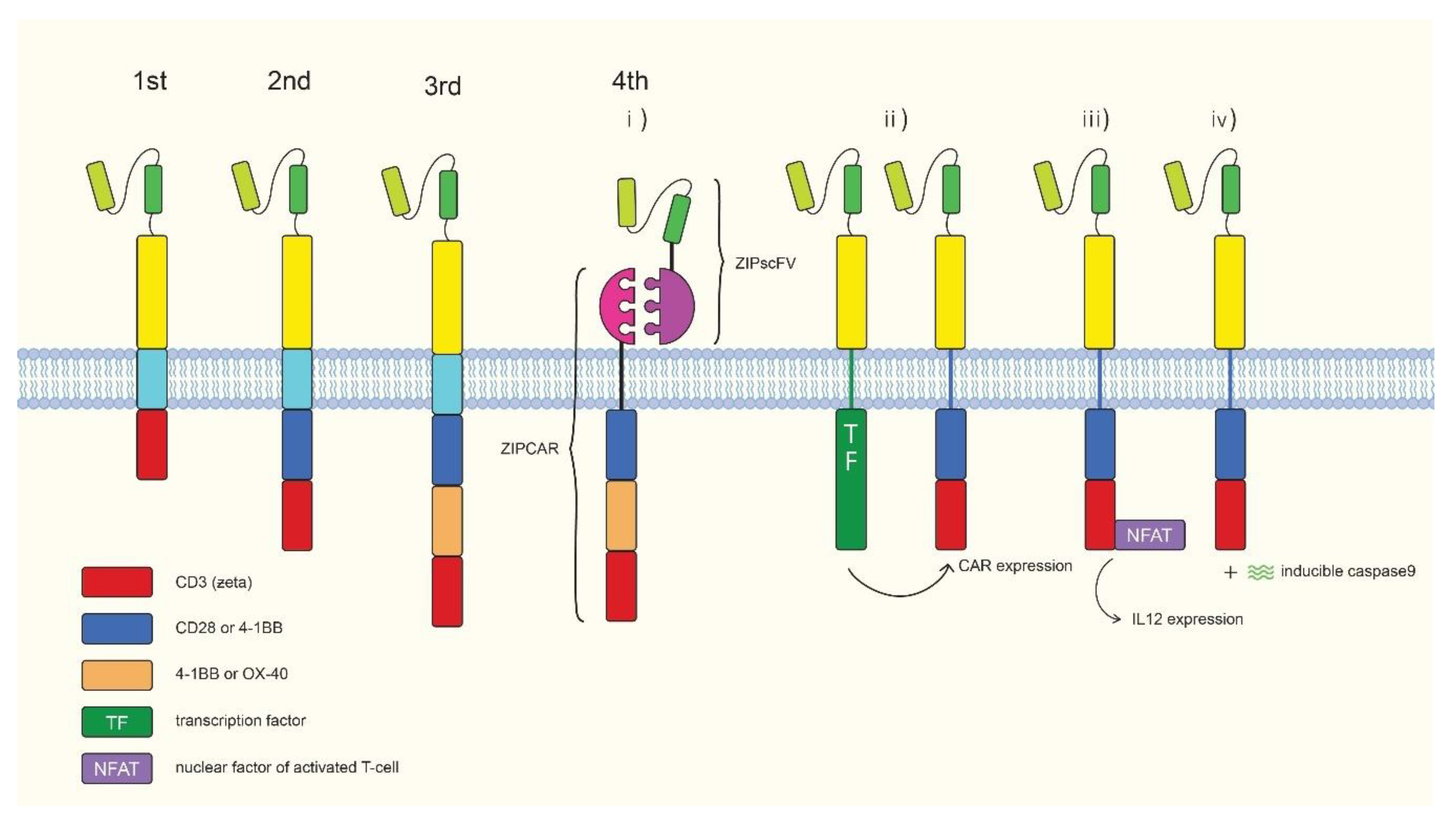
3. Chimeric Antigen Receptor
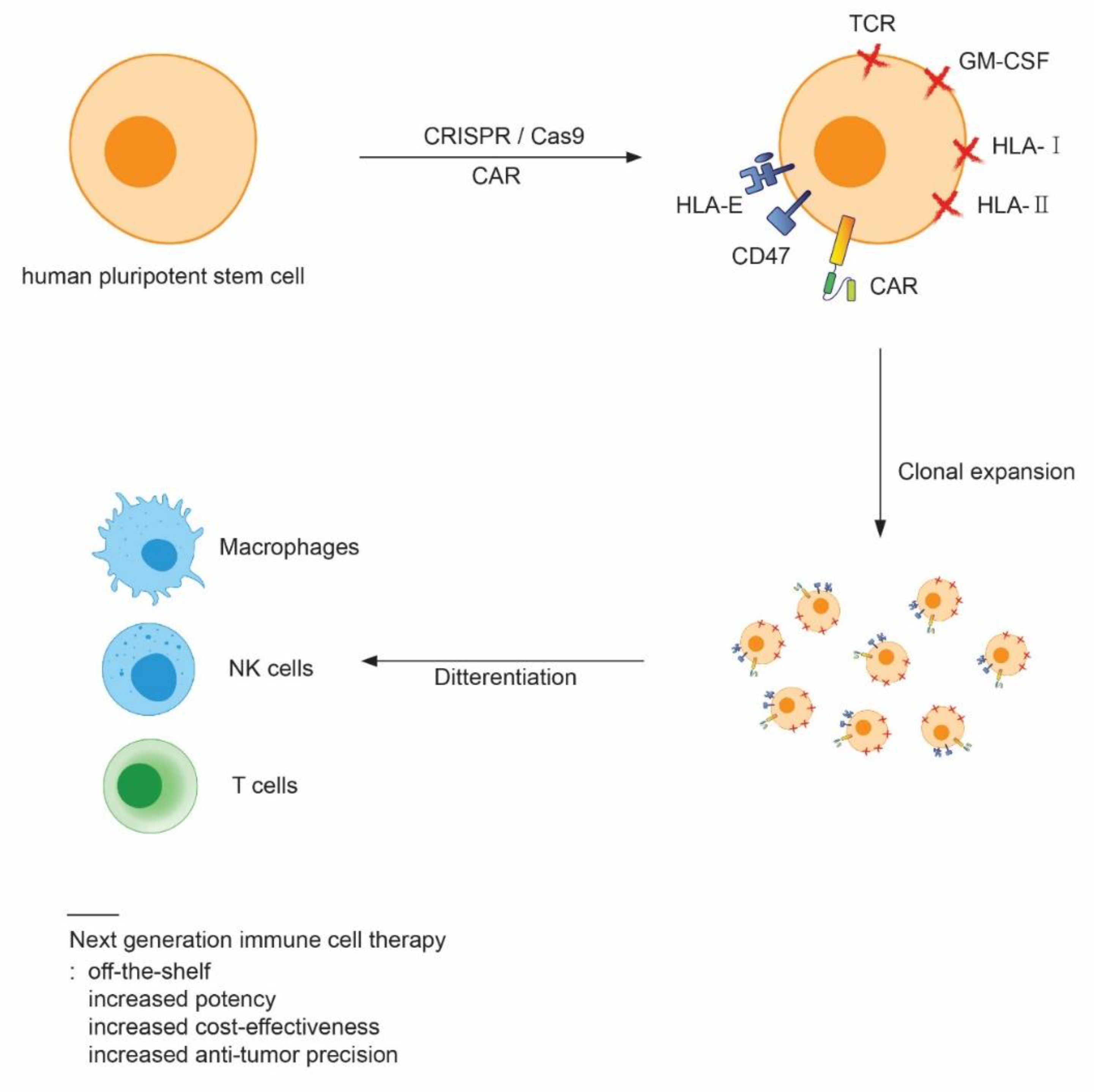
4. Stem Cell-Derived CAR Immune Cells
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Zakrzewski, W.; Dobrzynski, M.; Szymonowicz, M.; Rybak, Z. Stem cells: Past, present, and future. Stem Cell Res. Ther. 2019, 10, 68. [Google Scholar] [CrossRef]
- Mikkilineni, L.; Kochenderfer, J.N. Chimeric antigen receptor T-cell therapies for multiple myeloma. Blood 2017, 130, 2594–2602. [Google Scholar] [CrossRef] [Green Version]
- Susanibar Adaniya, S.P.; Cohen, A.D.; Garfall, A.L. Chimeric antigen receptor T cell immunotherapy for multiple myeloma: A review of current data and potential clinical applications. Am. J. Hematol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Kim, C.H. Evolution of chimeric antigen receptor (CAR) T cell therapy: Current status and future perspectives. Arch. Pharm. Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.P.; Kros, J.M.; Li, J. Approved CAR T cell therapies: Ice bucket challenges on glaring safety risks and long-term impacts. Drug Discov. Today 2018, 23, 1175–1182. [Google Scholar] [CrossRef]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Benda, C.; Duzinger, S.; Huang, Y.; Li, X.; Li, Y.; Guo, X.; Cao, G.; Chen, S.; Hao, L.; et al. Generation of induced pluripotent stem cells from urine. J. Am. Soc. Nephrol. 2011, 22, 1221–1228. [Google Scholar] [CrossRef]
- Sugimura, R.; Jha, D.K.; Han, A.; Soria-Valles, C.; da Rocha, E.L.; Lu, Y.F.; Goettel, J.A.; Serrao, E.; Rowe, R.G.; Malleshaiah, M.; et al. Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature 2017, 545, 432–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokkinaki, M.; Sahibzada, N.; Golestaneh, N. Human induced pluripotent stem-derived retinal pigment epithelium (RPE) cells exhibit ion transport, membrane potential, polarized vascular endothelial growth factor secretion, and gene expression pattern similar to native rpe. Stem Cells 2011, 29, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Millman, J.R.; Xie, C.; Van Dervort, A.; Gurtler, M.; Pagliuca, F.W.; Melton, D.A. Generation of stem cell-derived beta-cells from patients with type 1 diabetes. Nat. Commun. 2016, 7, 11463. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Lal, G. The molecular mechanism of natural killer cells function and its importance in cancer immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef]
- Guillerey, C.; Huntington, N.D.; Smyth, M.J. Targeting natural killer cells in cancer immunotherapy. Nat. Immunol. 2016, 17, 1025–1036. [Google Scholar] [CrossRef]
