Design and Synthesis of a New Soluble Natural β-Carboline Derivative for Preclinical Study by Intravenous Injection


Abstract
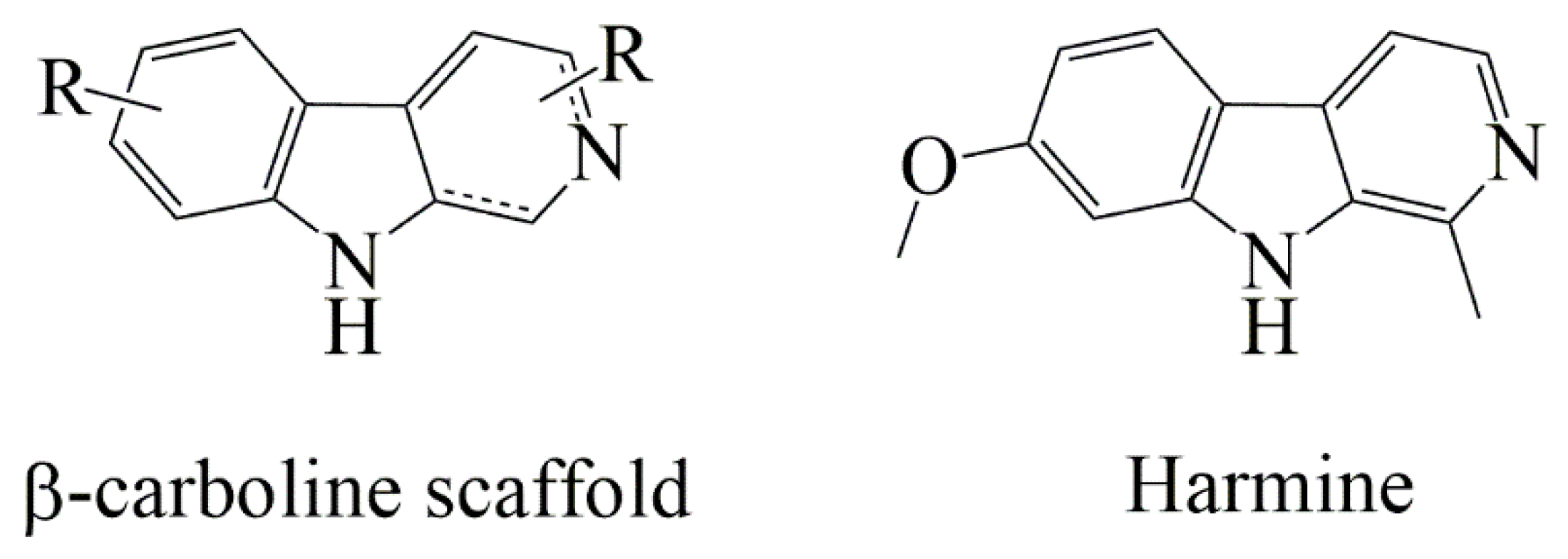
1. Introduction
2. Results
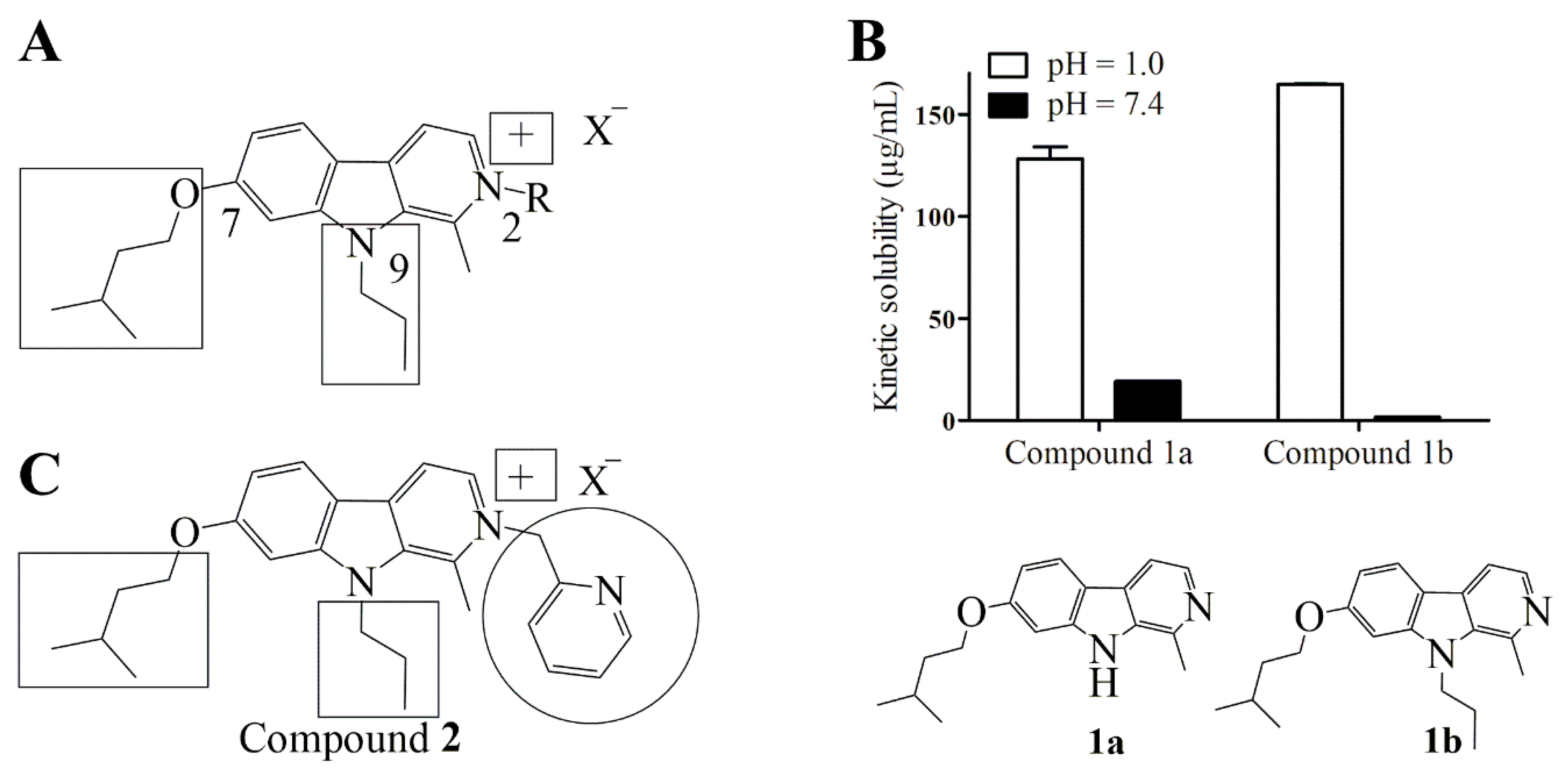
2.1. Design
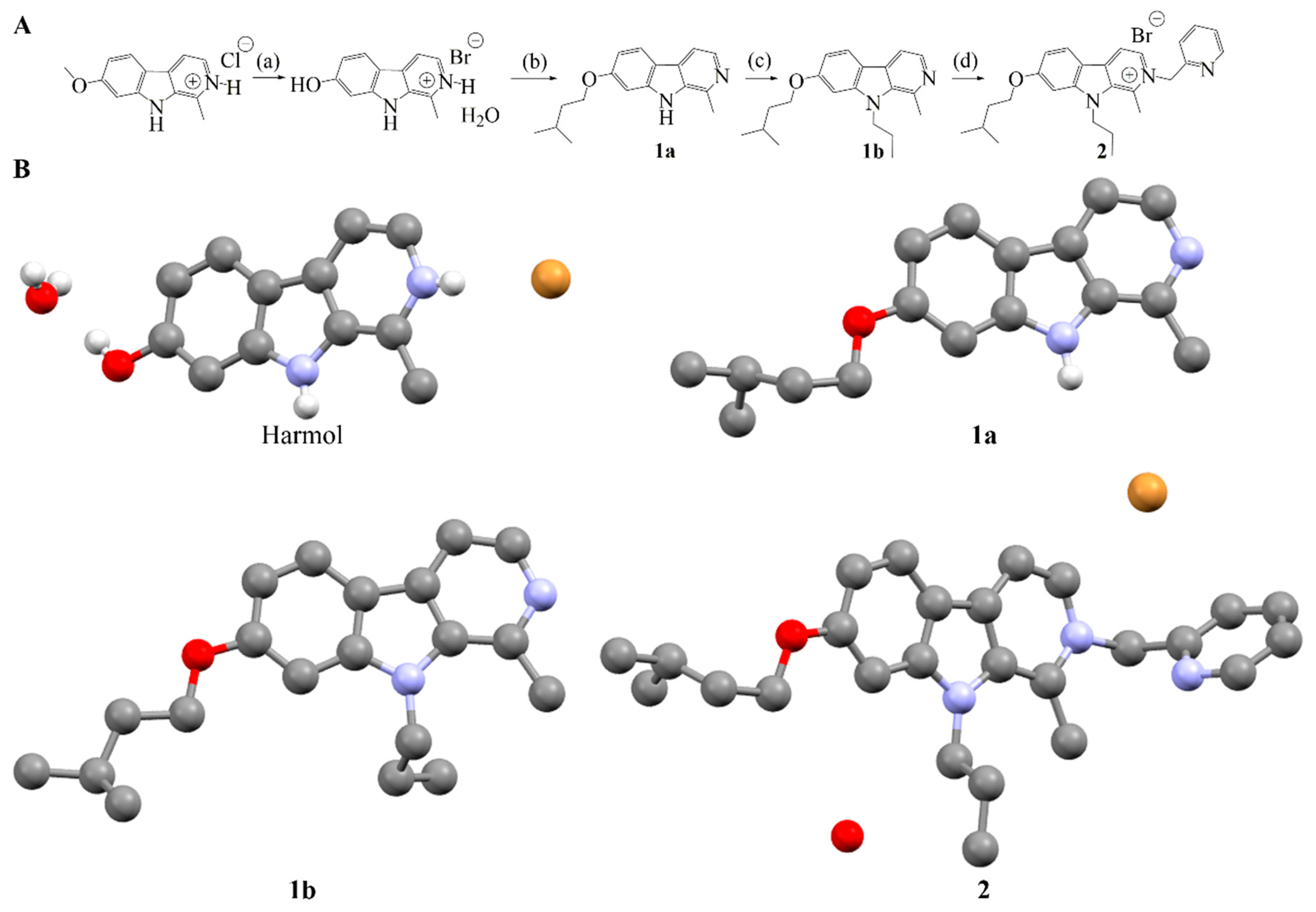
2.2. Synthesis of Compound 2
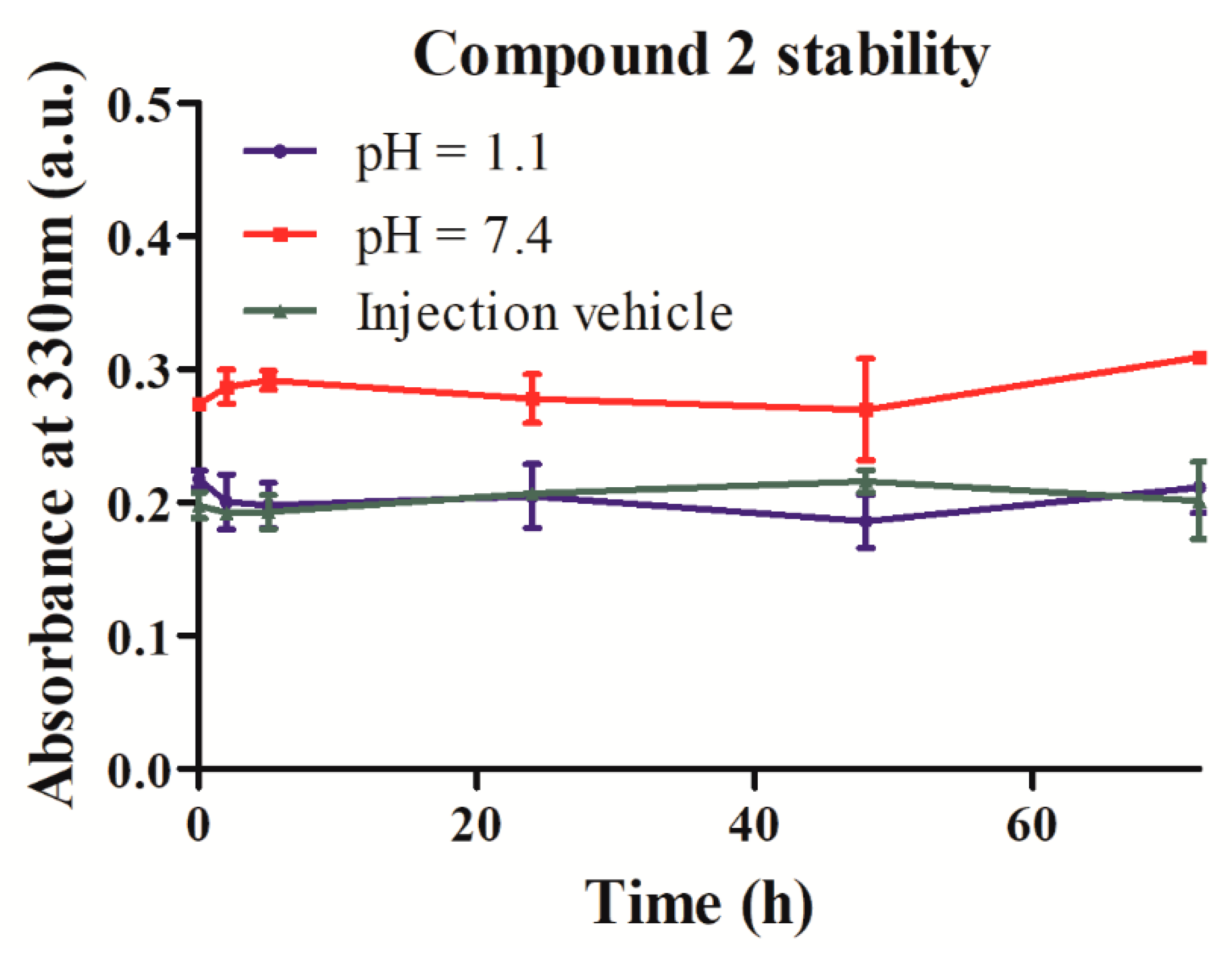
2.3. Stability and Solubility of Compound 2 in Three Different Media
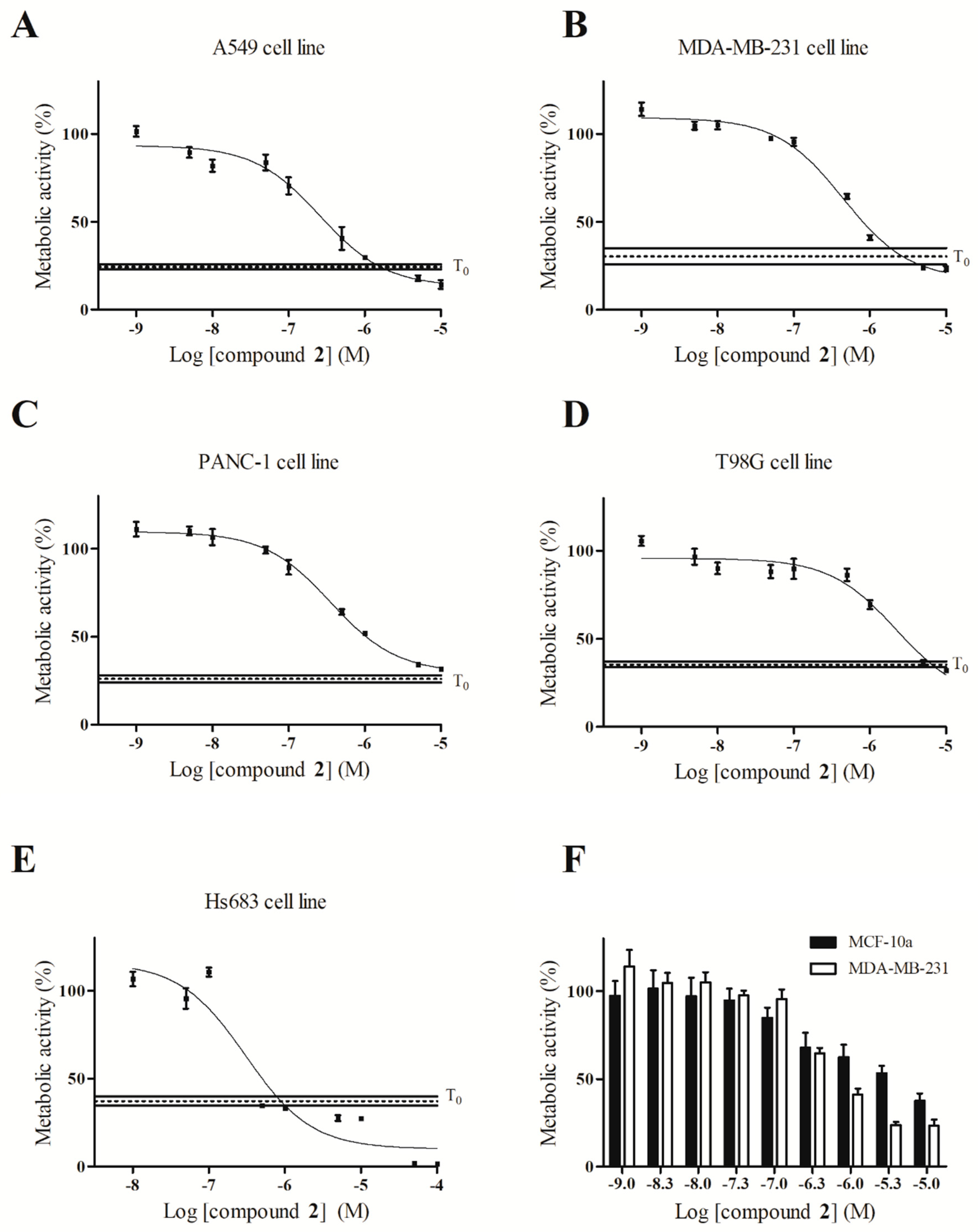
2.4. Antiproliferative Activity Determination of Compound 2 on Five Different Cancer Cell Lines
3. Discussion and Conclusions
4. Materials and Methods
4.1. Cell Culture
4.2. Synthesis
4.2.1. Solvents and Reagents
4.2.2. Nuclear Magnetic Resonance
4.2.3. Liquid Chromatography Coupled with Mass Spectrometry
4.2.4. Elemental Analysis
4.2.5. Thin-Layer Chromatography and Flash Column Chromatography
4.2.6. Mechanochemistry
4.2.7. Melting Point Measurement
4.2.8. Single-Crystal X-ray Diffraction (SCXRD)
4.2.9. Synthesis of 1-methyl-9H-pyrido[3,4-b]indol-7-ol (harmol)
4.2.10. Synthesis of 1-methyl-7-(3-methylbutoxy)-9H-pyrido[3,4-b]indole (1a)
4.2.11. Synthesis of 1-methyl-7-(3-methylbutoxy)-9-propyl-9H-pyrido[3,4-b]indole (1b)
4.2.12. Synthesis of 1-methyl-7-(3-methylbutoxy)-9-propyl-2-[(pyridin-2-yl)methyl]-9H-pyrido[3,4-b]indol-2-ium bromide (2)
4.3. Determination of Kinetic Solubility of Compounds 1a and 1b at Physiological and Acidic pH
4.4. Stability and Solubility Study of Compound 2
4.4.1. Evaluation of Compound 2 Stability at Physiological pH and in Simulated Injection Vehicle
4.4.2. Evaluation of the Thermodynamic Solubility of Compound 2
4.5. Determination of Antiproliferative Activity by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| LAG | Liquid-assisted grinding |
