Current Status and Future Prospects of Clinically Exploiting Cancer-specific Metabolism—Why Is Tumor Metabolism Not More Extensively Translated into Clinical Targets and Biomarkers?

Abstract
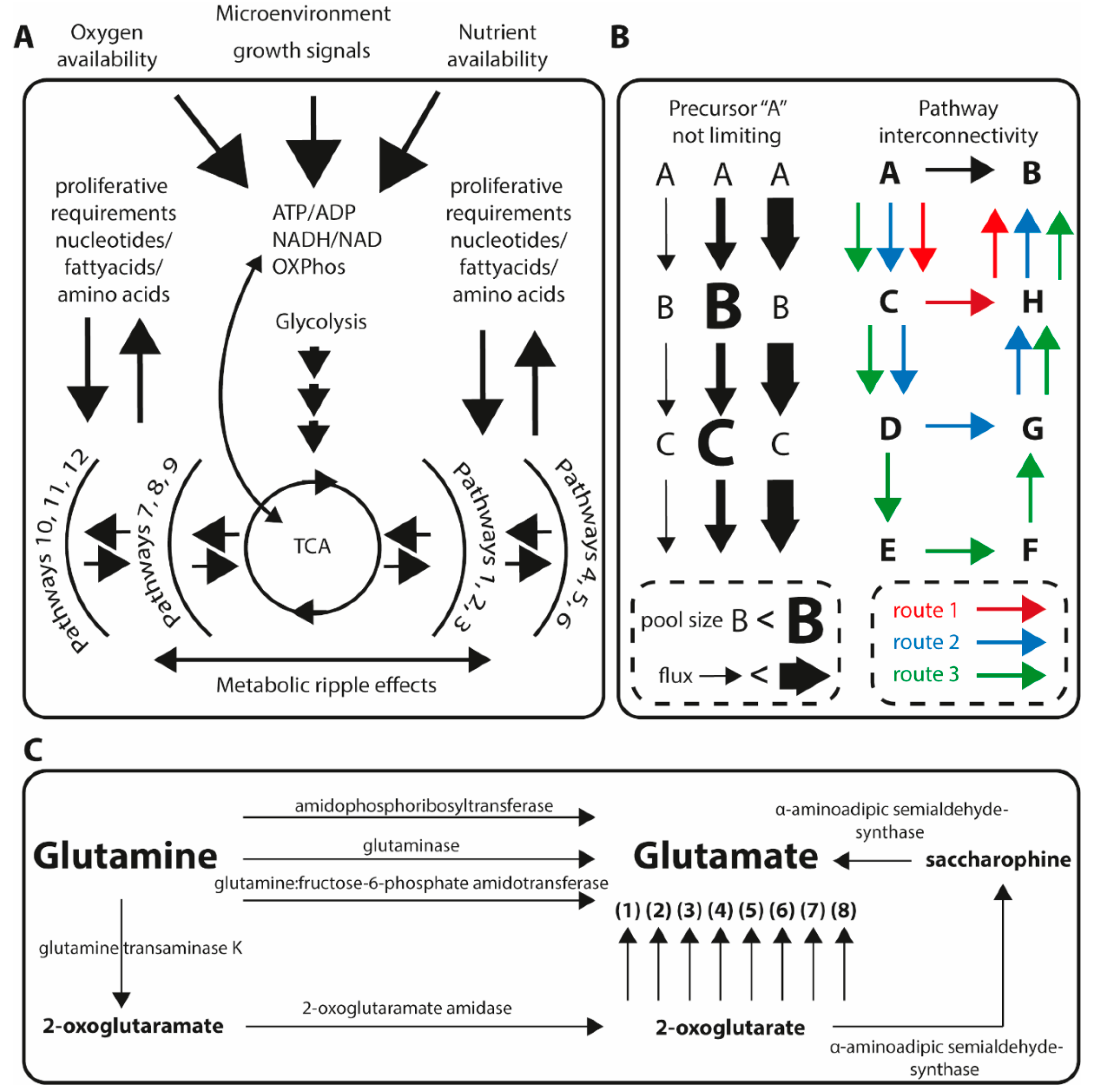
1. Metabolic Homeostasis and Implications for Studying Quantitative Metabolic Profiles in Tumor Biology
2. Cancer-Specific Metabolism as a Drug Target Opportunity at Present and What We Can Learn from Historical Tumor Metabolic Drug Target Discovery?
3. Does Cancer Cell Metabolism Leave a Disease Specific Metabolic Imprint That Can Be Used as a Biomarker?
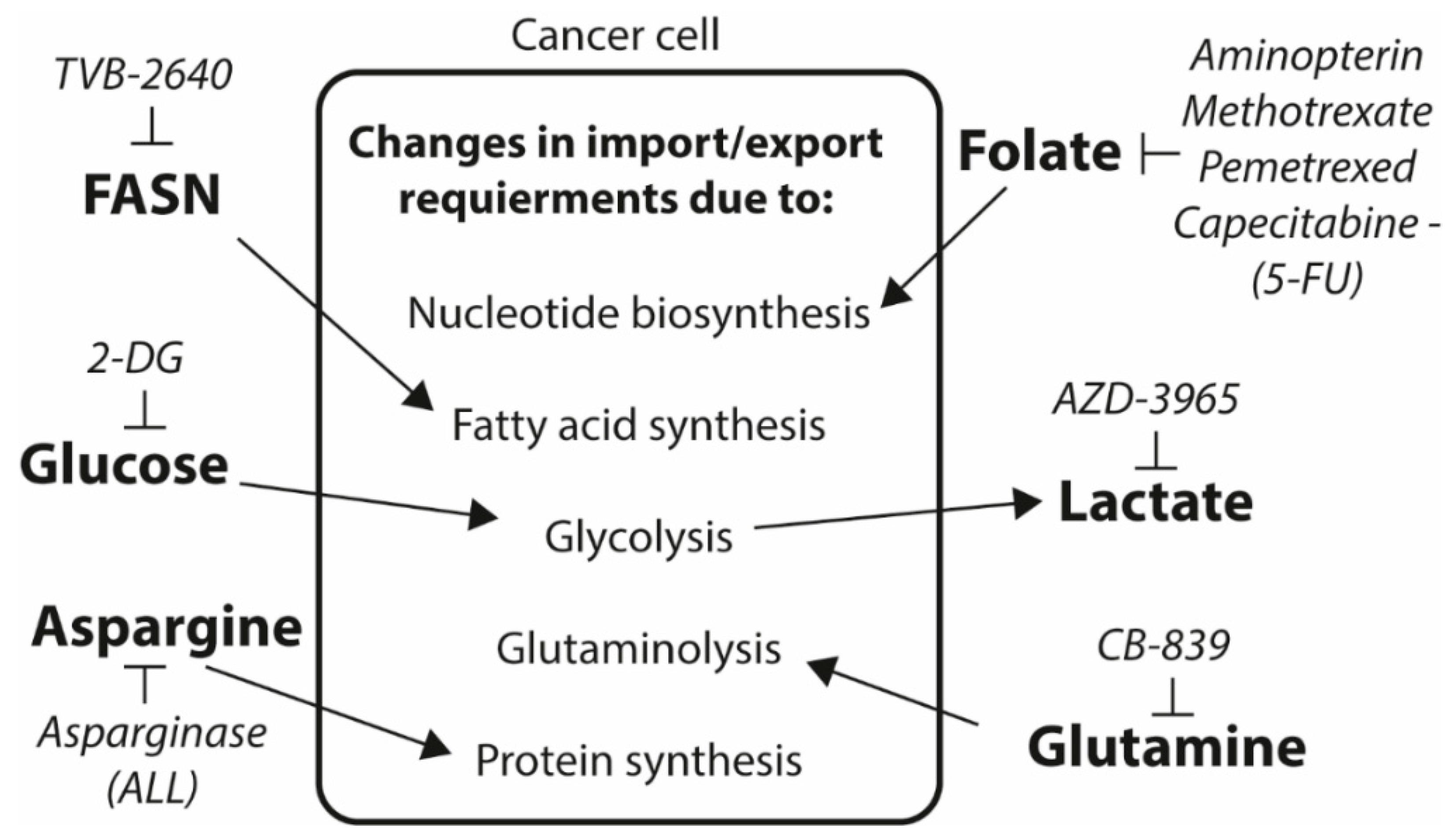
4. Future Prospects for Exploiting the Altered Cancer Metabolism as Drug Target and Biomarker
Funding
Conflicts of Interest
References
- Westerterp, K.R.; Verboeket-van de Venne, W.P.; Bouten, C.V.; de Graaf, C.; van het Hof, K.H.; Weststrate, J.A. Energy expenditure and physical activity in subjects consuming full-or reduced-fat products as part of their normal diet. Br. J. Nutr. 1996, 76, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Westerterp, K.R. Physical activity and physical activity induced energy expenditure in humans: Measurement, determinants, and effects. Front. Physiol. 2013, 4, 90. [Google Scholar] [CrossRef] [PubMed]
- Zaslaver, A.; Mayo, A.E.; Rosenberg, R.; Bashkin, P.; Sberro, H.; Tsalyuk, M.; Surette, M.G.; Alon, U. Just-in-time transcription program in metabolic pathways. Nat. Genet. 2004, 36, 486–491. [Google Scholar] [CrossRef]
- Cederbaum, S. Phenylketonuria: An update. Curr. Opin. Pediatr. 2002, 14, 702–706. [Google Scholar] [CrossRef]
- Emmett, M. Acetaminophen toxicity and 5-oxoproline (pyroglutamic acid): A tale of two cycles, one an ATP-depleting futile cycle and the other a useful cycle. Clin. J. Am. Soc. Nephrol. 2014, 9, 191–200. [Google Scholar] [CrossRef]
- Papazyan, R.; Sun, Z.; Kim, Y.H.; Titchenell, P.M.; Hill, D.A.; Lu, W.; Damle, M.; Wan, M.; Zhang, Y.; Briggs, E.R.; et al. Physiological Suppression of Lipotoxic Liver Damage by Complementary Actions of HDAC3 and SCAP/SREBP. Cell Metab. 2016, 24, 863–874. [Google Scholar] [CrossRef]
- Staubert, C.; Broom, O.J.; Nordstrom, A. Hydroxycarboxylic acid receptors are essential for breast cancer cells to control their lipid/fatty acid metabolism. Oncotarget 2015, 6, 19706–19720. [Google Scholar] [CrossRef] [PubMed]
- Garbarino, J.; Sturley, S.L. Saturated with fat: New perspectives on lipotoxicity. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.F. Regulation of cellular metabolism: Programming and maintaining metabolic homeostasis. J. Appl. Physiol. (1985) 2013, 115, 1583–1588. [Google Scholar] [CrossRef]
- Hochachka, P.W.; McClelland, G.B. Cellular metabolic homeostasis during large-scale change in ATP turnover rates in muscles. J. Exp. Biol. 1997, 200 Pt 2, 381–386. [Google Scholar]
- Lieberthal, W.; Menza, S.A.; Levine, J.S. Graded ATP depletion can cause necrosis or apoptosis of cultured mouse proximal tubular cells. Am. J. Physiol. 1998, 274 Pt 2, F315–F327. [Google Scholar] [CrossRef]
- Yoon, M.J.; Lee, H.J.; Kim, J.H.; Kim, D.K. Extracellular ATP induces apoptotic signaling in human monocyte leukemic cells, HL-60 and F-36P. Arch. Pharm. Res. 2006, 29, 1032–1041. [Google Scholar] [CrossRef]
- Eguchi, Y.; Shimizu, S.; Tsujimoto, Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997, 57, 1835–1840. [Google Scholar]
- Wilson, D.F.; Stubbs, M.; Veech, R.L.; Erecinska, M.; Krebs, H.A. Equilibrium relations between the oxidation-reduction reactions and the adenosine triphosphate synthesis in suspensions of isolated liver cells. Biochem. J. 1974, 140, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Owen, O.E.; Kalhan, S.C.; Hanson, R.W. The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 2002, 277, 30409–30412. [Google Scholar] [CrossRef]
- Gibala, M.J.; Young, M.E.; Taegtmeyer, H. Anaplerosis of the citric acid cycle: Role in energy metabolism of heart and skeletal muscle. Acta Physiol. Scand. 2000, 168, 657–665. [Google Scholar] [CrossRef]
- Schwartz, L.; Supuran, C.T.; Alfarouk, K.O. The Warburg Effect and the Hallmarks of Cancer. Anticancer Agents Med. Chem. 2017, 17, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Ahn, E.; Kumar, P.; Mukha, D.; Tzur, A.; Shlomi, T. Temporal fluxomics reveals oscillations in TCA cycle flux throughout the mammalian cell cycle. Mol. Syst. Biol. 2017, 13, 953. [Google Scholar] [CrossRef]
- Reina-Campos, M.; Moscat, J.; Diaz-Meco, M. Metabolism shapes the tumor microenvironment. Curr. Opin. Cell Biol. 2017, 48, 47–53. [Google Scholar] [CrossRef]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef]
- Dalgliesh, C.E.; Horning, E.C.; Horning, M.G.; Knox, K.L.; Yarger, K. A gas-liquid-chromatographic procedure for separating a wide range of metabolites occuring in urine or tissue extracts. Biochem. J. 1966, 101, 792–810. [Google Scholar] [CrossRef] [PubMed]
- Horning, E.C.; Horning, M.G. Metabolic profiles: Gas-phase methods for analysis of metabolites. Clin. Chem. 1971, 17, 802–809. [Google Scholar] [PubMed]
- Pauling, L.; Robinson, A.B.; Teranishi, R.; Cary, P. Quantitative analysis of urine vapor and breath by gas-liquid partition chromatography. Proc. Natl. Acad. Sci. USA 1971, 68, 2374–2376. [Google Scholar] [CrossRef] [PubMed]
- Oliver, S.G.; Winson, M.K.; Kell, D.B.; Baganz, F. Systematic functional analysis of the yeast genome. Trends Biotechnol. 1998, 16, 373–378. [Google Scholar] [CrossRef]