- Brown, M.G.; Dokun, A.O.; Heusel, J.W.; Smith, H.R.; Beckman, D.L.; Blattenberger, E.A.; Dubbelde, C.E.; Stone, L.R.; Scalzo, A.A.; Yokoyama, W.M. Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science 2001, 292, 934–937. [Google Scholar] [CrossRef]
- Mandelboim, O.; Lieberman, N.; Lev, M.; Paul, L.; Arnon, T.I.; Bushkin, Y.; Davis, D.M.; Strominger, J.L.; Yewdell, J.W.; Porgador, A. Recognition of haemagglutinins on virus-infected cells by nkp46 activates lysis by human NK cells. Nature 2001, 409, 1055–1060. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, A.M.; Diefenbach, A.; McMahon, C.W.; Xiong, N.; Carlyle, J.R.; Raulet, D.H. The role of the nkg2d immunoreceptor in immune cell activation and natural killing. Immunity 2002, 17, 19–29. [Google Scholar] [CrossRef]
- Lee, S.H.; Miyagi, T.; Biron, C.A. Keeping NK cells in highly regulated antiviral warfare. Trends Immunol. 2007, 28, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Bluman, E.M.; Bartynski, K.J.; Avalos, B.R.; Caligiuri, M.A. Human natural killer cells produce abundant macrophage inflammatory protein-1 alpha in response to monocyte-derived cytokines. J. Clin. Investig. 1996, 97, 2722–2727. [Google Scholar] [CrossRef]
- Roda, J.M.; Parihar, R.; Magro, C.; Nuovo, G.J.; Tridandapani, S.; Carson, W.E., 3rd. Natural killer cells produce T cell-recruiting chemokines in response to antibody-coated tumor cells. Cancer Res. 2006, 66, 517–526. [Google Scholar] [CrossRef]
- Fauriat, C.; Long, E.O.; Ljunggren, H.G.; Bryceson, Y.T. Regulation of human nk-cell cytokine and chemokine production by target cell recognition. Blood 2010, 115, 2167–2176. [Google Scholar] [CrossRef]
- Lettau, M.; Schmidt, H.; Kabelitz, D.; Janssen, O. Secretory lysosomes and their cargo in T and NK cells. Immunol. Lett. 2007, 108, 10–19. [Google Scholar] [CrossRef]
- Pipkin, M.E.; Lieberman, J. Delivering the kiss of death: Progress on understanding how perforin works. Curr. Opin. Immunol. 2007, 19, 301–308. [Google Scholar] [CrossRef]
- Trapani, J.A.; Bird, P.I. A renaissance in understanding the multiple and diverse functions of granzymes? Immunity 2008, 29, 665–667. [Google Scholar] [CrossRef]
- Colucci, F.; Caligiuri, M.A.; Di Santo, J.P. What does it take to make a natural killer? Nat. Rev. Immunol. 2003, 3, 413–425. [Google Scholar] [CrossRef]
- Woll, P.S.; Martin, C.H.; Miller, J.S.; Kaufman, D.S. Human embryonic stem cell-derived NK cells acquire functional receptors and cytolytic activity. J. Immunol. 2005, 175, 5095–5103. [Google Scholar] [CrossRef]
- Woll, P.S.; Grzywacz, B.; Tian, X.; Marcus, R.K.; Knorr, D.A.; Verneris, M.R.; Kaufman, D.S. Human embryonic stem cells differentiate into a homogeneous population of natural killer cells with potent in vivo antitumor activity. Blood 2009, 113, 6094–6101. [Google Scholar] [CrossRef] [Green Version]