| HAc | Acetic acid |
| QSAR | Quantitative structure-activity relationship |
| SCXRD | Single-crystal X-ray diffraction |
| DMF | N,N-dimethylformamide |
| DCM | Dichloromethane |
| EtOH | Ethanol |
| MeOH | Methanol |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
References
- Song, H.; Liu, Y.; Liu, Y.; Wang, L.; Wang, Q. Synthesis and Antiviral and Fungicidal Activity Evaluation of -Carboline, Dihydro--carboline, Tetrahydro--carboline Alkaloids and Their Derivatives. J. Agric. Food Chem. 2014, 62, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Reniers, J.; Robert, S.; Frederick, R.; Masereel, B.; Vincent, S.; Wouters, J. Synthesis and evaluation of β-carboline derivatives as potential monoamine oxidase inhibitors. Bioorg. Med. Chem. 2011, 19, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Peng, W.; Wang, Z.; Xu, A. -Carboline alkaloids: biochemical and pharmacological functions. Curr. Med. Chem. 2007, 14, 479–500. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Gadewar, M.; Tripathi, R.; Prasad, S.; Patel, D.K. A review on medicinal importance, pharmacological activity and bioanalytical aspects of beta-carboline alkaloid “Harmine”. Asian Pac. J. Trop. Biomed. 2012, 2, 660–664. [Google Scholar] [CrossRef]
- Vignoni, M.; Erra-Balsells, R.; Epe, B.; Cabrerizo, F.M. Intra- and extra-cellular DNA damage by harmine and 9-methyl-harmine. J. Photochem. Photobiol. B 2014, 132C, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Nafisi, S.; Bonsaii, M.; Maali, P.; Khalilzadeh, M.A.; Manouchehri, F. Beta-carboline alkaloids bind DNA. J. Photochem. Photobiol. B 2010, 100, 84–91. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, K.; Ding, J.; Xu, H.; Zhu, L.; Zhang, K.; Li, X.; Sun, W. Harmine induces apoptosis and inhibits tumor cell proliferation, migration and invasion through down-regulation of cyclooxygenase-2 expression in gastric cancer. Phytomedicine 2014, 21, 348–355. [Google Scholar] [CrossRef]