- Fiehn, O.; Kopka, J.; Dormann, P.; Altmann, T.; Trethewey, R.N.; Willmitzer, L. Metabolite profiling for plant functional genomics. Nat. Biotechnol. 2000, 18, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Raamsdonk, L.M.; Teusink, B.; Broadhurst, D.; Zhang, N.; Hayes, A.; Walsh, M.C.; Berden, J.A.; Brindle, K.M.; Kell, D.B.; Rowland, J.J.; et al. A functional genomics strategy that uses metabolome data to reveal the phenotype of silent mutations. Nature biotechnology 2001, 19, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Hakimi, A.A.; Reznik, E.; Lee, C.H.; Creighton, C.J.; Brannon, A.R.; Luna, A.; Aksoy, B.A.; Liu, E.M.; Shen, R.; Lee, W.; et al. An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell 2016, 29, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Makarov, A.; Denisov, E.; Kholomeev, A.; Balschun, W.; Lange, O.; Strupat, K.; Horning, S. Performance evaluation of a hybrid linear ion trap/orbitrap mass spectrometer. Anal. Chem. 2006, 78, 2113–2120. [Google Scholar] [CrossRef]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef]
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; et al. The BioCyc collection of microbial genomes and metabolic pathways. Brief Bioinform. 2017. [Google Scholar] [CrossRef]
- Guijas, C.; Montenegro-Burke, J.R.; Domingo-Almenara, X.; Palermo, A.; Warth, B.; Hermann, G.; Koellensperger, G.; Huan, T.; Uritboonthai, W.; Aisporna, A.E.; et al. METLIN: A Technology Platform for Identifying Knowns and Unknowns. Anal. Chem. 2018, 90, 3156–3164. [Google Scholar] [CrossRef]
- Otto, A.M. Warburg effect(s)-a biographical sketch of Otto Warburg and his impacts on tumor metabolism. Cancer Metab. 2016, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Eagle, H. The minimum vitamin requirements of the L and HeLa cells in tissue culture, the production of specific vitamin deficiencies, and their cure. J. Exp. Med. 1955, 102, 595–600. [Google Scholar] [CrossRef]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef] [PubMed]
- Eagle, H.; Oyama, V.I.; Levy, M.; Horton, C.L.; Fleischman, R. The growth response of mammalian cells in tissue culture to L-glutamine and L-glutamic acid. J. Biol. Chem. 1956, 218, 607–616. [Google Scholar] [PubMed]
- Medes, G.; Friedmann, B.; Weinhouse, S. Fatty acid metabolism. VIII. Acetate metabolism in vitro during hepatocarcinogenesis by p-dimethylaminoazobenzene. Cancer Res. 1956, 16, 57–62. [Google Scholar]
- Schug, Z.T.; Vande Voorde, J.; Gottlieb, E. The metabolic fate of acetate in cancer. Nat. Rev. Cancer 2016, 16, 708–717. [Google Scholar] [CrossRef]
- Li, K.; Naviaux, J.C.; Bright, A.T.; Wang, L.; Naviaux, R.K. A robust, single-injection method for targeted, broad-spectrum plasma metabolomics. Metabolomics 2017, 13, 122. [Google Scholar] [CrossRef] [PubMed]
- Yip, W.C.Y.; Sequeira, I.R.; Plank, L.D.; Poppitt, S.D. Prevalence of Pre-Diabetes across Ethnicities: A Review of Impaired Fasting Glucose (IFG) and Impaired Glucose Tolerance (IGT) for Classification of Dysglycaemia. Nutrients 2017, 9, 1273. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Peter, A.; Fritsche, J.; Elcnerova, M.; Fritsche, A.; Haring, H.U.; Schleicher, E.D.; Xu, G.; Lehmann, R. Changes of the plasma metabolome during an oral glucose tolerance test: Is there more than glucose to look at? Am. J. Physiol. Endocrinol. Metab. 2009, 296, E384–E393. [Google Scholar] [CrossRef]
- Buescher, J.M.; Antoniewicz, M.R.; Boros, L.G.; Burgess, S.C.; Brunengraber, H.; Clish, C.B.; DeBerardinis, R.J.; Feron, O.; Frezza, C.; Ghesquiere, B.; et al. A roadmap for interpreting (13)C metabolite labeling patterns from cells. Curr. Opin. Biotechnol. 2015, 34, 189–201. [Google Scholar] [CrossRef]
- Jang, C.; Chen, L.; Rabinowitz, J.D. Metabolomics and Isotope Tracing. Cell 2018, 173, 822–837. [Google Scholar] [CrossRef]
- Wiechert, W. 13C metabolic flux analysis. Metab. Eng. 2001, 3, 195–206. [Google Scholar] [CrossRef]
- Sun, J.; Wei, Q.; Zhou, Y.; Wang, J.; Liu, Q.; Xu, H. A systematic analysis of FDA-approved anticancer drugs. BMC Syst. Biol. 2017, 11 (Suppl. 5), 87. [Google Scholar] [CrossRef]
- Semba, R.D. The discovery of the vitamins. Int. J. Vitam. Nutr. Res. 2012, 82, 310–315. [Google Scholar] [CrossRef]
- Laszlo, D.; Leuchtenberger, C. Inositol a Tumor Growth Inhibitor. Science 1943, 97, 515. [Google Scholar] [CrossRef]
- Leuchtenberger, C.; Lewisohn, R.; Laszlo, D.; Leuchtenberger, R. “Folic Acid” a Tumor Growth Inhibitor. Exp. Biol. Med. 1944, 55, 204–205. [Google Scholar] [CrossRef]
- Leuchtenberger, R.; Leuchtenberger, C.; Laszlo, D.; Lewisohn, R. The Influence of “Folic Acid” on Spontaneous Breast Cancers in Mice. Science 1945, 101, 46. [Google Scholar] [CrossRef]
- Farber, S.; Cutler, E.C.; Hawkins, J.W.; Harrison, J.H.; Peirce, E.C., 2nd; Lenz, G.G. The Action of Pteroylglutamic Conjugates on Man. Science 1947, 106, 619–621. [Google Scholar] [CrossRef]
- Farber, S.; Diamond, L.K. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N. Engl. J. Med. 1948, 238, 787–793. [Google Scholar] [CrossRef]
- Bonadonna, G.; Brusamolino, E.; Valagussa, P.; Rossi, A.; Brugnatelli, L.; Brambilla, C.; De Lena, M.; Tancini, G.; Bajetta, E.; Musumeci, R.; et al. Combination chemotherapy as an adjuvant treatment in operable breast cancer. N. Engl. J. Med. 1976, 294, 405–410. [Google Scholar] [CrossRef]
- Smolen, J.S.; Landewe, R.; Bijlsma, J.; Burmester, G.; Chatzidionysiou, K.; Dougados, M.; Nam, J.; Ramiro, S.; Voshaar, M.; van Vollenhoven, R.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann. Rheum. Dis. 2017, 76, 960–977. [Google Scholar] [CrossRef]