- Ni, Z.; Knorr, D.A.; Clouser, C.L.; Hexum, M.K.; Southern, P.; Mansky, L.M.; Park, I.H.; Kaufman, D.S. Human pluripotent stem cells produce natural killer cells that mediate anti-hiv-1 activity by utilizing diverse cellular mechanisms. J. Virol. 2011, 85, 43–50. [Google Scholar] [CrossRef]
- Ni, Z.; Knorr, D.A.; Kaufman, D.S. Hematopoietic and nature killer cell development from human pluripotent stem cells. Methods Mol. Biol. 2013, 1029, 33–41. [Google Scholar]
- Knorr, D.A.; Ni, Z.; Hermanson, D.; Hexum, M.K.; Bendzick, L.; Cooper, L.J.; Lee, D.A.; Kaufman, D.S. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl. Med. 2013, 2, 274–283. [Google Scholar] [CrossRef]
- Bock, A.M.; Knorr, D.; Kaufman, D.S. Development, expansion, and in vivo monitoring of human NK cells from human embryonic stem cells (hESCs) and and induced pluripotent stem cells (iPSCs). J. Vis. Exp. 2013, e50337. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.; Pollard, J.W.; et al. A lineage of myeloid cells independent of myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef]
- Serbina, N.V.; Jia, T.; Hohl, T.M.; Pamer, E.G. Monocyte-mediated defense against microbial pathogens. Annu. Rev. Immunol. 2008, 26, 421–452. [Google Scholar] [CrossRef]
- Chen, S.; Tisch, N.; Kegel, M.; Yerbes, R.; Hermann, R.; Hudalla, H.; Zuliani, C.; Gulculer, G.S.; Zwadlo, K.; von Engelhardt, J.; et al. Cns macrophages control neurovascular development via CD95l. Cell Rep. 2017, 19, 1378–1393. [Google Scholar] [CrossRef]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Wulfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages facilitate electrical conduction in the heart. Cell 2017, 169, 510–522.e20. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Weiss, G.; Schaible, U.E. Macrophage defense mechanisms against intracellular bacteria. Immunol. Rev. 2015, 264, 182–203. [Google Scholar] [CrossRef] [Green Version]
- Lachmann, N.; Ackermann, M.; Frenzel, E.; Liebhaber, S.; Brennig, S.; Happle, C.; Hoffmann, D.; Klimenkova, O.; Luttge, D.; Buchegger, T.; et al. Large-scale hematopoietic differentiation of human induced pluripotent stem cells provides granulocytes or macrophages for Cell Rep.lacement therapies. Stem Cell Rep. 2015, 4, 282–296. [Google Scholar] [CrossRef]
- van Wilgenburg, B.; Browne, C.; Vowles, J.; Cowley, S.A. Efficient, long term production of monocyte-derived macrophages from human pluripotent stem cells under partly-defined and fully-defined conditions. PLoS ONE 2013, 8, e71098. [Google Scholar] [CrossRef]