- Chen, Q.; Chao, R.; Chen, H.; Hou, X.; Yan, H.; Zhou, S.; Peng, W.; Xu, A. Antitumor and neurotoxic effects of novel harmine derivatives and structure-activity relationship analysis. Int. J. Cancer 2005, 114, 675–682. [Google Scholar] [CrossRef]
- Cao, R.; Chen, H.; Peng, W.; Ma, Y.; Hou, X.; Guan, H.; Liu, X.; Xu, A. Design, synthesis and in vitro and in vivo antitumor activities of novel beta-carboline derivatives. Eur. J. Med. Chem. 2005, 40, 991–1001. [Google Scholar] [CrossRef]
- Cao, M.-R.; Li, Q.; Liu, Z.-L.; Liu, H.-H.; Wang, W.; Liao, X.-L.; Pan, Y.-L.; Jiang, J.-W. Harmine induces apoptosis in HepG2 cells via mitochondrial signaling pathway. Hepatobiliary Pancreat. Dis. Int. 2011, 10, 599–604. [Google Scholar] [CrossRef]
- Liu, H.; Han, D.; Liu, Y.; Hou, X.; Wu, J.; Li, H.; Yang, J.; Shen, C.; Yang, G.; Fu, C.; et al. Harmine hydrochloride inhibits Akt phosphorylation and depletes the pool of cancer stem-like cells of glioblastoma. J. Neurooncol. 2013, 112, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.F.; Sun, R.Q.; Jia, Y.F.; Chen, Q.; Tu, R.F.; Li, K.K.; Zhang, X.D.; Du, R.L.; Cao, R.H. Synthesis and mechanisms of action of novel harmine derivatives as potential antitumor agents. Sci. Rep. 2016, 6, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Frédérick, R.; Bruyère, C.; Vancraeynest, C.; Reniers, J.; Meinguet, C.; Pochet, L.; Backlund, A.; Masereel, B.; Kiss, R.; Wouters, J. Novel trisubstituted harmine derivatives with original in vitro anticancer activity. J. Med. Chem. 2012, 55, 6489–6501. [Google Scholar]