- Kidd, J.G. Regression of transplanted lymphomas induced in vivo by means of normal guinea pig serum. I. Course of transplanted cancers of various kinds in mice and rats given guinea pig serum, horse serum, or rabbit serum. J. Exp. Med. 1953, 98, 565–582. [Google Scholar] [CrossRef]
- Broome, J.D. Evidence that the L-asparaginase of guinea pig serum is responsible for its antilymphoma effects. I. Properties of the L-asparaginase of guinea pig serum in relation to those of the antilymphoma substance. J. Exp. Med. 1963, 118, 99–120. [Google Scholar] [CrossRef]
- Broome, J.D. Evidence that the L-asparaginase of guinea pig serum is responsible for its antilymphoma effects. II. Lymphoma 6C3HED cells cultured in a medium devoid of L-asparagine lose their susceptibility to the effects of guinea pig serum in vivo. J. Exp. Med. 1963, 118, 121–148. [Google Scholar] [CrossRef]
- Ralser, M.; Wamelink, M.M.; Struys, E.A.; Joppich, C.; Krobitsch, S.; Jakobs, C.; Lehrach, H. A catabolic block does not sufficiently explain how 2-deoxy-D-glucose inhibits cell growth. Proc. Natl. Acad. Sci. USA 2008, 105, 17807–17811. [Google Scholar] [CrossRef]
- Mohanti, B.K.; Rath, G.K.; Anantha, N.; Kannan, V.; Das, B.S.; Chandramouli, B.A.; Banerjee, A.K.; Das, S.; Jena, A.; Ravichandran, R.; et al. Improving cancer radiotherapy with 2-deoxy-D-glucose: Phase I/II clinical trials on human cerebral gliomas. Int. J. Radiat. Oncol. Biol. Phys. 1996, 35, 103–111. [Google Scholar] [CrossRef]
- Singh, D.; Banerji, A.K.; Dwarakanath, B.S.; Tripathi, R.P.; Gupta, J.P.; Mathew, T.L.; Ravindranath, T.; Jain, V. Optimizing cancer radiotherapy with 2-deoxy-d-glucose dose escalation studies in patients with glioblastoma multiforme. Strahlenther. Onkol. 2005, 181, 507–514. [Google Scholar] [CrossRef]
- Raez, L.E.; Papadopoulos, K.; Ricart, A.D.; Chiorean, E.G.; Dipaola, R.S.; Stein, M.N.; Rocha Lima, C.M.; Schlesselman, J.J.; Tolba, K.; Langmuir, V.K.; et al. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 523–530. [Google Scholar] [CrossRef]
- Curtis, N.J.; Mooney, L.; Hopcroft, L.; Michopoulos, F.; Whalley, N.; Zhong, H.; Murray, C.; Logie, A.; Revill, M.; Byth, K.F.; et al. Pre-clinical pharmacology of AZD3965, a selective inhibitor of MCT1: DLBCL, NHL and Burkitt’s lymphoma anti-tumor activity. Oncotarget 2017, 8, 69219–69236. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Qin, S.; Ryoo, B.Y.; Lu, S.N.; Yen, C.J.; Feng, Y.H.; Lim, H.Y.; Izzo, F.; Colombo, M.; Sarker, D.; et al. Phase III randomized study of second line ADI-PEG 20 plus best supportive care versus placebo plus best supportive care in patients with advanced hepatocellular carcinoma. Ann. Oncol. 2018, 29, 1402–1408. [Google Scholar] [CrossRef]
- Tsai, H.J.; Jiang, S.S.; Hung, W.C.; Borthakur, G.; Lin, S.F.; Pemmaraju, N.; Jabbour, E.; Bomalaski, J.S.; Chen, Y.P.; Hsiao, H.H.; et al. A Phase II Study of Arginine Deiminase (ADI-PEG20) in Relapsed/Refractory or Poor-Risk Acute Myeloid Leukemia Patients. Sci. Rep. 2017, 7, 11253. [Google Scholar] [CrossRef]
- Lowery, M.A.; Yu, K.H.; Kelsen, D.P.; Harding, J.J.; Bomalaski, J.S.; Glassman, D.C.; Covington, C.M.; Brenner, R.; Hollywood, E.; Barba, A.; et al. A phase 1/1B trial of ADI-PEG 20 plus nab-paclitaxel and gemcitabine in patients with advanced pancreatic adenocarcinoma. Cancer 2017, 123, 4556–4565. [Google Scholar] [CrossRef]
- Lee, D.; Xu, I.M.; Chiu, D.K.; Lai, R.K.; Tse, A.P.; Lee, L.L.; Law, C.T.; Tsang, F.H.; Wei, L.L.; Chan, C.Y.; et al. Folate cycle enzyme MTHFD1L confers metabolic advantages in hepatocellular carcinoma. J. Clin. Investig. 2017, 127, 1856–1872. [Google Scholar] [CrossRef]
- Touzart, A.; Lengline, E.; Latiri, M.; Belhocine, M.; Smith, C.; Thomas, X.; Spicuglia, S.; Puthier, D.; Pflumio, F.; Leguay, T.; et al. Epigenetic silencing affects L-asparaginase sensitivity and predicts outcome in T-ALL. Clin. Cancer. Res. 2019. [Google Scholar] [CrossRef]
- Yizhak, K.; Gaude, E.; Le Devedec, S.; Waldman, Y.Y.; Stein, G.Y.; van de Water, B.; Frezza, C.; Ruppin, E. Phenotype-based cell-specific metabolic modeling reveals metabolic liabilities of cancer. Elife 2014, 3. [Google Scholar] [CrossRef]
- Apaolaza, I.; San Jose-Eneriz, E.; Tobalina, L.; Miranda, E.; Garate, L.; Agirre, X.; Prosper, F.; Planes, F.J. An in-silico approach to predict and exploit synthetic lethality in cancer metabolism. Nat. Commun. 2017, 8, 459. [Google Scholar] [CrossRef]
- Pey, J.; San Jose-Eneriz, E.; Ochoa, M.C.; Apaolaza, I.; de Atauri, P.; Rubio, A.; Cendoya, X.; Miranda, E.; Garate, L.; Cascante, M.; et al. In-silico gene essentiality analysis of polyamine biosynthesis reveals APRT as a potential target in cancer. Sci. Rep. 2017, 7, 14358. [Google Scholar] [CrossRef]
- Thiele, I.; Swainston, N.; Fleming, R.M.; Hoppe, A.; Sahoo, S.; Aurich, M.K.; Haraldsdottir, H.; Mo, M.L.; Rolfsson, O.; Stobbe, M.D.; et al. A community-driven global reconstruction of human metabolism. Nat. Biotechnol. 2013, 31, 419–425. [Google Scholar] [CrossRef]
- Lv, W.W.; Liu, D.; Liu, X.C.; Feng, T.N.; Li, L.; Qian, B.Y.; Li, W.X. Effects of PKM2 on global metabolic changes and prognosis in hepatocellular carcinoma: From gene expression to drug discovery. BMC Cancer 2018, 18, 1150. [Google Scholar] [CrossRef]
- Cantor, J.R.; Abu-Remaileh, M.; Kanarek, N.; Freinkman, E.; Gao, X.; Louissaint, A., Jr.; Lewis, C.A.; Sabatini, D.M. Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell 2017, 169, 258–272.e17. [Google Scholar] [CrossRef]
- Li, X.; Chung, A.C.K.; Li, S.; Wu, L.; Xu, J.; Yu, J.; Wong, C.; Cai, Z. LC-MS-based metabolomics revealed SLC25A22 as an essential regulator of aspartate-derived amino acids and polyamines in KRAS-mutant colorectal cancer. Oncotarget 2017, 8, 101333–101344. [Google Scholar] [CrossRef]