- Takata, K.; Kozaki, T.; Lee, C.Z.W.; Thion, M.S.; Otsuka, M.; Lim, S.; Utami, K.H.; Fidan, K.; Park, D.S.; Malleret, B.; et al. Induced-pluripotent-stem-cell-derived primitive macrophages provide a platform for modeling tissue-resident macrophage differentiation and function. Immunity 2017, 47, 183–198. [Google Scholar] [CrossRef]
- Zhu, J.; Paul, W.E. CD4 T cells: Fates, functions, and faults. Blood 2008, 112, 1557–1569. [Google Scholar] [CrossRef]
- Alcover, A.; Alarcon, B.; Di Bartolo, V. Cell biology of T cell receptor expression and regulation. Annu. Rev. Immunol. 2018, 36, 103–125. [Google Scholar] [CrossRef]
- Zhang, N.; Bevan, M.J. CD8+ T cells: Foot soldiers of the immune system. Immunity 2011, 35, 161–168. [Google Scholar] [CrossRef]
- Kennedy, M.; Awong, G.; Sturgeon, C.M.; Ditadi, A.; LaMotte-Mohs, R.; Zuniga-Pflucker, J.C.; Keller, G. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Rep. 2012, 2, 1722–1735. [Google Scholar] [CrossRef]
- Timmermans, F.; Velghe, I.; Vanwalleghem, L.; De Smedt, M.; Van Coppernolle, S.; Taghon, T.; Moore, H.D.; Leclercq, G.; Langerak, A.W.; Kerre, T.; et al. Generation of T cells from human embryonic stem cell-derived hematopoietic zones. J. Immunol. 2009, 182, 6879–6888. [Google Scholar] [CrossRef]
- Lin, C.; Zhang, J. Chimeric antigen receptor engineered innate immune cells in cancer immunotherapy. Sci. China Life Sci. 2019. [Google Scholar] [CrossRef]
- Kumar, A.; Lee, J.H.; Suknuntha, K.; D’Souza, S.S.; Thakur, A.S.; Slukvin, II. Notch activation at the hematovascular mesoderm stage facilitates efficient generation of T cells with high proliferation potential from human pluripotent stem cells. J. Immunol. 2019, 202, 770–776. [Google Scholar] [CrossRef]
- Nishimura, T.; Kaneko, S.; Kawana-Tachikawa, A.; Tajima, Y.; Goto, H.; Zhu, D.; Nakayama-Hosoya, K.; Iriguchi, S.; Uemura, Y.; Shimizu, T.; et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell 2013, 12, 114–126. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-t-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and t-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef]
- Finney, H.M.; Lawson, A.D.; Bebbington, C.R.; Weir, A.N. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J. Immunol. 1998, 161, 2791–2797. [Google Scholar]
- June, C.H.; Sadelain, M. Chimeric antigen receptor therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Liu, B.; Song, Y.; Liu, D. Clinical trials of CAR-T cells in China. J. Hematol. Oncol. 2017, 10, 166. [Google Scholar] [CrossRef] [Green Version]
- Carpenito, C.; Milone, M.C.; Hassan, R.; Simonet, J.C.; Lakhal, M.; Suhoski, M.M.; Varela-Rohena, A.; Haines, K.M.; Heitjan, D.F.; Albelda, S.M.; et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl. Acad. Sci. USA 2009, 106, 3360–3365. [Google Scholar] [CrossRef] [Green Version]