- Guo, L.; Chen, W.; Cao, R.; Fan, W.; Ma, Q.; Zhang, J.; Dai, B. Synthesis and structure-activity relationships of asymmetric dimeric β-carboline derivatives as potential antitumor agents. Eur. J. Med. Chem. 2018, 147, 253–265. [Google Scholar] [CrossRef]
- Shi, B.; Cao, R.; Fan, W.; Guo, L.; Ma, Q.; Chen, X.; Zhang, G.; Qiu, L.; Song, H. Design, synthesis and in vitro and in vivo antitumor activities of novel bivalent β-carbolines. Eur. J. Med. Chem. 2013, 60, 10–22. [Google Scholar] [CrossRef]
- Cao, R.; Fan, W.; Guo, L.; Ma, Q.; Zhang, G.; Li, J.; Chen, X.; Ren, Z.; Qiu, L. Synthesis and structure-activity relationships of harmine derivatives as potential antitumor agents. Eur. J. Med. Chem. 2013, 60, 135–143. [Google Scholar] [CrossRef]
- Zhang, G.; Cao, R.; Guo, L.; Ma, Q.; Fan, W.; Chen, X.; Li, J.; Shao, G.; Qiu, L.; Ren, Z. Synthesis and structure-activity relationships of N2-alkylated quaternary β-carbolines as novel antitumor agents. Eur. J. Med. Chem. 2013, 65, 21–31. [Google Scholar] [CrossRef]
- Cao, R.; Chen, Q.; Hou, X.; Chen, H.; Guan, H.; Ma, Y.; Peng, W.; Xu, A. Synthesis, acute toxicities, and antitumor effects of novel 9-substituted beta-carboline derivatives. Bioorg. Med. Chem. 2004, 12, 4613–4623. [Google Scholar] [CrossRef]
- Hamsa, T.P.; Kuttan, G. Harmine inhibits tumour specific neo-vessel formation by regulating VEGF, MMP, TIMP and pro-inflammatory mediators both in vivo and in vitro. Eur. J. Pharmacol. 2010, 649, 64–73. [Google Scholar] [CrossRef]
- Li, S.; Wang, A.; Gu, F.; Wang, Z.; Tian, C.; Qian, Z.; Tang, L.; Gu, Y. Novel harmine derivatives for tumor targeted therapy. Oncotarget 2015, 6, 8988–9001. [Google Scholar] [CrossRef][Green Version]
- van de Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef]