- Birsoy, K.; Possemato, R.; Lorbeer, F.K.; Bayraktar, E.C.; Thiru, P.; Yucel, B.; Wang, T.; Chen, W.W.; Clish, C.B.; Sabatini, D.M. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 2014, 508, 108–112. [Google Scholar] [CrossRef]
- Lee, J.; Shin, D.; Roh, J.L. Development of an in vitro cell-sheet cancer model for chemotherapeutic screening. Theranostics 2018, 8, 3964–3973. [Google Scholar] [CrossRef] [PubMed]
- Gunda, V.; Yu, F.; Singh, P.K. Validation of Metabolic Alterations in Microscale Cell Culture Lysates Using Hydrophilic Interaction Liquid Chromatography (HILIC)-Tandem Mass Spectrometry-Based Metabolomics. PLoS ONE 2016, 11, e0154416. [Google Scholar] [CrossRef]
- Kus, K.; Kij, A.; Zakrzewska, A.; Jasztal, A.; Stojak, M.; Walczak, M.; Chlopicki, S. Alterations in arginine and energy metabolism, structural and signalling lipids in metastatic breast cancer in mice detected in plasma by targeted metabolomics and lipidomics. Breast Cancer Res. 2018, 20, 148. [Google Scholar] [CrossRef]
- Teoh, S.T.; Ogrodzinski, M.P.; Ross, C.; Hunter, K.W.; Lunt, S.Y. Sialic Acid Metabolism: A Key Player in Breast Cancer Metastasis Revealed by Metabolomics. Front. Oncol. 2018, 8, 174. [Google Scholar] [CrossRef]
- Momcilovic, M.; Bailey, S.T.; Lee, J.T.; Fishbein, M.C.; Braas, D.; Go, J.; Graeber, T.G.; Parlati, F.; Demo, S.; Li, R.; et al. The GSK3 Signaling Axis Regulates Adaptive Glutamine Metabolism in Lung Squamous Cell Carcinoma. Cancer Cell 2018, 33, 905–921.e5. [Google Scholar] [CrossRef]
- Pore, N.; Jalla, S.; Liu, Z.; Higgs, B.; Sorio, C.; Scarpa, A.; Hollingsworth, R.; Tice, D.A.; Michelotti, E. In Vivo Loss of Function Screening Reveals Carbonic Anhydrase IX as a Key Modulator of Tumor Initiating Potential in Primary Pancreatic Tumors. Neoplasia 2015, 17, 473–480. [Google Scholar] [CrossRef]
- Lindahl, A.; Forshed, J.; Nordstrom, A. Overlap in serum metabolic profiles between non-related diseases: Implications for LC-MS metabolomics biomarker discovery. Biochem. Biophys. Res. Commun. 2016, 478, 1472–1477. [Google Scholar] [CrossRef]
- Anderson, B.W.; Suh, Y.S.; Choi, B.; Lee, H.J.; Yab, T.C.; Taylor, W.R.; Dukek, B.A.; Berger, C.K.; Cao, X.; Foote, P.H.; et al. Detection of Gastric Cancer with Novel Methylated DNA Markers: Discovery, Tissue Validation, and Pilot Testing in Plasma. Clin. Cancer. Res. 2018, 24, 5724–5734. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [PubMed]
- Ahlquist, D.A. Universal cancer screening: Revolutionary, rational, and realizable. NPJ Precis. Oncol. 2018, 2, 23. [Google Scholar] [CrossRef]
- Lopez-Lopez, A.; Lopez-Gonzalvez, A.; Barker-Tejeda, T.C.; Barbas, C. A review of validated biomarkers obtained through metabolomics. Expert Rev. Mol. Diagn. 2018, 18, 557–575. [Google Scholar] [CrossRef] [PubMed]
- Kalita, J.; Misra, U.K.; Kumar, V.; Parashar, V. Predictors of seizure in Wilson disease: A clinico-radiological and biomarkers study. Neurotoxicology 2018, 71, 87–92. [Google Scholar] [CrossRef]
- Davidson, J.A.; Pfeifer, Z.; Frank, B.; Tong, S.; Urban, T.T.; Wischmeyer, P.A.; Mourani, P.; Landeck, B.; Christians, U.; Klawitter, J. Metabolomic Fingerprinting of Infants Undergoing Cardiopulmonary Bypass: Changes in Metabolic Pathways and Association With Mortality and Cardiac Intensive Care Unit Length of Stay. J. Am. Heart Assoc. 2018, 7, e010711. [Google Scholar] [CrossRef] [PubMed]
- Madeira, C.; Vargas-Lopes, C.; Brandao, C.O.; Reis, T.; Laks, J.; Panizzutti, R.; Ferreira, S.T. Elevated Glutamate and Glutamine Levels in the Cerebrospinal Fluid of Patients With Probable Alzheimer’s Disease and Depression. Front. Psychiatry 2018, 9, 561. [Google Scholar] [CrossRef] [PubMed]
- Callejon-Leblic, B.; Pereira-Vega, A.; Vazquez-Gandullo, E.; Sanchez-Ramos, J.L.; Gomez-Ariza, J.L.; Garcia-Barrera, T. Study of the metabolomic relationship between lung cancer and chronic obstructive pulmonary disease based on direct infusion mass spectrometry. Biochimie 2018, 157, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Yin, X.; Edden, R.A.E.; Evans, A.C.; Xu, J.; Cao, G.; Li, H.; Li, M.; Zhao, B.; Wang, J.; et al. Altered hippocampal GABA and glutamate levels and uncoupling from functional connectivity in multiple sclerosis. Hippocampus 2018, 28, 813–823. [Google Scholar] [CrossRef]
- Asiago, V.M.; Alvarado, L.Z.; Shanaiah, N.; Gowda, G.A.; Owusu-Sarfo, K.; Ballas, R.A.; Raftery, D. Early detection of recurrent breast cancer using metabolite profiling. Cancer Res. 2010, 70, 8309–8318. [Google Scholar] [CrossRef] [PubMed]
- Budczies, J.; Pfitzner, B.M.; Gyorffy, B.; Winzer, K.J.; Radke, C.; Dietel, M.; Fiehn, O.; Denkert, C. Glutamate enrichment as new diagnostic opportunity in breast cancer. Int. J. Cancer 2015, 136, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Li, M.; Zha, W.; Zhao, Q.; Gu, R.; Liu, L.; Shi, J.; Zhou, J.; Zhou, F.; Wu, X.; et al. Metabolomic approach to evaluating adriamycin pharmacodynamics and resistance in breast cancer cells. Metabolomics 2013, 9, 960–973. [Google Scholar] [CrossRef]
- Hart, C.D.; Vignoli, A.; Tenori, L.; Uy, G.L.; Van To, T.; Adebamowo, C.; Hossain, S.M.; Biganzoli, L.; Risi, E.; Love, R.R.; et al. Serum Metabolomic Profiles Identify ER-Positive Early Breast Cancer Patients at Increased Risk of Disease Recurrence in a Multicenter Population. Clin. Cancer. Res. 2017, 23, 1422–1431. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Chong, N.; Lewis, N.E.; Jia, W.; Xie, G.; Garmire, L.X. Novel personalized pathway-based metabolomics models reveal key metabolic pathways for breast cancer diagnosis. Genome Med. 2016, 8, 34. [Google Scholar] [CrossRef]
