The Genetic Background of Mice Influences the Effects of Cigarette Smoke on Onset and Severity of Experimental Autoimmune Encephalomyelitis

 and
and
Abstract
1. Introduction
2. Results
2.1. Disease Incidence of Spontaneous EAE in the RR Mouse Model is too Low to Study Effects of Cigarette Smoke on Disease Development
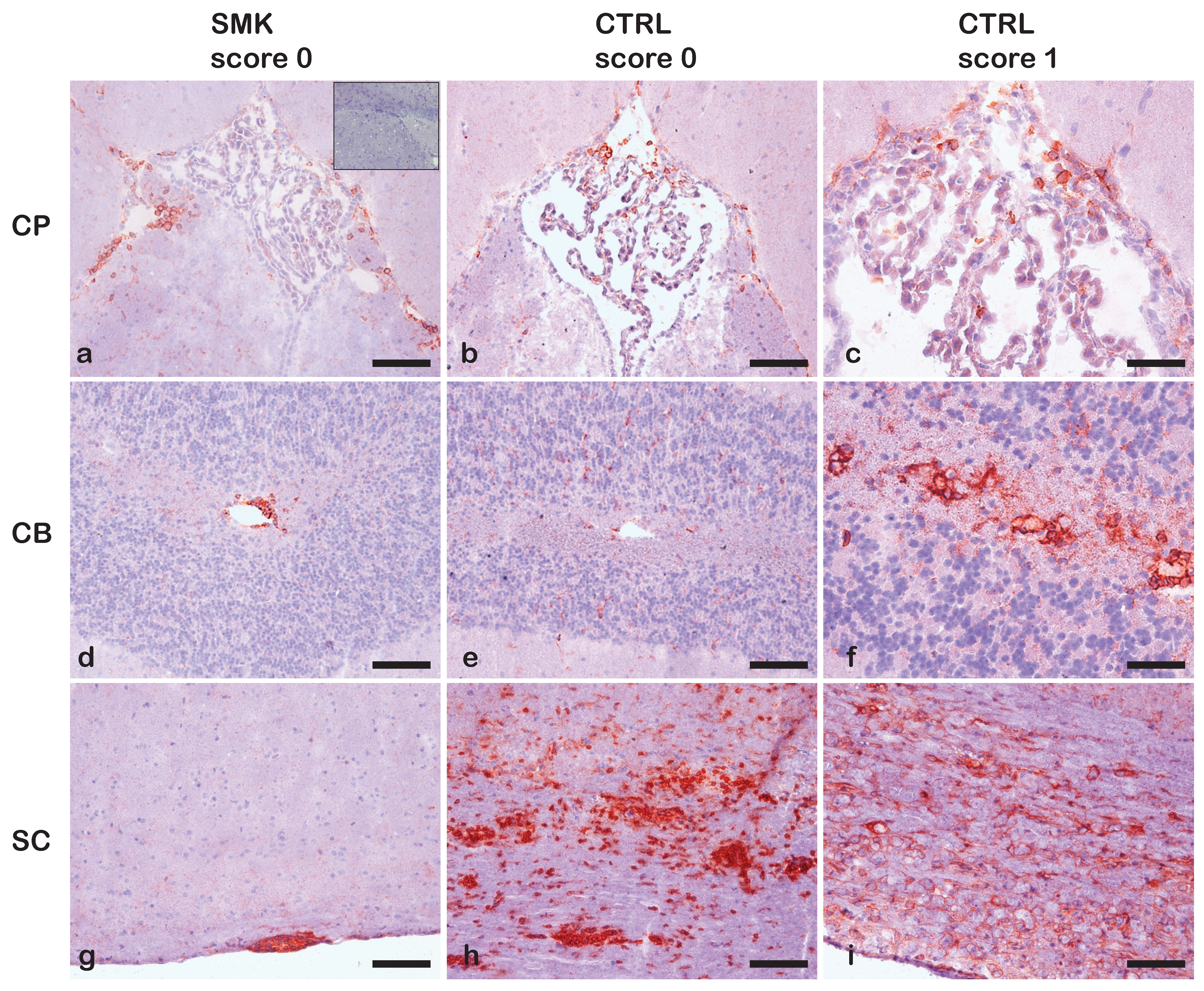
2.2. Early Onset of Spontaneous EAE in the OSE Model in the C57BL/6J Background Prevents Studying the Effect of Cigarette Smoke
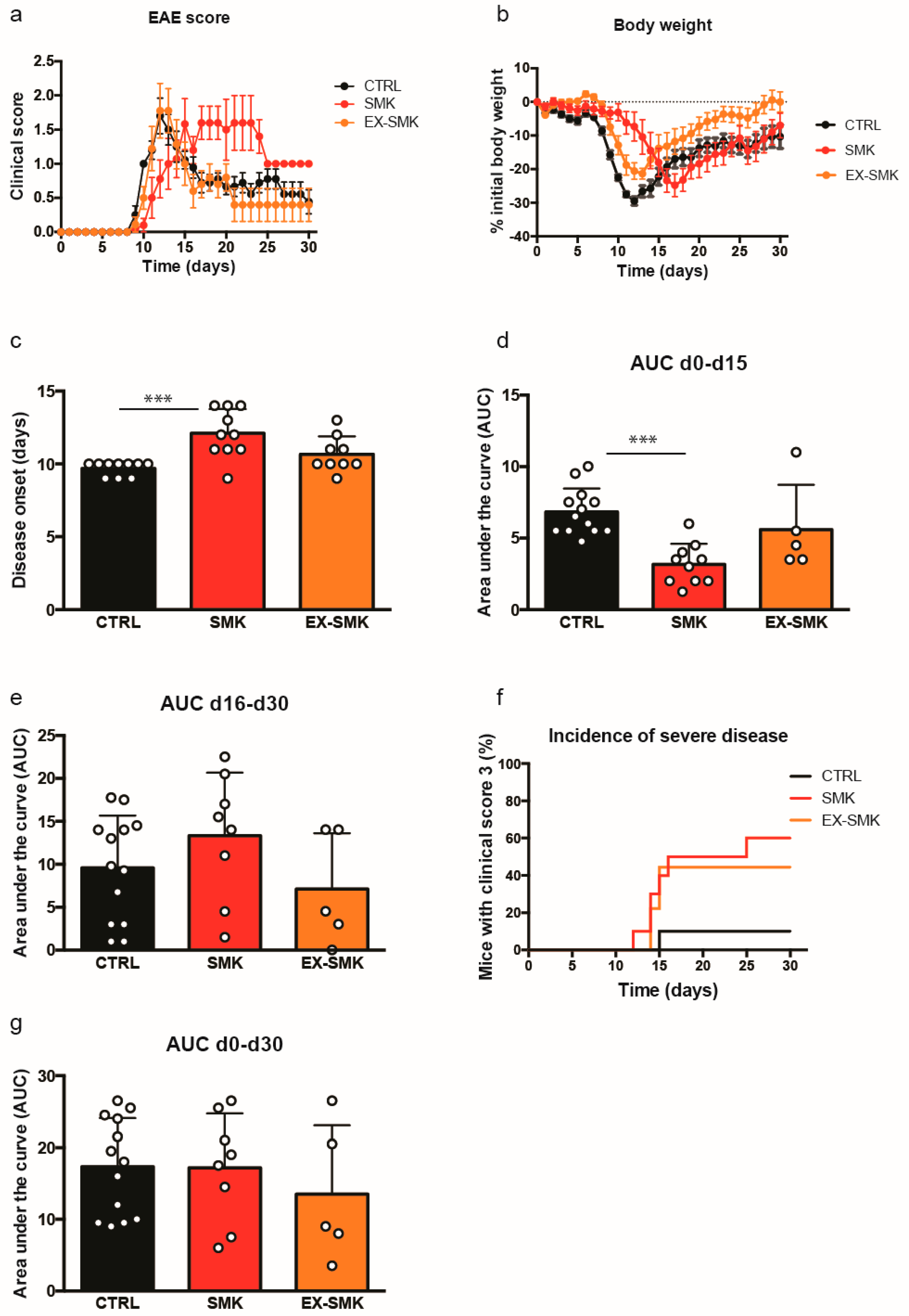
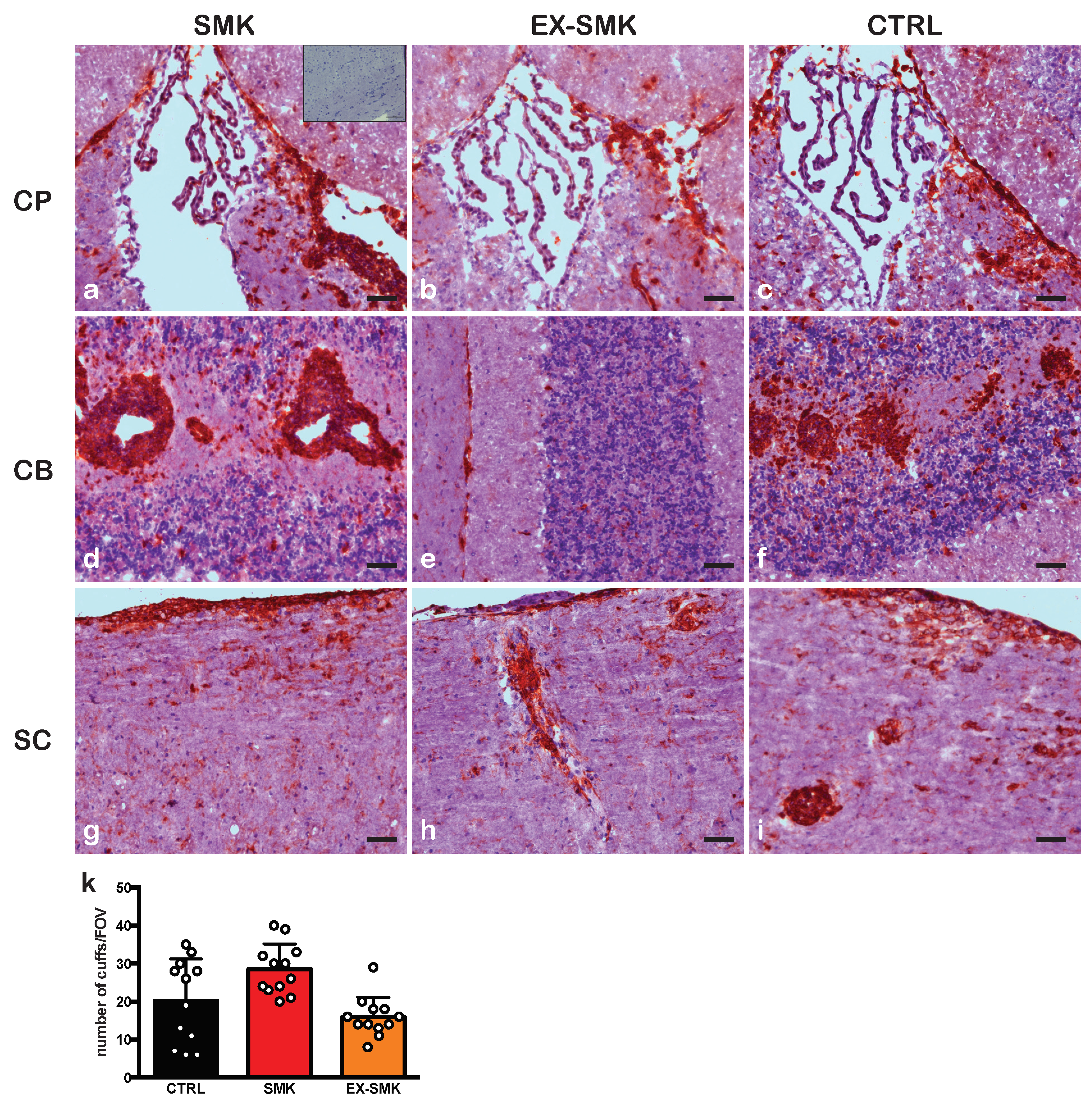
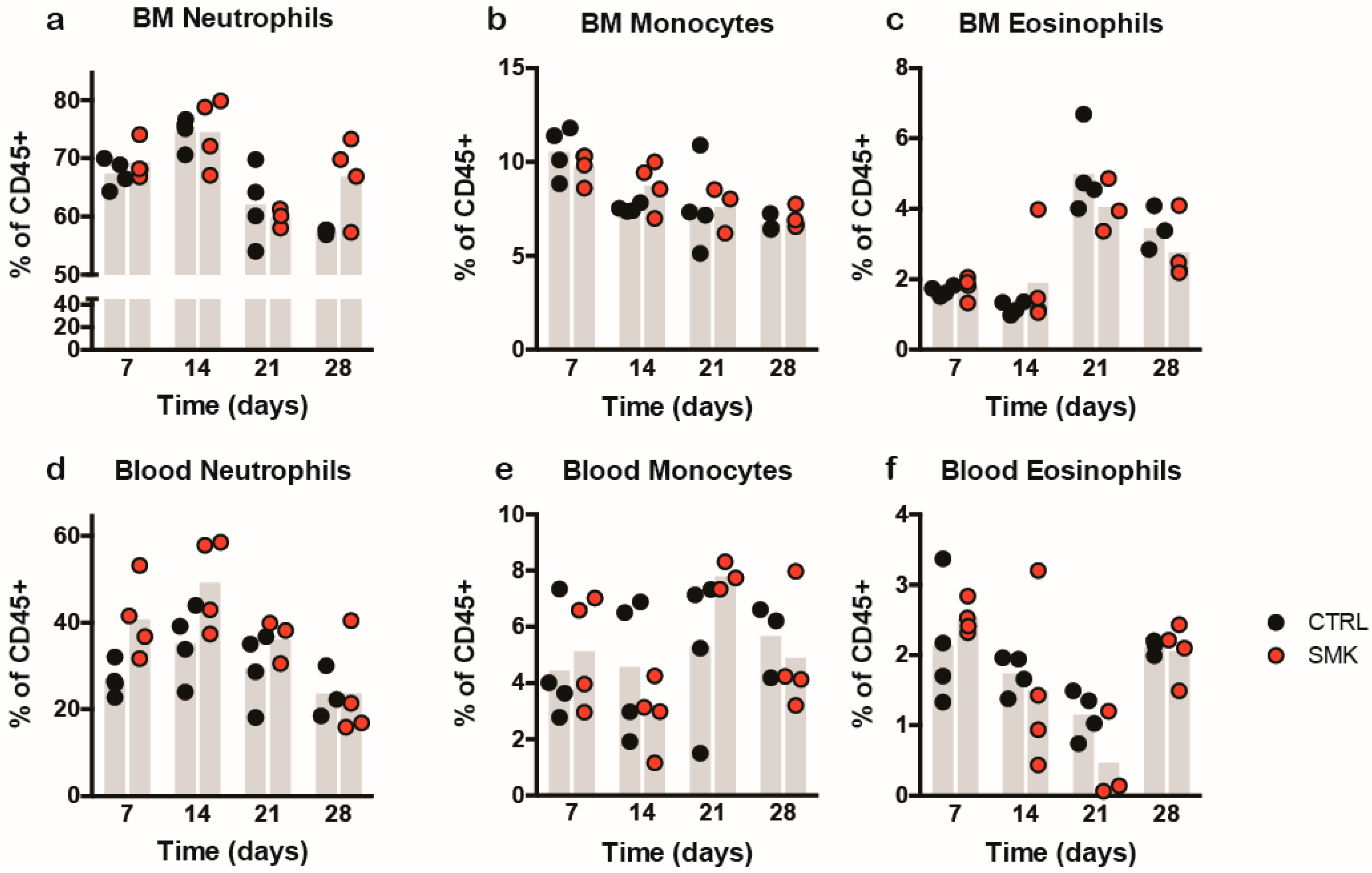
2.3. Cigarette Smoke Exposure Induces Protracted Disease with a Delayed Onset in PLPaa139–153 - Induced EAE in SJL Mice
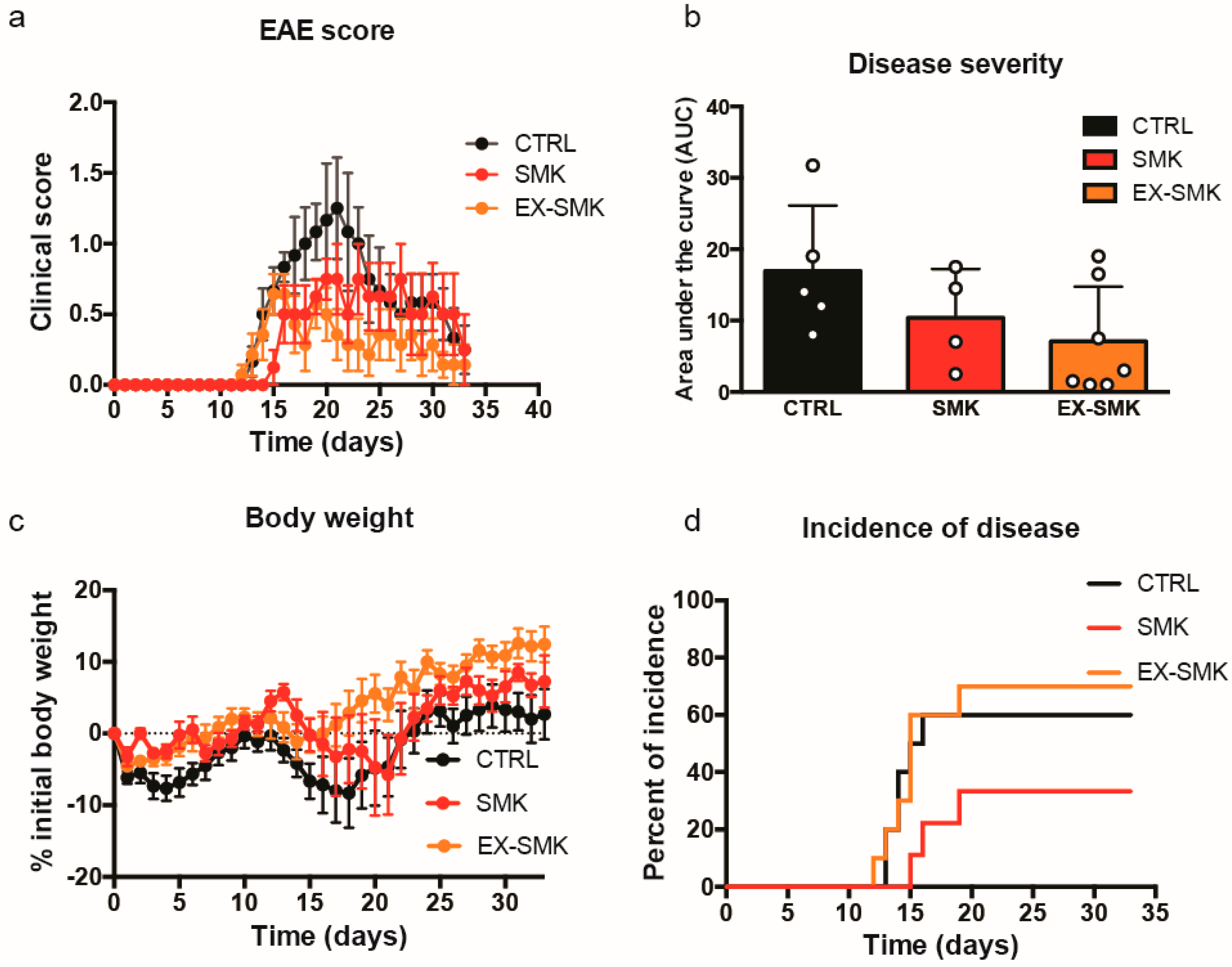
2.4. Cigarette Smoke Exposure Ameliorates MOGaa35–55 Induced EAE in C57BL/6J Mice
3. Discussion
3.1. Spontaneous EAE Models
3.2. Actively Induced EAE Models
4. Material and Methods
4.1. Experimental Autoimmune Encephalomyelitis
4.2. Statistics
4.3. Smoke Exposure
4.4. Tissue Processing and Immunohistology
4.5. Flow Cytometry Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Compliance with Ethical Standards
References
- Axisa, P.P.; Hafler, D.A. Multiple sclerosis: Genetics, biomarkers, treatments. Curr. Opin. Neurol. 2016, 29, 345–353. [Google Scholar] [CrossRef]
- Mitrovič, M.; Patsopoulos, N.A.; Beecham, A.H.; Dankowski, T.; Goris, A.; Dubois, B.; D’hooghe, M.B.; Lemmens, R.; Van Damme, P.; Søndergaard, H.B.; et al. Low-Frequency and Rare-Coding Variation Contributes to Multiple Sclerosis Risk. Cell 2018, 175, 1679–1687. [Google Scholar] [CrossRef]
