HDAC Inhibitors: Therapeutic Potential in Fibrosis-Associated Human Diseases


Abstract
1. Introduction
1.1. Fibrosis
1.2. HDAC and HDAC Inhibitors
1.3. Functional Relevance of HDAC in Fibrogenesis
1.4. HDAC Inhibitors and Their Therapeutic Potential
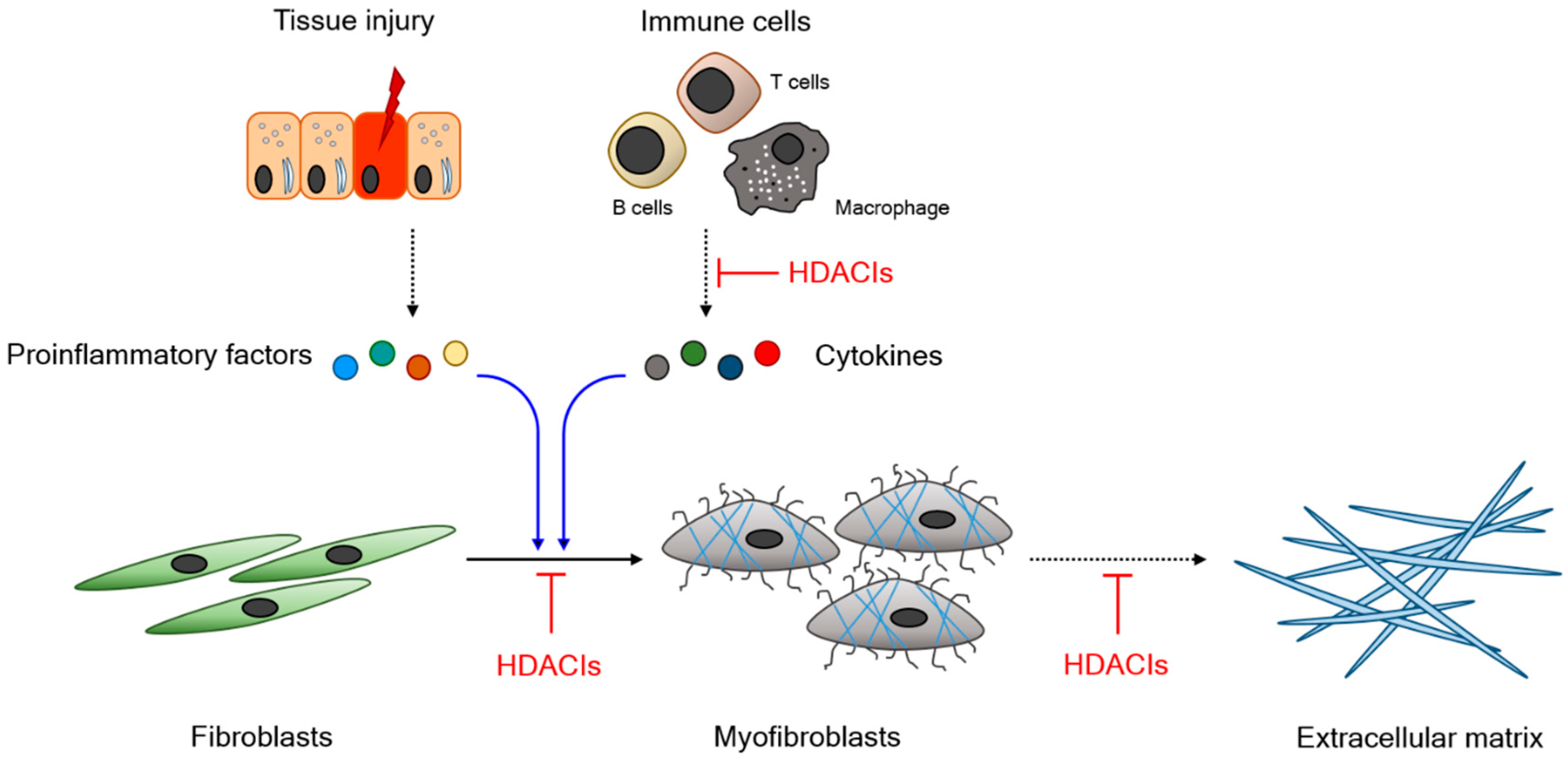
2. Experimental Outcomes of HDAC Inhibitors in Animal Models of Fibrosis-Associated Disease
2.1. Liver Cirrhosis
2.2. Cardiac Fibrosis
2.3. Pulmonary Fibrosis
2.4. Renal Fibrosis
2.5. Miscellaneous Diseases
3. Limitations and Future Perspectives
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AF | Atrial fibrillation |
| BDL | Bile duct ligation |
| CFTR | Cystic fibrosis transmembrane conductance regulator |
| HAT | Histone acetyltransferase |
| HDAC | Histone deacetylase |
| HDACI | Histone deacetylase inhibitor |
| HSC | Hepatic stellate cells |
| IPF | Idiopathic pulmonary fibrosis |
| NHLF | Normal human lung fibroblast |
| PKD | Polycystic kidney disease |
| PTCL | Percutaneous T cell lymphoma |
| STAT | Signal transducer and activator of transcription |
| STZ | Streptozotocin |
| TGF-β | Transforming growth factor β |
| TNF-α | Tumor necrosis factor α |
| TSA | Trichostatin A |
| UUO | Unilateral ureteral obstruction |
References
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Pardali, E.; Sanchez-Duffhues, G.; Gomez-Puerto, M.C.; Ten Dijke, P. TGF-beta-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases. Int. J. Mol. Sci. 2017, 18, 2157. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Cong, M.; Paik, Y.; Scholten, D.; Jiang, C.; Benner, C.; Iwaisako, K.; Moore-Morris, T.; Scott, B.; Tsukamoto, H.; et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Nat. Acad. Sic. USA 2012, 109, 9448–9453. [Google Scholar] [CrossRef] [PubMed]
- Mehrad, B.; Burdick, M.D.; Wandersee, N.J.; Shahir, K.S.; Zhang, L.; Simpson, P.M.; Strieter, R.M.; Field, J.J. Circulating fibrocytes as biomarkers of impaired lung function in adults with sickle cell disease. Blood Adv. 2017, 1, 2217–2224. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.M.; Seki, E. TNFalpha in liver fibrosis. Curr. Pathobiol. Rep. 2015, 3, 253–261. [Google Scholar] [CrossRef]
- Klinkhammer, B.M.; Floege, J.; Boor, P. PDGF in organ fibrosis. Mol. Asp. Med. 2018, 62, 44–62. [Google Scholar] [CrossRef]
- Strutz, F.; Zeisberg, M.; Hemmerlein, B.; Sattler, B.; Hummel, K.; Becker, V.; Muller, G.A. Basic fibroblast growth factor expression is increased in human renal fibrogenesis and may mediate autocrine fibroblast proliferation. Kidney Int. 2000, 57, 1521–1538. [Google Scholar] [CrossRef]
- Toda, N.; Mukoyama, M.; Yanagita, M.; Yokoi, H. CTGF in kidney fibrosis and glomerulonephritis. Inflamm. Regen. 2018, 38, 14. [Google Scholar] [CrossRef]
- Yazdani, S.; Bansal, R.; Prakash, J. Drug targeting to myofibroblasts: Implications for fibrosis and cancer. Adv. Drug Deliv. Rev. 2017, 121, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Walton, K.L.; Johnson, K.E.; Harrison, C.A. Targeting TGF-beta Mediated SMAD Signaling for the Prevention of Fibrosis. Front. Pharmacol. 2017, 8, 461. [Google Scholar] [CrossRef]
- Lawrence, J.; Nho, R. The Role of the Mammalian Target of Rapamycin (mTOR) in Pulmonary Fibrosis. Int. J. Mol. Sci. 2018, 19, 778. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell. Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Eom, G.H.; Kook, H. Posttranslational modifications of histone deacetylases: Implications for cardiovascular diseases. Pharm. Ther. 2014, 143, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Eom, G.H.; Kook, H. Role of histone deacetylase 2 and its posttranslational modifications in cardiac hypertrophy. BMB Rep. 2015, 48, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Hammond, C.M.; Stromme, C.B.; Huang, H.; Patel, D.J.; Groth, A. Histone chaperone networks shaping chromatin function. Nat. Rev. Mol. Cell. Biol. 2017, 18, 141–158. [Google Scholar] [CrossRef]