- Jobard, E.; Pontoizeau, C.; Blaise, B.J.; Bachelot, T.; Elena-Herrmann, B.; Tredan, O. A serum nuclear magnetic resonance-based metabolomic signature of advanced metastatic human breast cancer. Cancer Lett. 2014, 343, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Maria, R.M.; Altei, W.F.; Andricopulo, A.D.; Becceneri, A.B.; Cominetti, M.R.; Venancio, T.; Colnago, L.A. Characterization of metabolic profile of intact non-tumor and tumor breast cells by high-resolution magic angle spinning nuclear magnetic resonance spectroscopy. Anal. Biochem. 2015, 488, 14–18. [Google Scholar] [CrossRef]
- Richard, V.; Conotte, R.; Mayne, D.; Colet, J.M. Does the 1H-NMR plasma metabolome reflect the host-tumor interactions in human breast cancer? Oncotarget 2017, 8, 49915–49930. [Google Scholar] [CrossRef]
- Simpson, N.E.; Tryndyak, V.P.; Beland, F.A.; Pogribny, I.P. An in vitro investigation of metabolically sensitive biomarkers in breast cancer progression. Breast Cancer Res. Treat. 2012, 133, 959–968. [Google Scholar] [CrossRef]
- Suman, S.; Sharma, R.K.; Kumar, V.; Sinha, N.; Shukla, Y. Metabolic fingerprinting in breast cancer stages through (1)H NMR spectroscopy-based metabolomic analysis of plasma. J. Pharm. Biomed. Anal. 2018, 160, 38–45. [Google Scholar] [CrossRef]
- Tenori, L.; Oakman, C.; Morris, P.G.; Gralka, E.; Turner, N.; Cappadona, S.; Fornier, M.; Hudis, C.; Norton, L.; Luchinat, C.; et al. Serum metabolomic profiles evaluated after surgery may identify patients with oestrogen receptor negative early breast cancer at increased risk of disease recurrence. Results from a retrospective study. Mol. Oncol. 2015, 9, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Schafferer, S.; Tang, Q.; Scheffler, M.; Nees, J.; Heil, J.; Schott, S.; Golatta, M.; Wallwiener, M.; Sohn, C.; et al. A plasma metabolite panel as biomarkers for early primary breast cancer detection. Int. J. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Xu, H.; Liu, R.; Gao, P.; Yang, X.; Li, P.; Wang, X.; Zhang, Y.; Bi, K.; Li, Q. Highly Sensitive Quantification Method for Amine Submetabolome Based on AQC-Labeled-LC-Tandem-MS and Multiple Statistical Data Mining: A Potential Cancer Screening Approach. Anal. Chem. 2018, 90, 11941–11948. [Google Scholar] [CrossRef] [PubMed]
- Giskeodegard, G.F.; Lundgren, S.; Sitter, B.; Fjosne, H.E.; Postma, G.; Buydens, L.M.; Gribbestad, I.S.; Bathen, T.F. Lactate and glycine-potential MR biomarkers of prognosis in estrogen receptor-positive breast cancers. NMR Biomed. 2012, 25, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Maria, R.M.; Altei, W.F.; Selistre-de-Araujo, H.S.; Colnago, L.A. Impact of chemotherapy on metabolic reprogramming: Characterization of the metabolic profile of breast cancer MDA-MB-231 cells using (1)H HR-MAS NMR spectroscopy. J. Pharm. Biomed. Anal. 2017, 146, 324–328. [Google Scholar] [CrossRef]
- Martineau, E.; Tea, I.; Akoka, S.; Giraudeau, P. Absolute quantification of metabolites in breast cancer cell extracts by quantitative 2D (1) H INADEQUATE NMR. NMR Biomed. 2012, 25, 985–992. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Prisco, M.; Ertel, A.; Tsirigos, A.; Lin, Z.; Pavlides, S.; Wang, C.; Flomenberg, N.; Knudsen, E.S.; Howell, A.; et al. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: Achieving personalized medicine via Metabolo-Genomics. Cell Cycle 2011, 10, 1271–1286. [Google Scholar] [CrossRef]
- Sitter, B.; Bathen, T.F.; Singstad, T.E.; Fjosne, H.E.; Lundgren, S.; Halgunset, J.; Gribbestad, I.S. Quantification of metabolites in breast cancer patients with different clinical prognosis using HR MAS MR spectroscopy. NMR Biomed. 2010, 23, 424–431. [Google Scholar] [CrossRef]
- Slupsky, C.M.; Steed, H.; Wells, T.H.; Dabbs, K.; Schepansky, A.; Capstick, V.; Faught, W.; Sawyer, M.B. Urine metabolite analysis offers potential early diagnosis of ovarian and breast cancers. Clin. Cancer. Res. 2010, 16, 5835–5841. [Google Scholar] [CrossRef]
- Tredwell, G.D.; Miller, J.A.; Chow, H.H.; Thompson, P.A.; Keun, H.C. Metabolomic characterization of nipple aspirate fluid by (1)H NMR spectroscopy and GC-MS. J. Proteome Res. 2014, 13, 883–889. [Google Scholar] [CrossRef]
- Weljie, A.M.; Bondareva, A.; Zang, P.; Jirik, F.R. (1)H NMR metabolomics identification of markers of hypoxia-induced metabolic shifts in a breast cancer model system. J. Biomol. NMR 2011, 49, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Bathen, T.F.; Geurts, B.; Sitter, B.; Fjosne, H.E.; Lundgren, S.; Buydens, L.M.; Gribbestad, I.S.; Postma, G.; Giskeodegard, G.F. Feasibility of MR metabolomics for immediate analysis of resection margins during breast cancer surgery. PLoS ONE 2013, 8, e61578. [Google Scholar] [CrossRef] [PubMed]
- Brockmoller, S.F.; Bucher, E.; Muller, B.M.; Budczies, J.; Hilvo, M.; Griffin, J.L.; Oresic, M.; Kallioniemi, O.; Iljin, K.; Loibl, S.; et al. Integration of metabolomics and expression of glycerol-3-phosphate acyltransferase (GPAM) in breast cancer-link to patient survival, hormone receptor status, and metabolic profiling. J. Proteome Res. 2012, 11, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, T.; Floegel, A.; Sookthai, D.; Johnson, T.; Rolle-Kampczyk, U.; Otto, W.; von Bergen, M.; Boeing, H.; Kaaks, R. Higher plasma levels of lysophosphatidylcholine 18:0 are related to a lower risk of common cancers in a prospective metabolomics study. BMC Med. 2016, 14, 13. [Google Scholar] [CrossRef] [PubMed]