- Belbasis, L.; Bellou, V.; Evangelou, E.; Ioannidis, J.P.; Tzoulaki, I. Environmental risk factors and multiple sclerosis: An umbrella review of systematic reviews and meta-analyses. Lancet Neurol 2015, 14, 263–273. [Google Scholar] [CrossRef]
- Hedstrom, A.K.; Olsson, T.; Alfredsson, L. The Role of Environment and Lifestyle in Determining the Risk of Multiple Sclerosis. Curr. Top. Behav. Neurosci. 2015, 26, 87–104. [Google Scholar] [CrossRef]
- Wingerchuk, D.M. Smoking: Effects on multiple sclerosis susceptibility and disease progression. Ther. Adv. Neurol. Disord. 2012, 5, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Pekmezovic, T.; Drulovic, J.; Milenkovic, M.; Jarebinski, M.; Stojsavljevic, N.; Mesaros, S.; Kisic, D.; Kostic, J. Lifestyle factors and multiple sclerosis: A case-control study in Belgrade. Neuroepidemiology 2006, 27, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Hedstrom, A.K.; Baarnhielm, M.; Olsson, T.; Alfredsson, L. Tobacco smoking, but not Swedish snuff use, increases the risk of multiple sclerosis. Neurology 2009, 73, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Van der Mei, I.A.; Simpson, S., Jr.; Stankovich, J.; Taylor, B.V. Individual and joint action of environmental factors and risk of MS. Neurol. Clin. 2011, 29, 233–255. [Google Scholar] [CrossRef] [PubMed]
- Hedstrom, A.K.; Bomfim, I.L.; Barcellos, L.F.; Briggs, F.; Schaefer, C.; Kockum, I.; Olsson, T.; Alfredsson, L. Interaction between passive smoking and two HLA genes with regard to multiple sclerosis risk. Int. J. Epidemiol. 2014, 43, 1791–1798. [Google Scholar] [CrossRef]
- Carlens, C.; Hergens, M.P.; Grunewald, J.; Ekbom, A.; Eklund, A.; Hoglund, C.O.; Askling, J. Smoking, use of moist snuff, and risk of chronic inflammatory diseases. Am. J. Respir. Crit. Care Med. 2010, 181, 1217–1222. [Google Scholar] [CrossRef]