- Meinguet, C.; Masereel, B.; Wouters, J. Preparation and characterization of a new harmine-based antiproliferative compound in complex with cyclodextrin: Increasing solubility while maintaining biological activity. Eur. J. Pharm. Sci. 2015, 77, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Guan, X.; Shi, B.; Chen, Z.; Ren, Z.; Peng, W.; Song, H. Design, synthesis and 3D-QSAR of beta-carboline derivatives as potent antitumor agents. Eur. J. Med. Chem. 2010, 45, 2503–2515. [Google Scholar] [CrossRef]
- Meinguet, C.; Bruyère, C.; Frédérick, R.; Mathieu, V.; Vancraeynest, C.; Pochet, L.; Laloy, J.; Mortier, J.; Wolber, G.; Kiss, R.; et al. 3D-QSAR, design, synthesis and characterization of trisubstituted harmine derivatives with in vitro antiproliferative properties. Eur. J. Med. Chem. 2015, 94, 45–55. [Google Scholar] [CrossRef]
- Tran, T.D.; Pryde, D.C.; Jones, P.; Adam, F.M.; Benson, N.; Bish, G.; Calo, F.; Ciaramella, G.; Dixon, R.; Duckworth, J.; et al. Design and optimisation of orally active TLR7 agonists for the treatment of hepatitis C virus infection. Bioorganic Med. Chem. Lett. 2011, 21, 2389–2393. [Google Scholar] [CrossRef] [PubMed]
- Gfesser, G.A.; Bayburt, E.K.; Cowart, M.; DiDomenico, S.; Gomtsyan, A.; Lee, C.H.; Stewart, A.O.; Jarvis, M.F.; Kowaluk, E.A.; Bhagwat, S.S. Synthesis and structure-activity relationships of 5-heteroatom-substituted pyridopyrimidines as adenosine kinase inhibitors. Eur. J. Med. Chem. 2003, 38, 245–252. [Google Scholar] [CrossRef]