- Hull, E.E.; Montgomery, M.R.; Leyva, K.J. HDAC Inhibitors as Epigenetic Regulators of the Immune System: Impacts on Cancer Therapy and Inflammatory Diseases. Biomed. Res. Int. 2016, 2016, 8797206. [Google Scholar] [CrossRef]
- Carta, S.; Tassi, S.; Semino, C.; Fossati, G.; Mascagni, P.; Dinarello, C.A.; Rubartelli, A. Histone deacetylase inhibitors prevent exocytosis of interleukin-1beta-containing secretory lysosomes: Role of microtubules. Blood 2006, 108, 1618–1626. [Google Scholar] [CrossRef]
- Makki, M.S.; Haqqi, T.M. Histone deacetylase inhibitor vorinostat (SAHA, MK0683) perturb miR-9-MCPIP1 axis to block IL-1beta-induced IL-6 expression in human OA chondrocytes. Connect. Tissue Res. 2017, 58, 64–75. [Google Scholar] [CrossRef]
- Glauben, R.; Sonnenberg, E.; Wetzel, M.; Mascagni, P.; Siegmund, B. Histone deacetylase inhibitors modulate interleukin 6-dependent CD4+ T cell polarization in vitro and in vivo. J. Biol. Chem. 2014, 289, 6142–6151. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Shan, L.; Schiller, P.W.; Mai, A.; Peng, T. Histone deacetylase-3 activation promotes tumor necrosis factor-alpha (TNF-alpha) expression in cardiomyocytes during lipopolysaccharide stimulation. J. Biol. Chem. 2010, 285, 9429–9436. [Google Scholar] [CrossRef] [PubMed]
- Klampfer, L.; Huang, J.; Swaby, L.A.; Augenlicht, L. Requirement of histone deacetylase activity for signaling by STAT1. J. Biol. Chem. 2004, 279, 30358–30368. [Google Scholar] [CrossRef] [PubMed]
- Angiolilli, C.; Kabala, P.A.; Grabiec, A.M.; Van Baarsen, I.M.; Ferguson, B.S.; Garcia, S.; Malvar Fernandez, B.; McKinsey, T.A.; Tak, P.P.; Fossati, G.; et al. Histone deacetylase 3 regulates the inflammatory gene expression programme of rheumatoid arthritis fibroblast-like synoviocytes. Ann. Rheum. Dis. 2017, 76, 277–285. [Google Scholar] [CrossRef]
- Guo, W.; Shan, B.; Klingsberg, R.C.; Qin, X.; Lasky, J.A. Abrogation of TGF-beta1-induced fibroblast-myofibroblast differentiation by histone deacetylase inhibition. Am. J. Phys. Lung Cell. Mol. Phys. 2009, 297, L864–L870. [Google Scholar] [CrossRef]
- Glenisson, W.; Castronovo, V.; Waltregny, D. Histone deacetylase 4 is required for TGFbeta1-induced myofibroblastic differentiation. Biochim. Biophys. Acta. 2007, 1773, 1572–1582. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Zhuang, Y.; Shan, B.; Danchuk, S.; Luo, F.; Korfei, M.; Guenther, A.; Lasky, J.A. Tubastatin ameliorates pulmonary fibrosis by targeting the TGFbeta-PI3K-Akt pathway. PLoS ONE 2017, 12, e0186615. [Google Scholar] [CrossRef]
- Zheng, Y.; Khan, Z.; Zanfagnin, V.; Correa, L.F.; Delaney, A.A.; Daftary, G.S. Epigenetic Modulation of Collagen 1A1: Therapeutic Implications in Fibrosis and Endometriosis. Biol. Reprod. 2016, 94, 87. [Google Scholar] [CrossRef]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam. Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef]
- Zhao, H.; Yu, Z.; Zhao, L.; He, M.; Ren, J.; Wu, H.; Chen, Q.; Yao, W.; Wei, M. HDAC2 overexpression is a poor prognostic factor of breast cancer patients with increased multidrug resistance-associated protein expression who received anthracyclines therapy. Jpn. J. Clin. Oncol. 2016, 46, 893–902. [Google Scholar] [CrossRef]
- Zhou, L.; Xu, X.; Liu, H.; Hu, X.; Zhang, W.; Ye, M.; Zhu, X. Prognosis Analysis of Histone Deacetylases mRNA Expression in Ovarian Cancer Patients. J. Cancer 2018, 9, 4547–4555. [Google Scholar] [CrossRef] [PubMed]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef]