- Usuda, K.; Konno, K.; Kono, K.; Tamashiro, H. WHO’s Framework Convention on Tobacco Control potential impact on tobacco control in Japan. Nihon Koshu Eisei Zasshi 2002, 49, 236–245. [Google Scholar]
- Maghzi, A.H.; Ghazavi, H.; Ahsan, M.; Etemadifar, M.; Mousavi, S.; Khorvash, F.; Minagar, A. Increasing female preponderance of multiple sclerosis in Isfahan, Iran: A population-based study. Mult. Scler. J. 2010, 16, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Alrouji, M.; Manouchehrinia, A.; Gran, B.; Constantinescu, C.S. Effects of cigarette smoke on immunity, neuroinflammation and multiple sclerosis. J. Neuroimmunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Moretto, N.; Bertolini, S.; Iadicicco, C.; Marchini, G.; Kaur, M.; Volpi, G.; Patacchini, R.; Singh, D.; Facchinetti, F. Cigarette smoke and its component acrolein augment IL-8/CXCL8 mRNA stability via p38 MAPK/MK2 signaling in human pulmonary cells. Am. J. Physiol. Lung. Cell Mol. Physiol. 2012, 303, L929–L938. [Google Scholar] [CrossRef] [PubMed]
- Hellermann, G.R.; Nagy, S.B.; Kong, X.; Lockey, R.F.; Mohapatra, S.S. Mechanism of cigarette smoke condensate-induced acute inflammatory response in human bronchial epithelial cells. Respir. Res. 2002, 3, 22. [Google Scholar] [CrossRef] [PubMed]
- Adler, K.B.; Fischer, B.M.; Wright, D.T.; Cohn, L.A.; Becker, S. Interactions between respiratory epithelial cells and cytokines: Relationships to lung inflammation. Ann. N. Y. Acad. Sci. 1994, 725, 128–145. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Megiovanni, A.M.; Sanchez, F.; Robledo-Sarmiento, M.; Morel, C.; Gluckman, J.C.; Boudaly, S. Polymorphonuclear neutrophils deliver activation signals and antigenic molecules to dendritic cells: A new link between leukocytes upstream of T lymphocytes. J. Leukoc. Biol. 2006, 79, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Van Gisbergen, K.P.; Sanchez-Hernandez, M.; Geijtenbeek, T.B.; van Kooyk, Y. Neutrophils mediate immune modulation of dendritic cells through glycosylation-dependent interactions between Mac-1 and DC-SIGN. J. Exp. Med. 2005, 201, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Rumble, J.M.; Huber, A.K.; Krishnamoorthy, G.; Srinivasan, A.; Giles, D.A.; Zhang, X.; Wang, L.; Segal, B.M. Neutrophil-related factors as biomarkers in EAE and MS. J. Exp. Med. 2015, 212, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Pollinger, B.; Krishnamoorthy, G.; Berer, K.; Lassmann, H.; Bosl, M.R.; Dunn, R.; Domingues, H.S.; Holz, A.; Kurschus, F.C.; Wekerle, H. Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells. J. Exp. Med. 2009, 206, 1303–1316. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, G.; Lassmann, H.; Wekerle, H.; Holz, A. Spontaneous opticospinal encephalomyelitis in a double-transgenic mouse model of autoimmune T cell/B cell cooperation. J. Clin. Investig. 2006, 116, 2385–2392. [Google Scholar] [CrossRef] [PubMed]
- Cremona, T.P.; Tschanz, S.A.; von Garnier, C.; Benarafa, C. SerpinB1 deficiency is not associated with increased susceptibility to pulmonary emphysema in mice. Am. J. Physiol. Lung. Cell Mol. Physiol. 2013, 305, L981–L989. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.J.; Andrews, N.; Ball, D.; Bellantuono, I.; Gray, J.; Hachoumi, L.; Holmes, A.; Latcham, J.; Petrie, A.; Potter, P.; et al. Does age matter? The impact of rodent age on study outcomes. Lab. Anim. 2017, 51, 160–169. [Google Scholar] [CrossRef]