- Haso, W.; Lee, D.W.; Shah, N.N.; Stetler-Stevenson, M.; Yuan, C.M.; Pastan, I.H.; Dimitrov, D.S.; Morgan, R.A.; FitzGerald, D.J.; Barrett, D.M.; et al. Anti-CD22-chimeric antigen receptors targeting b-cell precursor acute lymphoblastic leukemia. Blood 2013, 121, 1165–1174. [Google Scholar] [CrossRef]
- Tang, X.Y.; Sun, Y.; Zhang, A.; Hu, G.L.; Cao, W.; Wang, D.H.; Zhang, B.; Chen, H. Third-generation CD28/4-1BB chimeric antigen receptor T cells for chemotherapy relapsed or refractory acute lymphoblastic leukaemia: A non-randomised, open-label phase i trial protocol. BMJ Open 2016, 6, e013904. [Google Scholar] [CrossRef]
- Zhong, X.S.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol. Ther. 2010, 18, 413–420. [Google Scholar] [CrossRef]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef]
- Diaconu, I.; Ballard, B.; Zhang, M.; Chen, Y.; West, J.; Dotti, G.; Savoldo, B. Inducible caspase-9 selectively modulates the toxicities of CD19-specific chimeric antigen receptor-modified T cells. Mol. Ther. 2017, 25, 580–592. [Google Scholar] [CrossRef]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef]
- Cho, J.H.; Collins, J.J.; Wong, W.W. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell 2018, 173, 1426–1438. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Wilson, W.H.; Janik, J.E.; Dudley, M.E.; Stetler-Stevenson, M.; Feldman, S.A.; Maric, I.; Raffeld, M.; Nathan, D.A.; Lanier, B.J.; et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010, 116, 4099–4102. [Google Scholar] [CrossRef] [Green Version]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra138. [Google Scholar] [CrossRef]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef]
- Knochelmann, H.M.; Smith, A.S.; Dwyer, C.J.; Wyatt, M.M.; Mehrotra, S.; Paulos, C.M. CAR T cells in solid tumors: Blueprints for building effective therapies. Front. Immunol. 2018, 9, 1740. [Google Scholar] [CrossRef]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef]
- Zhang, E.; Gu, J.; Xu, H. Prospects for chimeric antigen receptor-modified T cell therapy for solid tumors. Mol. Cancer 2018, 17, 7. [Google Scholar] [CrossRef] [Green Version]
- Jamal-Hanjani, M.; Quezada, S.A.; Larkin, J.; Swanton, C. Translational implications of tumor heterogeneity. Clin. Cancer Res. 2015, 21, 1258–1266. [Google Scholar] [CrossRef]
- Singh, N.; Perazzelli, J.; Grupp, S.A.; Barrett, D.M. Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Sci. Transl. Med. 2016, 8, 320ra323. [Google Scholar] [CrossRef]
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global manufacturing of CAR T cell therapy. Mol. Ther. Methods Clin. Dev. 2017, 4, 92–101. [Google Scholar] [CrossRef]