- Findeisen, H.M.; Gizard, F.; Zhao, Y.; Qing, H.; Heywood, E.B.; Jones, K.L.; Cohn, D.; Bruemmer, D. Epigenetic regulation of vascular smooth muscle cell proliferation and neointima formation by histone deacetylase inhibition. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hu, X.; Jiang, H. HDAC inhibition: A novel therapeutic target for attenuating myocardial ischemia and reperfusion injury by reversing cardiac remodeling. Int. J. Cardiol. 2015, 190, 126–127. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.S.; Zhang, R.; Wang, G.; Zhang, Y.F. The development prospection of HDAC inhibitors as a potential therapeutic direction in Alzheimer’s disease. Transl. Neurodegener. 2017, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, H.; Kobayashi, T.; Azuma, A. Idiopathic Pulmonary Fibrosis: Treatment and Prognosis. Clin. Med. Insights Circ. Respir. Pulm. Med. 2015, 9, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Park, K.C.; Park, J.H.; Jeon, J.Y.; Kim, S.Y.; Kim, J.M.; Lim, C.Y.; Lee, T.H.; Kim, H.K.; Lee, H.G.; Kim, S.M.; et al. A new histone deacetylase inhibitor improves liver fibrosis in BDL rats through suppression of hepatic stellate cells. Br. J. Pharmacol. 2014, 171, 4820–4830. [Google Scholar] [CrossRef]
- Mannaerts, I.; Eysackers, N.; Onyema, O.O.; Van Beneden, K.; Valente, S.; Mai, A.; Odenthal, M.; van Grunsven, L.A. Class II HDAC inhibition hampers hepatic stellate cell activation by induction of microRNA-29. PLoS ONE 2013, 8, e55786. [Google Scholar] [CrossRef]
- Aher, J.S.; Khan, S.; Jain, S.; Tikoo, K.; Jena, G. Valproate ameliorates thioacetamide-induced fibrosis by hepatic stellate cell inactivation. Hum. Exp. Toxicol. 2015, 34, 44–55. [Google Scholar] [CrossRef]
- Kee, H.J.; Sohn, I.S.; Nam, K.I.; Park, J.E.; Qian, Y.R.; Yin, Z.; Ahn, Y.; Jeong, M.H.; Bang, Y.J.; Kim, N.; et al. Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation 2006, 113, 51–59. [Google Scholar] [CrossRef]
- Gallo, P.; Latronico, M.V.; Gallo, P.; Grimaldi, S.; Borgia, F.; Todaro, M.; Jones, P.; Gallinari, P.; De Francesco, R.; Ciliberto, G.; et al. Inhibition of class I histone deacetylase with an apicidin derivative prevents cardiac hypertrophy and failure. Cardiovasc. Res. 2008, 80, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Tannous, P.; Lu, G.; Berenji, K.; Rothermel, B.A.; Olson, E.N.; Hill, J.A. Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation 2006, 113, 2579–2588. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.K.; Eom, G.H.; Kee, H.J.; Kim, H.S.; Choi, W.Y.; Nam, K.I.; Ma, J.S.; Kook, H. Sodium valproate, a histone deacetylase inhibitor, but not captopril, prevents right ventricular hypertrophy in rats. Circ. J. 2010, 74, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Levin, M.D.; Petrenko, N.B.; Lu, M.M.; Wang, T.; Yuan, L.J.; Stout, A.L.; Epstein, J.A.; Patel, V.V. Histone-deacetylase inhibition reverses atrial arrhythmia inducibility and fibrosis in cardiac hypertrophy independent of angiotensin. J. Mol. Cell. Cardiol. 2008, 45, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; LaCanna, R.; Powers, J.C.; Vrakas, C.; Liu, F.; Berretta, R.; Chacko, G.; Holten, J.; Jadiya, P.; Wang, T.; et al. Class I Histone Deacetylase Inhibition for the Treatment of Sustained Atrial Fibrillation. J. Pharmacol. Exp. Ther. 2016, 358, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Sanders, Y.Y.; Hagood, J.S.; Liu, H.; Zhang, W.; Ambalavanan, N.; Thannickal, V.J. Histone deacetylase inhibition promotes fibroblast apoptosis and ameliorates pulmonary fibrosis in mice. Eur. Respir. J. 2014, 43, 1448–1458. [Google Scholar] [CrossRef]