- Shi, F.D.; Piao, W.H.; Kuo, Y.P.; Campagnolo, D.I.; Vollmer, T.L.; Lukas, R.J. Nicotinic attenuation of central nervous system inflammation and autoimmunity. J. Immunol. 2009, 182, 1730–1739. [Google Scholar] [CrossRef] [PubMed]
- Nizri, E.; Irony-Tur-Sinai, M.; Lory, O.; Orr-Urtreger, A.; Lavi, E.; Brenner, T. Activation of the cholinergic anti-inflammatory system by nicotine attenuates neuroinflammation via suppression of Th1 and Th17 responses. J. Immunol. 2009, 183, 6681–6688. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Simard, A.R.; Turner, G.H.; Wu, J.; Whiteaker, P.; Lukas, R.J.; Shi, F.D. Attenuation of CNS inflammatory responses by nicotine involves alpha7 and non-alpha7 nicotinic receptors. Exp. Neurol. 2011, 227, 110–119. [Google Scholar] [CrossRef]
- Gomes, J.P.; Watad, A.; Shoenfeld, Y. Nicotine and autoimmunity: The lotus’ flower in tobacco. Pharmacol. Res. 2018, 128, 101–109. [Google Scholar] [CrossRef]
- Gao, Z.; Nissen, J.C.; Ji, K.; Tsirka, S.E. The experimental autoimmune encephalomyelitis disease course is modulated by nicotine and other cigarette smoke components. PLoS ONE 2014, 9, e107979. [Google Scholar] [CrossRef]
- Gao, Z.; Nissen, J.C.; Legakis, L.; Tsirka, S.E. Nicotine modulates neurogenesis in the central canal during experimental autoimmune encephalomyelitis. Neuroscience 2015, 297, 11–21. [Google Scholar] [CrossRef]
- Simard, A.R.; Gan, Y.; St-Pierre, S.; Kousari, A.; Patelm, V.; Whiteaker, P.; Morley, B.J.; Lukas, R.J.; Shi, F.D. Differential modulation of EAE by alpha9*- and beta2*-nicotinic acetylcholine receptors. Immunol. Cell Biol. 2013, 91, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Hide, I.; Matsubara, A.; Hama, C.; Harada, K.; Miyano, K.; Andra, M.; Matsubayashi, H.; Sakai, N.; Kohsaka, S.; et al. Microglial alpha7 nicotinic acetylcholine receptors drive a phospholipase C/IP3 pathway and modulate the cell activation toward a neuroprotective role. J. Neurosci. Res. 2006, 83, 1461–1470. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; St-Pierre, S.; Roy, P.; Morley, B.J.; Hao, J.; Simard, A.R. Infiltration of CCR2+Ly6Chigh Proinflammatory Monocytes and Neutrophils into the Central Nervous System Is Modulated by Nicotinic Acetylcholine Receptors in a Model of Multiple Sclerosis. J. Immunol. 2016, 196, 2095–2108. [Google Scholar] [CrossRef] [PubMed]
- Litzenburger, T.; Fassler, R.; Bauer, J.; Lassmann, H.; Linington, C.; Wekerle, H.; Iglesias, A. B lymphocytes producing demyelinating autoantibodies: Development and function in gene-targeted transgenic mice. J. Exp. Med. 1998, 188, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Pagany, M.; Weiner, H.L.; Linington, C.; Sobel, R.A.; Kuchroo, V.K. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J. Exp. Med. 2003, 197, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Tietz, S.M.; Zwahlen, M.; Haghayegh Jahromi, N.; Baden, P.; Lazarevic, I.; Enzmann, G.; Engelhardt, B. Refined clinical scoring in comparative EAE studies does not enhance the chance to observe statistically significant differences. Eur. J. Immunol. 2016, 46, 2481–2483. [Google Scholar] [CrossRef]
- Minten, C.; Alt, C.; Gentner, M.; Frei, E.; Deutsch, U.; Lyck, R.; Schaeren-Wiemers, N.; Rot, A.; Engelhardt, B. DARC shuttles inflammatory chemokines across the blood-brain barrier during autoimmune central nervous system inflammation. Brain 2014, 137, 1454–1469. [Google Scholar] [CrossRef]