- Gornalusse, G.G.; Hirata, R.K.; Funk, S.E.; Riolobos, L.; Lopes, V.S.; Manske, G.; Prunkard, D.; Colunga, A.G.; Hanafi, L.A.; Clegg, D.O.; et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat. Biotechnol. 2017, 35, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Deuse, T.; Hu, X.; Gravina, A.; Wang, D.; Tediashvili, G.; De, C.; Thayer, W.O.; Wahl, A.; Garcia, J.V.; Reichenspurner, H.; et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol. 2019, 37, 252–258. [Google Scholar] [CrossRef]
- Graham, C.; Jozwik, A.; Pepper, A.; Benjamin, R. Allogeneic CAR-T cells: More than ease of access? Cells 2018, 7, 155. [Google Scholar] [CrossRef]
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013, 31, 928–933. [Google Scholar] [CrossRef] [Green Version]
- Morvan, M.G.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2016, 16, 7–19. [Google Scholar] [CrossRef]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell 2018, 23, 181–192. [Google Scholar] [CrossRef]
- Morrissey, M.A.; Williamson, A.P.; Steinbach, A.M.; Roberts, E.W.; Kern, N.; Headley, M.B.; Vale, R.D. Chimeric antigen receptors that trigger phagocytosis. Elife 2018, 7, e36688. [Google Scholar] [CrossRef]
- Jung, I.Y.; Lee, J. Unleashing the therapeutic potential of CAR-T cell therapy using gene-editing technologies. Mol. Cells 2018, 41, 717–723. [Google Scholar]
- Hartmann, J.; Schussler-Lenz, M.; Bondanza, A.; Buchholz, C.J. Clinical development of CAR T cells-challenges and opportunities in translating innovative treatment concepts. EMBO Mol. Med. 2017, 9, 1183–1197. [Google Scholar] [CrossRef]
- Sachdeva, M.; Duchateau, P.; Depil, S.; Poirot, L.; Valton, J. Granulocyte-macrophage colony-stimulating factor inactivation in CAR T-cells prevents monocyte-dependent release of key cytokine release syndrome mediators. J. Biol. Chem. 2019, 294, 5430–5437. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| NCT Number | T-Cell | Indication | Country | Group | Features |
|---|---|---|---|---|---|
| NCT03399448 | NYESO-1 TCR-T | MM/Melanoma/Synovial Sarcoma/Myeloid/Round cell iposarcoma | US | U. Penn. | TCR/PD-1 KO |
| NCT03166878 | CD19 CAR-T | B-cell Leukemia/Lymphoma | China | Chinese PLA General Hospital | TCR/B2M KO |
| NCT03398967 | CD19 AND CD20/22 CAR-T | B-cell Leukemia/Lymphoma | China | Chinese PLA General Hospital | TCR KO |
| NCT03081715 | Primary T cell | Esophageal Cancer | China | Hangzhou Cancer Hospital Anhui Kedgene Biotechnology | PD-1 KO |
| NCT02863913 | Invasive Bladder Cancer Stage IV | China | Peking Univ./Cell Biotech | ||
| NCT02867345 | Hormone Refractory Prostate Cancer | China | Peking Univ./Cell Biotech | ||
| NCT02867332 | Metastatic Renal Cell Carcinoma | China | Peking Univ./Cell Biotech | ||
| NCT02793856 | Metastatic Non-small Cell Lung Cancer | China | Sichuan Univ./Chengdu MedGenCell | ||