- Coward, W.R.; Watts, K.; Feghali-Bostwick, C.A.; Knox, A.; Pang, L. Defective histone acetylation is responsible for the diminished expression of cyclooxygenase 2 in idiopathic pulmonary fibrosis. Mol. Cell. Biol. 2009, 29, 4325–4339. [Google Scholar] [CrossRef]
- Wu, W.P.; Tsai, Y.G.; Lin, T.Y.; Wu, M.J.; Lin, C.Y. The attenuation of renal fibrosis by histone deacetylase inhibitors is associated with the plasticity of FOXP3(+)IL-17(+) T cells. BMC Nephrol. 2017, 18, 225. [Google Scholar] [CrossRef]
- Tung, C.W.; Hsu, Y.C.; Cai, C.J.; Shih, Y.H.; Wang, C.J.; Chang, P.J.; Lin, C.L. Trichostatin A ameliorates renal tubulointerstitial fibrosis through modulation of the JNK-dependent Notch-2 signaling pathway. Sci. Rep. 2017, 7, 14495. [Google Scholar] [CrossRef]
- Marumo, T.; Hishikawa, K.; Yoshikawa, M.; Hirahashi, J.; Kawachi, S.; Fujita, T. Histone deacetylase modulates the proinflammatory and -fibrotic changes in tubulointerstitial injury. Am. J. Phys. Renal. Phys. 2010, 298, F133–F141. [Google Scholar] [CrossRef]
- Choi, H.S.; Song, J.H.; Kim, I.J.; Joo, S.Y.; Eom, G.H.; Kim, I.; Cha, H.; Cho, J.M.; Ma, S.K.; Kim, S.W.; et al. Histone deacetylase inhibitor, CG200745 attenuates renal fibrosis in obstructive kidney disease. Sci. Rep. 2018, 8, 11546. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.; Oh, E.Y.; Seo, J.Y.; Yu, M.R.; Kim, Y.O.; Ha, H.; Lee, H.B. Histone deacetylase-2 is a key regulator of diabetes- and transforming growth factor-beta1-induced renal injury. Am. J. Phys. Renal. Phys. 2009, 297, F729–F739. [Google Scholar] [CrossRef]
- Barrio, E.; Tome, S.; Rodriguez, I.; Gude, F.; Sanchez-Leira, J.; Perez-Becerra, E.; Gonzalez-Quintela, A. Liver disease in heavy drinkers with and without alcohol withdrawal syndrome. Alcohol. Clin. Exp. Res. 2004, 28, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, F. Alcoholic hepatitis. N. Engl. J. Med. 2009, 361, 1512–1513. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.J.; Yang, H.I.; Wu, H.C.; Liu, J.; Wang, L.Y.; Lu, S.N.; Lee, M.H.; Jen, C.L.; You, S.L.; Santella, R.M.; et al. Aflatoxin B1 exposure increases the risk of cirrhosis and hepatocellular carcinoma in chronic hepatitis B virus carriers. Int. J. Cancer 2017, 141, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Perz, J.F.; Armstrong, G.L.; Farrington, L.A.; Hutin, Y.J.; Bell, B.P. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J. Hepatol. 2006, 45, 529–538. [Google Scholar] [CrossRef]
- Francis Stuart, S.D.; De Jesus, N.M.; Lindsey, M.L.; Ripplinger, C.M. The crossroads of inflammation, fibrosis, and arrhythmia following myocardial infarction. J. Mol. Cell. Cardiol. 2016, 91, 114–122. [Google Scholar] [CrossRef]
- Everett, T.H.t.; Olgin, J.E. Atrial fibrosis and the mechanisms of atrial fibrillation. Heart Rhythm. 2007, 4, S24–S27. [Google Scholar] [CrossRef]
- Burstein, B.; Nattel, S. Atrial fibrosis: Mechanisms and clinical relevance in atrial fibrillation. J. Am. Coll. Cardiol. 2008, 51, 802–809. [Google Scholar] [CrossRef]
- January, C.T.; Wann, L.S.; Alpert, J.S.; Calkins, H.; Cigarroa, J.E.; Cleveland, J.C., Jr.; Conti, J.B.; Ellinor, P.T.; Ezekowitz, M.D.; Field, M.E.; et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: A report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the Heart Rhythm Society. Circulation 2014, 130, e199–e267. [Google Scholar] [CrossRef] [PubMed]
- Kotecha, D.; Piccini, J.P. Atrial fibrillation in heart failure: What should we do? Eur. Heart J. 2015, 36, 3250–3257. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Kook, T.; Min, H.K.; Kwon, D.H.; Cho, Y.K.; Kim, M.; Shin, S.; Joung, H.; Jeong, S.H.; Lee, S.; et al. PP2A negatively regulates the hypertrophic response by dephosphorylating HDAC2 S394 in the heart. Exp. Mol. Med. 2018, 50, 83. [Google Scholar] [CrossRef] [PubMed]