- Coisne, C.; Mao, W.; Engelhardt, B. Cutting edge: Natalizumab blocks adhesion but not initial contact of human T cells to the blood-brain barrier in vivo in an animal model of multiple sclerosis. J. Immunol. 2009, 182, 5909–5913. [Google Scholar] [CrossRef]
- Doring, A.; Wild, M.; Vestweber, D.; Deutsch, U.; Engelhardt, B. E- and P-selectin are not required for the development of experimental autoimmune encephalomyelitis in C57BL/6 and SJL mice. J. Immunol. 2007, 179, 8470–8479. [Google Scholar] [CrossRef]
- Reiss, Y.; Hoch, G.; Deutsch, U.; Engelhardt, B. T cell interaction with ICAM-1-deficient endothelium in vitro: Essential role for ICAM-1 and ICAM-2 in transendothelial migration of T cells. Eur. J. Immunol. 1998, 28, 3086–3099. [Google Scholar] [CrossRef]
- Engelhardt, B.; Laschinger, M.; Schulz, M.; Samulowitz, U.; Vestweber, D.; Hoch, G. The development of experimental autoimmune encephalomyelitis in the mouse requires alpha4-integrin but not alpha4beta7-integrin. J. Clin. Invest. 1998, 102, 2096–2105. [Google Scholar] [CrossRef]
- Daley, J.M.; Thomay, A.A.; Connolly, M.D.; Reichner, J.S.; Albina, J.E. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J. Leukoc. Biol. 2008, 83, 64–70. [Google Scholar] [CrossRef]
- Benarafa, C.; LeCuyer, T.E.; Baumann, M.; Stolley, J.M.; Cremona, T.P.; Remold-O’Donnell, E. SerpinB1 protects the mature neutrophil reserve in the bone marrow. J. Leukoc. Biol. 2011, 90, 21–29. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Score | Clinical Criteria |
|---|---|
| 0 | No symptom |
| 0.5 | Flaccid tail |
| 1 | Impaired righting reflex and/or irregular gait |
| 2 | Paraplegia |
| 3 | Paraplegia and incontinence |
| Score | Clinical Criteria |
|---|---|
| 0 | No symptom |