| NCT03044743 | EBV-CTL | Gastric Carcinoma/Nasopharyngeal Carcinoma/T-cell Lymphoma/Adult Hodgkin Lymphoma/Diffuse Large B-cell Lymphoma | China | Nanjing Drum Tower Hospital | |
| NCT02746952 | CD19 CAR-T | Relapsed/refractory B-All | US/Europe | Servier | TCR/CD52 KO |
| NCT03190278 | CD123 CAR-T | Acute Myeloid Leukemia | US | Cellectis | TCR KO |
| NCT03050190 | CD19 CAR-T | Relapsed and Refractory B cell Malignancies | China | Shenzhen Geno-Immune Medical Institute | Inducible apoptotic caspase 9 |
| NCT02247609 | High-risk and Refractory B cell Lymphomas | Peking Univ. | |||
| NCT02968472 | Relapsed and Refractory B cell Leukemia | The First People’s Hospital of Yunnan | |||
| NCT03098355 | CD19 and CD22 CAR-T | Refractory and/or Recurrent B cell Malignancies | Zhujiang Hospital | ||
| NCT02274584 | CD30 CAR-T | CD 30 Positive Lymphomas | Peking Univ. | ||
| NCT03185468 | PSMA, Fra CAR-T | Bladder Cancer | Shenzhen Gene-Immune Medical Institute |
| NCT Number | NK-Cell | Indication | Country | Group | Features |
|---|---|---|---|---|---|
| NCT03692767 | CD22 CAR-NK | Relapsed and Refractory B cell Leukemia | China | Allife Medical Science and Technology Co., Ltd | - |
| NCT03690310 | CD19 CAR-NK | Relapsed and Refractory B cell Leukemia | China | - | |
| NCT03398967 | Mesothelin CAR-NK | Epithelial Ovarian Cancer | China | - | |
| NCT03692663 | PSMA CAR-NK | Castration-Resistant Prostate Cancer | China | - | |
| NCT03824964 | CD19/CD22 CAR-NK | Relapsed and Refractory B cell Leukemia | China | - | |
| NCT03415100 | NKG2D-CAR-NK | Metastatic Solid Tumours | China | The Third Affiliated Hospital of Guangzhou Medical Univ. | - |
| NCT02944162 | CD33 CAR-NK | Relapsed/Refractory CD33+AML | China | PersonGen BioTherapeutics Co., Ltd | - |
| NCT02892695 | CD19 CAR-NK | CD19 Positive Leukemia and Lymphoma | China | - | |
| NCT03579927 | CD19 CAR-NK | B-cell Lymphoma | US | M.D. Anderson Cancer Center | Inducible apoptotic caspase 9 |
| NCT03056339 | CD19 CAR-NK | B Lymphoid Malignancies | US | Inducible apoptotic caspase 9 + IL15 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.M. When CAR Meets Stem Cells. Int. J. Mol. Sci. 2019, 20, 1825. https://doi.org/10.3390/ijms20081825
Lee JM. When CAR Meets Stem Cells. International Journal of Molecular Sciences. 2019; 20(8):1825. https://doi.org/10.3390/ijms20081825
Chicago/Turabian StyleLee, Jung Min. 2019. "When CAR Meets Stem Cells" International Journal of Molecular Sciences 20, no. 8: 1825. https://doi.org/10.3390/ijms20081825
APA StyleLee, J. M. (2019). When CAR Meets Stem Cells. International Journal of Molecular Sciences, 20(8), 1825. https://doi.org/10.3390/ijms20081825