- Eom, G.H.; Cho, Y.K.; Ko, J.H.; Shin, S.; Choe, N.; Kim, Y.; Joung, H.; Kim, H.S.; Nam, K.I.; Kee, H.J.; et al. Casein kinase-2alpha1 induces hypertrophic response by phosphorylation of histone deacetylase 2 S394 and its activation in the heart. Circulation 2011, 123, 2392–2403. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; McKinsey, T.A.; Zhang, C.L.; Richardson, J.A.; Hill, J.A.; Olson, E.N. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol. Cell. Biol. 2004, 24, 8467–8476. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.L.; Davis, C.A.; Potthoff, M.J.; Haberland, M.; Fielitz, J.; Qi, X.; Hill, J.A.; Richardson, J.A.; Olson, E.N. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007, 21, 1790–1802. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, F.; Kai, H.; Tokuda, K.; Kai, M.; Takeshita, A.; Egashira, K.; Imaizumi, T. Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation 2002, 106, 130–135. [Google Scholar] [CrossRef]
- Reed, A.L.; Tanaka, A.; Sorescu, D.; Liu, H.; Jeong, E.M.; Sturdy, M.; Walp, E.R.; Dudley, S.C., Jr.; Sutliff, R.L. Diastolic dysfunction is associated with cardiac fibrosis in the senescence-accelerated mouse. Am. J. Phys. 2011, 301, H824–H831. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.F.; Hammill, B.G.; O’Connor, C.M.; Schulman, K.A.; Curtis, L.H.; Fonarow, G.C. Clinical effectiveness of beta-blockers in heart failure: Findings from the OPTIMIZE-HF (Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients with Heart Failure) Registry. J. Am. Coll. Cardiol. 2009, 53, 184–192. [Google Scholar] [CrossRef]
- Yusuf, S.; Pfeffer, M.A.; Swedberg, K.; Granger, C.B.; Held, P.; McMurray, J.J.; Michelson, E.L.; Olofsson, B.; Ostergren, J.; Investigators, C.; et al. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: The CHARM-Preserved Trial. Lancet 2003, 362, 777–781. [Google Scholar] [CrossRef]
- Massie, B.M.; Carson, P.E.; McMurray, J.J.; Komajda, M.; McKelvie, R.; Zile, M.R.; Anderson, S.; Donovan, M.; Iverson, E.; Staiger, C.; et al. Irbesartan in patients with heart failure and preserved ejection fraction. N. Engl. J. Med. 2008, 359, 2456–2467. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Pfeffer, M.A.; Assmann, S.F.; Boineau, R.; Anand, I.S.; Claggett, B.; Clausell, N.; Desai, A.S.; Diaz, R.; Fleg, J.L.; et al. Spironolactone for heart failure with preserved ejection fraction. N. Engl. J. Med. 2014, 370, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.J.; van Ballegoij, J.J. Treatment of heart failure with normal ejection fraction: An inconvenient truth! J. Am. Coll. Cardiol. 2010, 55, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Meraviglia, V.; Bocchi, L.; Sacchetto, R.; Florio, M.C.; Motta, B.M.; Corti, C.; Weichenberger, C.X.; Savi, M.; D’Elia, Y.; Rosato-Siri, M.D.; et al. HDAC Inhibition Improves the Sarcoendoplasmic Reticulum Ca(2+)-ATPase Activity in Cardiac Myocytes. Int. J. Mol. Sci. 2018, 19, 419. [Google Scholar] [CrossRef] [PubMed]
- Eom, G.H.; Nam, Y.S.; Oh, J.G.; Choe, N.; Min, H.K.; Yoo, E.K.; Kang, G.; Nguyen, V.H.; Min, J.J.; Kim, J.K.; et al. Regulation of acetylation of histone deacetylase 2 by p300/CBP-associated factor/histone deacetylase 5 in the development of cardiac hypertrophy. Circ. Res. 2014, 114, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Kim, M.; Min, H.-K.; Lee, Y.-U.; Kwon, D.-H.; Lee, M.; Lee, S.; Kook, T.; Joung, H.; Nam, K.-I.; et al. Inhibition of heat shock protein 70 blocks the development of cardiac hypertrophy by modulating the phosphorylation of histone deacetylase 2. Cardiovasc. Res. 2018. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Hill, J.A. Inhibition of hypertrophy is a good therapeutic strategy in ventricular pressure overload. Circulation 2015, 131, 1435–1447. [Google Scholar] [CrossRef]