- Cerreia Vioglio, P.; Chierotti, M.R.; Gobetto, R. Pharmaceutical aspects of salt and cocrystal forms of APIs and characterization challenges. Adv. Drug Deliv. Rev. 2017, 117, 86–110. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, Z.; Chen, Y.; Chen, Z.; Chen, H.; Pui, Y.; Qian, F. Oral bioavailability enhancement of β-lapachone, a poorly soluble fast crystallizer, by cocrystal, amorphous solid dispersion, and crystalline solid dispersion. Eur. J. Pharm. Biopharm. 2018, 124, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: importance and enhancement techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef]
- Wouters, J.; Quere, L. Pharmaceutical Salts and Co-crystals; RSC Drug Discovery, Royal Society of Chemistry: Cambridge, UK, 2011; ISBN 978-1-84973-158-4. [Google Scholar]
- Centre Belge d’Information Pharmacotherapeutique (CBIP). Repertoire Commente Des Medicaments, 31st ed.; Christiaens, T., Ed.; CBIP: Gent, Belgium, 2018. [Google Scholar]
- Weaver, M.L.; Grossi, A.B.; Schützsack, J.; Parish, J.; Løgsted, J.; Bøgh, I.B.; Cameron, D.; Harvey, W.; Festag, M.; Downes, N.; et al. Vehicle Systems and Excipients Used in Minipig Drug Development Studies. Toxicol. Pathol. 2016, 44, 367–372. [Google Scholar] [CrossRef]
- Shimizu, S. Routes of Administration. In The Laboratory Mouse (Handbook of Experimental Animals); Hedrich, H., Ed.; Elsevier Academic Press: London, UK, 2004; p. 598. ISBN 0123364256. [Google Scholar]
- Li, C.; Wang, Y.; Wang, C.; Yi, X.; Li, M.; He, X. Phytomedicine Anticancer activities of harmine by inducing a pro-death autophagy and apoptosis in human gastric cancer cells. Phytomedicine 2017, 28, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, Q.; Liu, Z.; Lin, L.; Zhang, X.; Cao, M.; Jiang, J. Harmine induces cell cycle arrest and mitochondrial pathway-mediated cellular apoptosis in SW620 cells via inhibition of the Akt and ERK signaling pathways. Oncol Rep. 2016, 35, 3363–3370. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.; Chu, J.; Meinguet, C.; Kiss, R.; Vandenbussche, G.; Masereel, B.; Wouters, J.; Kornienko, A.; Pelletier, J.; Mathieu, V. A harmine-derived beta-carboline displays anti-cancer effects in vitro by targeting protein synthesis. Eur. J. Pharmacol. 2017, 805, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.; Chu, J.; Meinguet, C.; Kiss, R.; Vandenbussche, G.; Masereel, B.; Wouters, J.; Kornienko, A.; Pelletier, J.; Mathieu, V. Data in support of a harmine-derived beta-carboline in vitro effects in cancer cells through protein synthesis. Data Brief. 2017, 12, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: a Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef]
- Dolušić, E.; Larrieu, P.; Moineaux, L.; Stroobant, V.; Pilotte, L.; Colau, D.; Pochet, L.; van den Eynde, B.; Masereel, B.; Wouters, J.; et al. Tryptophan 2,3-Dioxygenase (TDO) Inhibitors. 3-(2-(Pyridyl)ethenyl) indoles as Potential Anticancer Immunomodulators. J. Med. Chem. 2011, 54, 5320–5334. [Google Scholar] [CrossRef]
- Wenlock, M.C.; Austin, R.P.; Potter, T.; Barton, P. A highly automated assay for determining the aqueous equilibrium solubility of drug discovery compounds. J. Lab. Autom. 2011, 16, 276–284. [Google Scholar] [CrossRef]
- Dumont, P.; Ingrassia, L.; Rouzeau, S.; Ribaucour, F.; Thomas, S.; Roland, I.; Darro, F.; Lefranc, F.; Kiss, R. The Amaryllidaceae isocarbostyril narciclasine induces apoptosis by activation of the death receptor and/or mitochondrial pathways in cancer cells but not in normal fibroblasts. Neoplasia 2007, 9, 766–776. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH 1.1 | pH 7.4 | Injection Vehicle | |
|---|---|---|---|
| Thermodynamic solubility at 37 °C (mg/mL) replicate 1 | 1.46 0.01 | 0.98 0.01 | 1.95 0.01 |