- Lodi, A.; Ronen, S.M. Magnetic resonance spectroscopy detectable metabolomic fingerprint of response to antineoplastic treatment. PLoS ONE 2011, 6, e26155. [Google Scholar] [CrossRef]
- Mimmi, M.C.; Finato, N.; Pizzolato, G.; Beltrami, C.A.; Fogolari, F.; Corazza, A.; Esposito, G. Absolute quantification of choline-related biomarkers in breast cancer biopsies by liquid chromatography electrospray ionization mass spectrometry. Anal. Cell. Pathol. (Amsterdam) 2013, 36, 71–83. [Google Scholar] [CrossRef]
- Qiu, Y.; Zhou, B.; Su, M.; Baxter, S.; Zheng, X.; Zhao, X.; Yen, Y.; Jia, W. Mass spectrometry-based quantitative metabolomics revealed a distinct lipid profile in breast cancer patients. Int. J. Mol. Sci. 2013, 14, 8047–8061. [Google Scholar] [CrossRef]
- van Asten, J.J.; Vettukattil, R.; Buckle, T.; Rottenberg, S.; van Leeuwen, F.; Bathen, T.F.; Heerschap, A. Increased levels of choline metabolites are an early marker of docetaxel treatment response in BRCA1-mutated mouse mammary tumors: An assessment by ex vivo proton magnetic resonance spectroscopy. J. Transl. Med. 2015, 13, 114. [Google Scholar] [CrossRef]
- Wang, S.; Chen, X.; Luan, H.; Gao, D.; Lin, S.; Cai, Z.; Liu, J.; Liu, H.; Jiang, Y. Matrix-assisted laser desorption/ionization mass spectrometry imaging of cell cultures for the lipidomic analysis of potential lipid markers in human breast cancer invasion. Rapid Commun. Mass Spectrom. 2016, 30, 533–542. [Google Scholar] [CrossRef]
- Zhong, L.; Cheng, F.; Lu, X.; Duan, Y.; Wang, X. Untargeted saliva metabonomics study of breast cancer based on ultra performance liquid chromatography coupled to mass spectrometry with HILIC and RPLC separations. Talanta 2016, 158, 351–360. [Google Scholar] [CrossRef]
- Chae, E.Y.; Shin, H.J.; Kim, S.; Baek, H.M.; Yoon, D.; Kim, S.; Shim, Y.E.; Kim, H.H.; Cha, J.H.; Choi, W.J.; et al. The Role of High-Resolution Magic Angle Spinning 1H Nuclear Magnetic Resonance Spectroscopy for Predicting the Invasive Component in Patients with Ductal Carcinoma In Situ Diagnosed on Preoperative Biopsy. PLoS ONE 2016, 11, e0161038. [Google Scholar] [CrossRef]
- Fan, Y.; Zhou, X.; Xia, T.S.; Chen, Z.; Li, J.; Liu, Q.; Alolga, R.N.; Chen, Y.; Lai, M.D.; Li, P.; et al. Human plasma metabolomics for identifying differential metabolites and predicting molecular subtypes of breast cancer. Oncotarget 2016, 7, 9925–9938. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Sharma, R.K.; Chagtoo, M.; Agarwal, G.; George, N.; Sinha, N.; Godbole, M.M. 1H NMR Metabolomics Reveals Association of High Expression of Inositol 1, 4, 5 Trisphosphate Receptor and Metabolites in Breast Cancer Patients. PLoS ONE 2017, 12, e0169330. [Google Scholar] [CrossRef] [PubMed]
- Vettukattil, R.; Hetland, T.E.; Florenes, V.A.; Kaern, J.; Davidson, B.; Bathen, T.F. Proton magnetic resonance metabolomic characterization of ovarian serous carcinoma effusions: Chemotherapy-related effects and comparison with malignant mesothelioma and breast carcinoma. Hum. Pathol. 2013, 44, 1859–1866. [Google Scholar] [CrossRef]
- Jove, M.; Collado, R.; Quiles, J.L.; Ramirez-Tortosa, M.C.; Sol, J.; Ruiz-Sanjuan, M.; Fernandez, M.; de la Torre Cabrera, C.; Ramirez-Tortosa, C.; Granados-Principal, S.; et al. A plasma metabolomic signature discloses human breast cancer. Oncotarget 2017, 8, 19522–19533. [Google Scholar] [CrossRef]
- Sugimoto, M.; Wong, D.T.; Hirayama, A.; Soga, T.; Tomita, M. Capillary electrophoresis mass spectrometry-based saliva metabolomics identified oral, breast and pancreatic cancer-specific profiles. Metabolomics 2010, 6, 78–95. [Google Scholar] [CrossRef] [PubMed]
- He, Y.U.; Li, Q.Q.; Guo, S.C. Taurine Attenuates Dimethylbenz[a]anthracene-induced Breast Tumorigenesis in Rats: A Plasma Metabolomic Study. Anticancer Res. 2016, 36, 533–543. [Google Scholar]
- Chen, Y.; Zhang, R.; Song, Y.; He, J.; Sun, J.; Bai, J.; An, Z.; Dong, L.; Zhan, Q.; Abliz, Z. RRLC-MS/MS-based metabonomics combined with in-depth analysis of metabolic correlation network: Finding potential biomarkers for breast cancer. The Analyst 2009, 134, 2003–2011. [Google Scholar] [CrossRef] [PubMed]
- Oakman, C.; Tenori, L.; Claudino, W.M.; Cappadona, S.; Nepi, S.; Battaglia, A.; Bernini, P.; Zafarana, E.; Saccenti, E.; Fornier, M.; et al. Identification of a serum-detectable metabolomic fingerprint potentially correlated with the presence of micrometastatic disease in early breast cancer patients at varying risks of disease relapse by traditional prognostic methods. Ann. Oncol. 2011, 22, 1295–1301. [Google Scholar] [CrossRef]
- Sun, Y.; Kim, J.H.; Vangipuram, K.; Hayes, D.F.; Smith, E.M.L.; Yeomans, L.; Henry, N.L.; Stringer, K.A.; Hertz, D.L. Pharmacometabolomics reveals a role for histidine, phenylalanine, and threonine in the development of paclitaxel-induced peripheral neuropathy. Breast Cancer Res. Treat. 2018, 171, 657–666. [Google Scholar] [CrossRef]
- Tenori, L.; Oakman, C.; Claudino, W.M.; Bernini, P.; Cappadona, S.; Nepi, S.; Biganzoli, L.; Arbushites, M.C.; Luchinat, C.; Bertini, I.; et al. Exploration of serum metabolomic profiles and outcomes in women with metastatic breast cancer: A pilot study. Mol. Oncol. 2012, 6, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Arceo, J.; Arnold, J.; Sreekumar, A.; Dovichi, N.J.; Li, J.; Littlepage, L.E. Metabolomics of oncogene-specific metabolic reprogramming during breast cancer. Cancer Metab. 2018, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Corona, G.; Polesel, J.; Fratino, L.; Miolo, G.; Rizzolio, F.; Crivellari, D.; Addobbati, R.; Cervo, S.; Toffoli, G. Metabolomics biomarkers of frailty in elderly breast cancer patients. J. Cell. Physiol. 2014, 229, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Wang, Q.; Chen, G. Serum metabolomics analysis reveals changes in signaling lipids in breast cancer patients. Biomed. Chromatogr. 2016, 30, 42–47. [Google Scholar] [CrossRef]