- Wilson, M.S.; Wynn, T.A. Pulmonary fibrosis: Pathogenesis, etiology and regulation. Mucosal. Immunol. 2009, 2, 103–121. [Google Scholar] [CrossRef]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers. 2017, 3, 17074. [Google Scholar] [CrossRef]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Timoshanko, J.R.; Kitching, A.R.; Semple, T.J.; Tipping, P.G.; Holdsworth, S.R. A pathogenetic role for mast cells in experimental crescentic glomerulonephritis. J. Am. Soc. Nephrol. 2006, 17, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, A.; Pires, M.J.; Oliveira, P.A. Pathophysiological Mechanisms of Renal Fibrosis: A Review of Animal Models and Therapeutic Strategies. In Vivo 2017, 31, 1–22. [Google Scholar] [CrossRef]
- Sakamoto-Ihara, T.; Suzuki, Y.; Kurusu, A.; Yamashita, M.; Horikoshi, S.; Tomino, Y. Possible involvement of mast cells in renal fibrosis in patients with IgA nephropathy. Inflamm. Res. 2007, 56, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Nakamura, T.; Noble, N.A.; Ruoslahti, E.; Border, W.A. Expression of transforming growth factor beta is elevated in human and experimental diabetic nephropathy. Proc. N. Acad. Sci. USA 1993, 90, 1814–1818. [Google Scholar] [CrossRef]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef]
- Duffield, J.S. Beyond EMT: Epithelial STAT3 as a Central Regulator of Fibrogenesis. J. Am. Soc. Nephrol. 2016, 27, 3502–3504. [Google Scholar] [CrossRef]
- Chevalier, R.L.; Forbes, M.S.; Thornhill, B.A. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 2009, 75, 1145–1152. [Google Scholar] [CrossRef]
- Pang, M.; Kothapally, J.; Mao, H.; Tolbert, E.; Ponnusamy, M.; Chin, Y.E.; Zhuang, S. Inhibition of histone deacetylase activity attenuates renal fibroblast activation and interstitial fibrosis in obstructive nephropathy. Am. J. Phys. Renal. Phys. 2009, 297, F996–F1005. [Google Scholar] [CrossRef]
- Wilson, P.D. Polycystic kidney disease. N. Engl. J. Med. 2004, 350, 151–164. [Google Scholar] [CrossRef]
- Kim, J.A.; Blumenfeld, J.D.; Chhabra, S.; Dutruel, S.P.; Thimmappa, N.D.; Bobb, W.O.; Donahue, S.; Rennert, H.E.; Tan, A.Y.; Giambrone, A.E.; et al. Pancreatic Cysts in Autosomal Dominant Polycystic Kidney Disease: Prevalence and Association with PKD2 Gene Mutations. Radiology 2016, 280, 762–770. [Google Scholar] [CrossRef]
- Mochizuki, T.; Wu, G.; Hayashi, T.; Xenophontos, S.L.; Veldhuisen, B.; Saris, J.J.; Reynolds, D.M.; Cai, Y.; Gabow, P.A.; Pierides, A.; et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 1996, 272, 1339–1342. [Google Scholar] [CrossRef] [PubMed]
- Boucher, C.; Sandford, R. Autosomal dominant polycystic kidney disease (ADPKD, MIM 173900, PKD1 and PKD2 genes, protein products known as polycystin-1 and polycystin-2). Eur. J. Hum. Gen. 2004, 12, 347–354. [Google Scholar] [CrossRef]
- Porath, B.; Gainullin, V.G.; Cornec-Le Gall, E.; Dillinger, E.K.; Heyer, C.M.; Hopp, K.; Edwards, M.E.; Madsen, C.D.; Mauritz, S.R.; Banks, C.J.; et al. Mutations in GANAB, Encoding the Glucosidase IIalpha Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease. Am. J. Hum. Gen. 2016, 98, 1193–1207. [Google Scholar] [CrossRef] [PubMed]