| Thermodynamic solubility at 37 °C (mg/mL) replicate 2 | 1.63 0.02 | 0.99 0.01 | 1.90 0.04 |
| Thermodynamic solubility at 37 °C (mg/mL) replicate 3 | 1.77 0.01 | 1.15 0.01 | 1.77 0.01 |
| Mean thermodynamic solubility at 37 °C (mg/mL) | 1.62 0.13 | 1.06 0.08 | 1.87 0.07 |
| Cell Lines | A549 | MDA-MB-231 | PANC-1 | T98G | Hs683 |
|---|---|---|---|---|---|
| R2 | 0.97 | 0.97 | 0.96 | 0.88 | 0.90 |
| IC50 (µM) | 0.261 | 0.449 | 0.350 | 2.190 | 0.305 |
| Confidence interval at 95% (µM) | 0.205 to 0.331 | 0.358 to 0.563 | 0.271 to 0.468 | 1.24 to 3.89 | 0.195 to 0.479 |
| Cytostatic at IC50 | Yes | Yes | No | No | Yes |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marx, S.; Bodart, L.; Tumanov, N.; Wouters, J. Design and Synthesis of a New Soluble Natural β-Carboline Derivative for Preclinical Study by Intravenous Injection. Int. J. Mol. Sci. 2019, 20, 1491. https://doi.org/10.3390/ijms20061491
Marx S, Bodart L, Tumanov N, Wouters J. Design and Synthesis of a New Soluble Natural β-Carboline Derivative for Preclinical Study by Intravenous Injection. International Journal of Molecular Sciences. 2019; 20(6):1491. https://doi.org/10.3390/ijms20061491
Chicago/Turabian StyleMarx, Sébastien, Laurie Bodart, Nikolay Tumanov, and Johan Wouters. 2019. "Design and Synthesis of a New Soluble Natural β-Carboline Derivative for Preclinical Study by Intravenous Injection" International Journal of Molecular Sciences 20, no. 6: 1491. https://doi.org/10.3390/ijms20061491
APA StyleMarx, S., Bodart, L., Tumanov, N., & Wouters, J. (2019). Design and Synthesis of a New Soluble Natural β-Carboline Derivative for Preclinical Study by Intravenous Injection. International Journal of Molecular Sciences, 20(6), 1491. https://doi.org/10.3390/ijms20061491