| 1 | Partly tilted body, feet fall into cage when walk on grid |
| 2 | Tilted body and tumbling |
| 3 | Heavily tilted body/head and moving in circles |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Enzmann, G.; Adelfio, R.; Godel, A.; Haghayegh Jahromi, N.; Tietz, S.; Burgener, S.S.; Deutsch, U.; Wekerle, H.; Benarafa, C.; Engelhardt, B. The Genetic Background of Mice Influences the Effects of Cigarette Smoke on Onset and Severity of Experimental Autoimmune Encephalomyelitis. Int. J. Mol. Sci. 2019, 20, 1433. https://doi.org/10.3390/ijms20061433
Enzmann G, Adelfio R, Godel A, Haghayegh Jahromi N, Tietz S, Burgener SS, Deutsch U, Wekerle H, Benarafa C, Engelhardt B. The Genetic Background of Mice Influences the Effects of Cigarette Smoke on Onset and Severity of Experimental Autoimmune Encephalomyelitis. International Journal of Molecular Sciences. 2019; 20(6):1433. https://doi.org/10.3390/ijms20061433
Chicago/Turabian StyleEnzmann, Gaby, Roberto Adelfio, Aurélie Godel, Neda Haghayegh Jahromi, Silvia Tietz, Sabrina S. Burgener, Urban Deutsch, Hartmut Wekerle, Charaf Benarafa, and Britta Engelhardt. 2019. "The Genetic Background of Mice Influences the Effects of Cigarette Smoke on Onset and Severity of Experimental Autoimmune Encephalomyelitis" International Journal of Molecular Sciences 20, no. 6: 1433. https://doi.org/10.3390/ijms20061433
APA StyleEnzmann, G., Adelfio, R., Godel, A., Haghayegh Jahromi, N., Tietz, S., Burgener, S. S., Deutsch, U., Wekerle, H., Benarafa, C., & Engelhardt, B. (2019). The Genetic Background of Mice Influences the Effects of Cigarette Smoke on Onset and Severity of Experimental Autoimmune Encephalomyelitis. International Journal of Molecular Sciences, 20(6), 1433. https://doi.org/10.3390/ijms20061433