- Cebotaru, L.; Liu, Q.; Yanda, M.K.; Boinot, C.; Outeda, P.; Huso, D.L.; Watnick, T.; Guggino, W.B.; Cebotaru, V. Inhibition of histone deacetylase 6 activity reduces cyst growth in polycystic kidney disease. Kidney Int. 2016, 90, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Yanda, M.K.; Liu, Q.; Cebotaru, V.; Guggino, W.B.; Cebotaru, L. Histone deacetylase 6 inhibition reduces cysts by decreasing cAMP and Ca(2+) in knock-out mouse models of polycystic kidney disease. J. Biol. Chem. 2017, 292, 17897–17908. [Google Scholar] [CrossRef]
- Cao, Y.; Semanchik, N.; Lee, S.H.; Somlo, S.; Barbano, P.E.; Coifman, R.; Sun, Z. Chemical modifier screen identifies HDAC inhibitors as suppressors of PKD models. Proc. N. Acad. Sci. USA 2009, 106, 21819–21824. [Google Scholar] [CrossRef] [PubMed]
- Cutting, G.R. Cystic fibrosis genetics: From molecular understanding to clinical application. Nat. Rev. Gen. 2015, 16, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Klimova, B.; Kuca, K.; Novotny, M.; Maresova, P. Cystic Fibrosis Revisited—A Review Study. Med. Chem. 2017, 13, 102–109. [Google Scholar] [CrossRef]
- Bodas, M.; Mazur, S.; Min, T.; Vij, N. Inhibition of histone-deacetylase activity rescues inflammatory cystic fibrosis lung disease by modulating innate and adaptive immune responses. Respir. Res. 2018, 19, 2. [Google Scholar] [CrossRef] [PubMed]
- Pankow, S.; Bamberger, C.; Calzolari, D.; Martinez-Bartolome, S.; Lavallee-Adam, M.; Balch, W.E.; Yates, J.R., 3rd. F508 CFTR interactome remodelling promotes rescue of cystic fibrosis. Nature 2015, 528, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Hutt, D.M.; Herman, D.; Rodrigues, A.P.; Noel, S.; Pilewski, J.M.; Matteson, J.; Hoch, B.; Kellner, W.; Kelly, J.W.; Schmidt, A.; et al. Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat. Chem. Biol. 2010, 6, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Diegelmann, R.F.; Cohen, I.K.; McCoy, B.J. Growth kinetics and collagen synthesis of normal skin, normal scar and keloid fibroblasts in vitro. J. Cell. Physiol. 1979, 98, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Khaw, P.T.; Chang, L.; Wong, T.T.; Mead, A.; Daniels, J.T.; Cordeiro, M.F. Modulation of wound healing after glaucoma surgery. Curr. Opin. Ophthalmol. 2001, 12, 143–148. [Google Scholar] [CrossRef]
- Jung, J.L.; Isida-Llerandi, C.G.; Lazcano-Gomez, G.; SooHoo, J.R.; Kahook, M.Y. Intraocular Pressure Control after Trabeculectomy, Phacotrabeculectomy and Phacoemulsification in a Hispanic Population. J. Curr. Glaucoma. Pract. 2014, 8, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Palanca-Capistrano, A.M.; Hall, J.; Cantor, L.B.; Morgan, L.; Hoop, J.; WuDunn, D. Long-term outcomes of intraoperative 5-fluorouracil versus intraoperative mitomycin C in primary trabeculectomy surgery. Ophthalmology 2009, 116, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Conlon, R.; Saheb, H.; Ahmed, I.I.K. Glaucoma treatment trends: A review. Can. J. Ophthalmol. 2017, 52, 114–124. [Google Scholar] [CrossRef]
- Sung, M.S.; Eom, G.H.; Kim, S.J.; Kim, S.Y.; Heo, H.; Park, S.W. Trichostatin A Ameliorates Conjunctival Fibrosis in a Rat Trabeculectomy Model. Investig. Ophthalmol. Vis. Sci. 2018, 59, 3115–3123. [Google Scholar] [CrossRef]
- Fitzgerald O’Connor, E.J.; Badshah, I.I.; Addae, L.Y.; Kundasamy, P.; Thanabalasingam, S.; Abioye, D.; Soldin, M.; Shaw, T.J. Histone deacetylase 2 is upregulated in normal and keloid scars. J. Investig. Dermatol. 2012, 132, 1293–1296. [Google Scholar] [CrossRef]
- Russell, S.B.; Russell, J.D.; Trupin, K.M.; Gayden, A.E.; Opalenik, S.R.; Nanney, L.B.; Broquist, A.H.; Raju, L.; Williams, S.M. Epigenetically altered wound healing in keloid fibroblasts. J. Investig. Dermatol. 2010, 130, 2489–2496. [Google Scholar] [CrossRef]