- Okamoto, Y.; Aoki, A.; Ueda, K.; Jinno, H. Metabolomic analysis uncovered an association of serum phospholipid levels with estrogen-induced mammary tumors in female ACI/Seg rats. Toxicol. Lett. 2018, 288, 65–70. [Google Scholar] [CrossRef]
- Zhang, R.; Zhuang, X.; Zong, L.; Liu, S.; Liu, Z.; Song, F. Investigations on the cell metabolomics basis of multidrug resistance from tumor cells by ultra-performance liquid chromatography-mass spectrometry. Anal. Bioanal. Chem. 2016, 408, 5843–5854. [Google Scholar] [CrossRef]
- Matos Do Canto, L.; Marian, C.; Varghese, R.S.; Ahn, J.; Da Cunha, P.A.; Willey, S.; Sidawy, M.; Rone, J.D.; Cheema, A.K.; Luta, G.; et al. Metabolomic profiling of breast tumors using ductal fluid. Int. J. Oncol. 2016, 49, 2245–2254. [Google Scholar] [CrossRef]
- Morvan, D. Functional metabolomics uncovers metabolic alterations associated to severe oxidative stress in MCF7 breast cancer cells exposed to ascididemin. Mar. Drugs 2013, 11, 3846–3860. [Google Scholar] [CrossRef]
- Wei, S.; Liu, L.; Zhang, J.; Bowers, J.; Gowda, G.A.; Seeger, H.; Fehm, T.; Neubauer, H.J.; Vogel, U.; Clare, S.E.; et al. Metabolomics approach for predicting response to neoadjuvant chemotherapy for breast cancer. Mol. Oncol. 2013, 7, 297–307. [Google Scholar] [CrossRef]
- Miolo, G.; Muraro, E.; Caruso, D.; Crivellari, D.; Ash, A.; Scalone, S.; Lombardi, D.; Rizzolio, F.; Giordano, A.; Corona, G. Pharmacometabolomics study identifies circulating spermidine and tryptophan as potential biomarkers associated with the complete pathological response to trastuzumab-paclitaxel neoadjuvant therapy in HER-2 positive breast cancer. Oncotarget 2016, 7, 39809–39822. [Google Scholar] [CrossRef]
- Geng, D.; Sun, D.; Zhang, L.; Zhang, W. The therapy of gefitinib towards breast cancer partially through reversing breast cancer biomarker arginine. Afr. Health Sci. 2015, 15, 594–597. [Google Scholar] [CrossRef]
- Tea, I.; Martineau, E.; Antheaume, I.; Lalande, J.; Mauve, C.; Gilard, F.; Barille-Nion, S.; Blackburn, A.C.; Tcherkez, G. (13)C and (15)N natural isotope abundance reflects breast cancer cell metabolism. Sci. Rep. 2016, 6, 34251. [Google Scholar] [CrossRef]
- Johnson, C.H.; Manna, S.K.; Krausz, K.W.; Bonzo, J.A.; Divelbiss, R.D.; Hollingshead, M.G.; Gonzalez, F.J. Global metabolomics reveals urinary biomarkers of breast cancer in a mcf-7 xenograft mouse model. Metabolites 2013, 3, 658–672. [Google Scholar] [CrossRef] [PubMed]
- Playdon, M.C.; Ziegler, R.G.; Sampson, J.N.; Stolzenberg-Solomon, R.; Thompson, H.J.; Irwin, M.L.; Mayne, S.T.; Hoover, R.N.; Moore, S.C. Nutritional metabolomics and breast cancer risk in a prospective study. Am. J. Clin. Nutr. 2017, 106, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Euceda, L.R.; Hill, D.K.; Stokke, E.; Hatem, R.; El Botty, R.; Bieche, I.; Marangoni, E.; Bathen, T.F.; Moestue, S.A. Metabolic Response to Everolimus in Patient-Derived Triple-Negative Breast Cancer Xenografts. J. Proteome Res. 2017, 16, 1868–1879. [Google Scholar] [CrossRef] [PubMed]
- Kanaan, Y.M.; Sampey, B.P.; Beyene, D.; Esnakula, A.K.; Naab, T.J.; Ricks-Santi, L.J.; Dasi, S.; Day, A.; Blackman, K.W.; Frederick, W.; et al. Metabolic profile of triple-negative breast cancer in African-American women reveals potential biomarkers of aggressive disease. Cancer Genom. Proteom. 2014, 11, 279–294. [Google Scholar]
- Cala, M.; Aldana, J.; Sanchez, J.; Guio, J.; Meesters, R.J.W. Urinary metabolite and lipid alterations in Colombian Hispanic women with breast cancer: A pilot study. J. Pharm. Biomed. Anal. 2018, 152, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Zhou, B.; Zhao, A.; Qiu, Y.; Zhao, X.; Garmire, L.; Shvetsov, Y.B.; Yu, H.; Yen, Y.; Jia, W. Lowered circulating aspartate is a metabolic feature of human breast cancer. Oncotarget 2015, 6, 33369–33381. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Gao, D.; Wang, Y.; Jin, F.; Wu, Q.; Liu, H. Application of metabolomics to investigate the antitumor mechanism of flavopiridol in MCF-7 breast cancer cells. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1025, 40–47. [Google Scholar] [CrossRef]
- Grankvist, N.; Watrous, J.D.; Lagerborg, K.A.; Lyutvinskiy, Y.; Jain, M.; Nilsson, R. Profiling the Metabolism of Human Cells by Deep (13)C Labeling. Cell Chem. Biol. 2018, 25, 1419–1427.e4. [Google Scholar] [CrossRef] [PubMed]
- Broekaert, D.; Fendt, S.M. Measuring In Vivo Tissue Metabolism Using (13)C Glucose Infusions in Mice. Methods Mol. Biol. 2019, 1862, 67–82. [Google Scholar] [PubMed]
- Staubert, C.; Krakowsky, R.; Bhuiyan, H.; Witek, B.; Lindahl, A.; Broom, O.; Nordstrom, A. Increased lanosterol turnover: A metabolic burden for daunorubicin-resistant leukemia cells. Med. Oncol. 2016, 33, 6. [Google Scholar] [CrossRef] [PubMed]
- Langenberg, M.L.; Tytgat, G.N.J.; Schipper, M.E.I.; Rietra, P.J.G.M.; Zanen, H.C. Campylobacter-like organisms in the stomach of patients and healthy individuals. Lancet 1984, 1, 1348–1349. [Google Scholar] [CrossRef]
- Graham, D.Y.; Klein, P.D.; Evans, D.J., Jr.; Evans, D.G.; Alpert, L.C.; Opekun, A.R.; Boutton, T.W. Campylobacter pylori detected noninvasively by the 13C-urea breath test. Lancet 1987, 1, 1174–1177. [Google Scholar] [CrossRef]