- Diao, J.S.; Xia, W.S.; Yi, C.G.; Yang, Y.; Zhang, X.; Xia, W.; Shu, M.G.; Wang, Y.M.; Gui, L.; Guo, S.Z. Histone deacetylase inhibitor reduces hypertrophic scarring in a rabbit ear model. Plast. Reconstr. Surg. 2013, 132, 61e–69e. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, M.C.; Kim, W.; Bariteau, M.; Davis, T.; Vempati, S.; Minehart, J.; Witkin, M.; Qi, J.; Krivtsov, A.V.; Bradner, J.E.; et al. Selective Inhibition of HDAC1 and HDAC2 as a Potential Therapeutic Option for B-ALL. Clin. Cancer Res. 2015, 21, 2348–2358. [Google Scholar] [CrossRef] [PubMed]
- Zimberlin, C.D.; Lancini, C.; Sno, R.; Rosekrans, S.L.; McLean, C.M.; Vlaming, H.; van den Brink, G.R.; Bots, M.; Medema, J.P.; Dannenberg, J.H. HDAC1 and HDAC2 collectively regulate intestinal stem cell homeostasis. FASEB J. 2015, 29, 2070–2080. [Google Scholar] [CrossRef] [PubMed]
- Butler, K.V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowski, A.P. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc. 2010, 132, 10842–10846. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.H.; Eom, G.H.; Ko, J.H.; Shin, S.; Joung, H.; Choe, N.; Nam, Y.S.; Min, H.K.; Kook, T.; Yoon, S.; et al. MDM2 E3 ligase-mediated ubiquitination and degradation of HDAC1 in vascular calcification. Nat. Commun. 2016, 7, 10492. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| HDAC Inhibitor | Selectivity | Model | Output (Except Fibrosis) | Reference | |
|---|---|---|---|---|---|
| Liver cirrhosis | SAHA HNHA | pan pan | Bile duct ligation | Improved hepatic function Survival ↑ | [37] |
| MC1568 | HDAC4/5/6 | CCl4 | HSC activation ↓ | [38] | |
| Valproate | pan | thioacetamide | HSC activation ↓ | [39] | |
| Cardiac fibrosis | TSA SK7041 | pan class I | Pressure overload | Heart failure ↓ Cardiac hypertrophy ↓ | [40] |
| Api-D | class I | Pressure overload | Heart failure ↓ Cardiac hypertrophy ↓ | [41] | |
| TSA Scriptaid | pan pan | Pressure overload | Heart failure ↓ Cardiac hypertrophy ↓ | [42] | |
| Valproate | pan | Pressure overload MCT | RV hypertrophy | [43] | |
| TSA | pan | TgHopX | Cx40 ↑ Normalized conduction | [44] | |
| Tacedinaline | class I | TgHopX pacing (dog) | Atrial fibrillation ↓ Immune cell infiltration ↓ | [45] | |
| Lung fibrosis | TSA | pan | TGF-β (NHLF cell) | Myofibroblast differentiation ↓ | [25] |
| SAHA | pan | Bleomycin | Lung compliance ↑ Airway resistance ↓ | [46] | |
| SAHA panobinostat | pan pan | Primary cells from IPF patient | Correction of epigenetic abnormality | [47] | |
| Renal fibrosis | TSA | pan | UUO | Immune cell infiltration ↓ | [48] |
| TSA | pan | UUO | Tubular cell apoptosis ↓ | [49] | |
| Valproate | pan | UUO | Macrophage infiltration ↓ | [50] | |
| CG200745 | pan | UUO | Serum NGAL level ↓ | [51] | |
| TSA Valproate SK7041 | pan pan class I | STZ | Urine protein/Cr ↓ EMT ↓ | [52] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoon, S.; Kang, G.; Eom, G.H. HDAC Inhibitors: Therapeutic Potential in Fibrosis-Associated Human Diseases. Int. J. Mol. Sci. 2019, 20, 1329. https://doi.org/10.3390/ijms20061329
Yoon S, Kang G, Eom GH. HDAC Inhibitors: Therapeutic Potential in Fibrosis-Associated Human Diseases. International Journal of Molecular Sciences. 2019; 20(6):1329. https://doi.org/10.3390/ijms20061329
Chicago/Turabian StyleYoon, Somy, Gaeun Kang, and Gwang Hyeon Eom. 2019. "HDAC Inhibitors: Therapeutic Potential in Fibrosis-Associated Human Diseases" International Journal of Molecular Sciences 20, no. 6: 1329. https://doi.org/10.3390/ijms20061329
APA StyleYoon, S., Kang, G., & Eom, G. H. (2019). HDAC Inhibitors: Therapeutic Potential in Fibrosis-Associated Human Diseases. International Journal of Molecular Sciences, 20(6), 1329. https://doi.org/10.3390/ijms20061329