- Guthrie, R. Blood Screening for Phenylketonuria. JAMA-J. Am. Med. Assoc. 1961, 178, 863. [Google Scholar] [CrossRef]
- Folling, I. The discovery of phenylketonuria. Acta Paediatr. Suppl. 1994, 407, 4–10. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Target | Drug | FDA Status in Cancer |
|---|---|---|
| Dihydrofolate reductase (DHFR) | Methotrexate Pemetrexed | Approved |
| 5-phosphoribosyl-1-pyrophosphatase (PRPP) amidotransferase | 6-mercaptopurine (6-MP) 6-thioguanine (6-TG) | Approved |
| Thymidylate synthase (TS) | 5-fluorouracil (5-FU) Capecitabine | Approved |
| DNA polymerase/ribonucleotide reductase (RnR) | Gemcitabine Cytarabine | Approved |
| Circulating asparagine | L-Asparaginase | Approved |
| mutant Isocitrate dehydrogenase 2 (IDH2) | Enasidenib (AG-221) | Approved |
| Target | Drug | Clinical Status | Cancer Type |
|---|---|---|---|
| Dihydroorotate dehydrogenase (DHODH) | Leflunomide | Phase II/III | Prostate Cancer |
| S6K, 4E-BP1, AMPK | Metformin | Phase II | Breast Cancer |
| Mutant Isocitrate dehydrogenase 2 (IDH2) | AG-120 IDH305 AG-881 | Phase I/II | Hematologic malignancies and Solid tumors |
| Glutaminase 1 | CB-839 | Phase I/II | Renal Cell Carcinoma (RCC), Melanoma, Non-Small Cell Lung Cancer (NSCLC) |
| Monocarboxylate transporter 1 (MCT1) | AZD3965 | Phase I/II | Advanced Solid Tumors, Diffuse large B cell lymphoma and Burkitt’s Lymphoma |
| Fatty Acid Synthase (FASN) | TVB-2640 | Phase II | Advanced HER2 positive Breast Cancer, Colon Cancer, Astrocytoma |
| Arginine Deiminase | ADI-PEG 20 | Phase I/II/III | Hepatocellular Carcinoma [62], Acute myeloid leukemia (AML) [63], advanced pancreatic adenocarcinoma [64] |
| α-Ketoglutarate dehydrogenase complex | CPI-613 | Phase II | Advanced and/or metastatic solid tumors |
| Metabolites (Deregulated) | Number of Reports | Sample Type | References |
|---|---|---|---|
| Glutamate | 13 | Plasma, Serum, Tissue, Cells | [91,92,93,94,95,96,97,98,99,100,101,102,103] |
| Lactate | 12 | Plasma, Serum, Tissue, Cells | [91,94,98,104,105,106,107,108,109,110,111] |
| Phosphatidylcholines (PCs) | 12 | Saliva, Blood, Plasma, Tissue, Cells | [97,105,108,112,113,114,115,116,117,118,119,120] |
| Alanine | 9 | Plasma, Serum, Blood, Tissue, Pleural and Peritoneal effusions, Cells | [94,95,97,103,106,121,122,123,124] |
| Taurine | 9 | Saliva, Serum, Plasma, Tissue, Cells | [93,95,97,105,108,112,125,126,127] |
| Phenylalanine | 7 | Serum, Urine | [94,96,98,101,128,129,130] |
| Histidine | 7 | Plasma, Serum, Whole blood | [91,96,98,103,106,130,131] |
| Choline | 6 | Serum, Tissue, Cells | [91,94,97,108,118,132] |
| Lysophosphatidylcholine (LPCs) | 6 | Saliva, Serum, Plasma | [117,120,122,133,134,135] |
| Creatine | 6 | Serum, Urine, Tissue, Cells | [94,97,108,109,118,136] |
| Glucose | 6 | Serum, Plasma, Tissue, Pleural and Peritoneal effusions | [100,101,112,124,129,131] |
| Glycerophosphocholine (GPC) | 6 | Serum, Ductal fluid, Tissue, Cells | [108,116,118,133,137,138] |
| Proline | 6 | Serum, Plasma, Cells | [91,94,103,122,129,136] |
| Glycine | 5 | Serum, Tissue | [94,98,104,108,112] |
| Threonine | 5 | Serum, Plasma, Cells | [97,102,106,130,139] |
| Tyrosine | 5 | Serum, Urine, Cells | [91,94,98,128,136] |
| Isoleucine | 4 | Serum, Cells | [94,98,106,139] |
| Lysine | 4 | Saliva, Serum, Plasma, Blood, Tissue | [100,123,126,129] |
| Tryptophan | 4 | Serum, Plasma, Urine | [102,128,133,140] |
| Acetate | 3 | Serum, Urine, Cells | [94,97,109] |
| Arginine | 3 | Serum, Plasma, Cells | [77,141,142] |
| Caproic acid | 3 | Blood, Plasma, Urine | [125,143,144] |
| Glutamine | 3 | Serum, Cells | [97,136,139] |
| Guanidinoacetate | 3 | Tissue, Cells | [97,105,145] |
| Sarcosine | 3 | Plasma, Tissue, Cells | [103,136,146] |
| Succinate | 3 | Tissue, Urine | [109,121,147] |
| Valine | 3 | Plasma, Cells | [98,106,122] |
| β-hydroxybutyrate | 2 | Serum | [91,94] |
| Aspartate | 2 | Serum, Plasma | [95,148] |
| Carnitine | 2 | Ductal fluid, Cells | [136,137] |
| Citrate | 2 | Serum, Cells | [94,138] |
| Cysteine | 2 | Serum, Cells | [133,136] |
| Formate | 2 | Serum | [91,94] |
| Leucine | 2 | Serum | [94,98] |
| Myo-inositol | 2 | Tissue, Cells | [97,108] |
| Pyruvate | 2 | Serum, Pleural and Peritoneal effusions | [96,124] |
| Serine | 2 | Serum, Cells | [133,136] |
| Spermidine | 2 | Serum, Cells | [140,149] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muthu, M.; Nordström, A. Current Status and Future Prospects of Clinically Exploiting Cancer-specific Metabolism—Why Is Tumor Metabolism Not More Extensively Translated into Clinical Targets and Biomarkers? Int. J. Mol. Sci. 2019, 20, 1385. https://doi.org/10.3390/ijms20061385
Muthu M, Nordström A. Current Status and Future Prospects of Clinically Exploiting Cancer-specific Metabolism—Why Is Tumor Metabolism Not More Extensively Translated into Clinical Targets and Biomarkers? International Journal of Molecular Sciences. 2019; 20(6):1385. https://doi.org/10.3390/ijms20061385
Chicago/Turabian StyleMuthu, Magesh, and Anders Nordström. 2019. "Current Status and Future Prospects of Clinically Exploiting Cancer-specific Metabolism—Why Is Tumor Metabolism Not More Extensively Translated into Clinical Targets and Biomarkers?" International Journal of Molecular Sciences 20, no. 6: 1385. https://doi.org/10.3390/ijms20061385
APA StyleMuthu, M., & Nordström, A. (2019). Current Status and Future Prospects of Clinically Exploiting Cancer-specific Metabolism—Why Is Tumor Metabolism Not More Extensively Translated into Clinical Targets and Biomarkers? International Journal of Molecular Sciences, 20(6), 1385. https://doi.org/10.3390/ijms20061385