Curcumin and its Potential for Systemic Targeting of Inflamm-Aging and Metabolic Reprogramming in Cancer

 ,
,  ,
,  ,
, 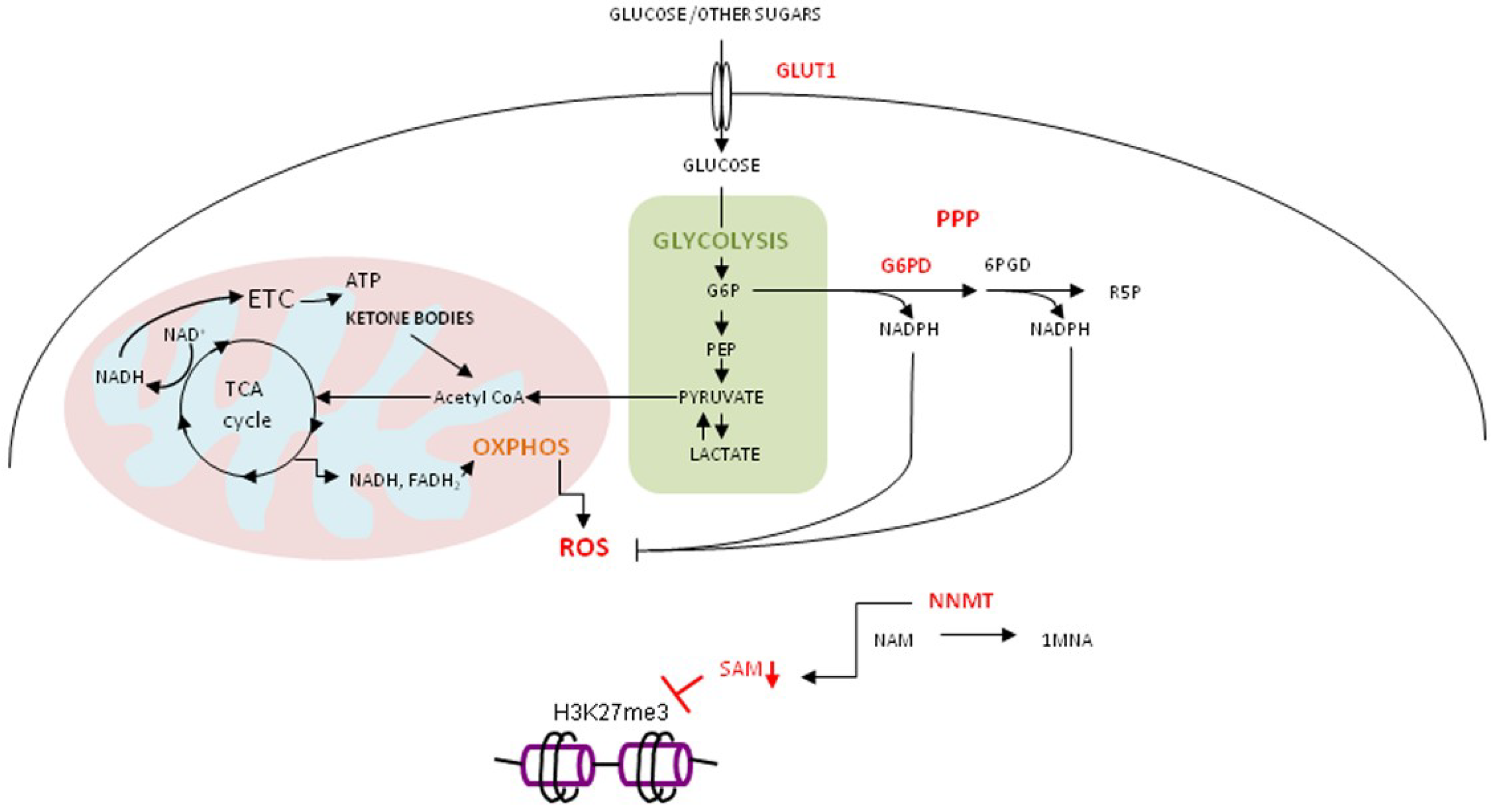{kind=link}
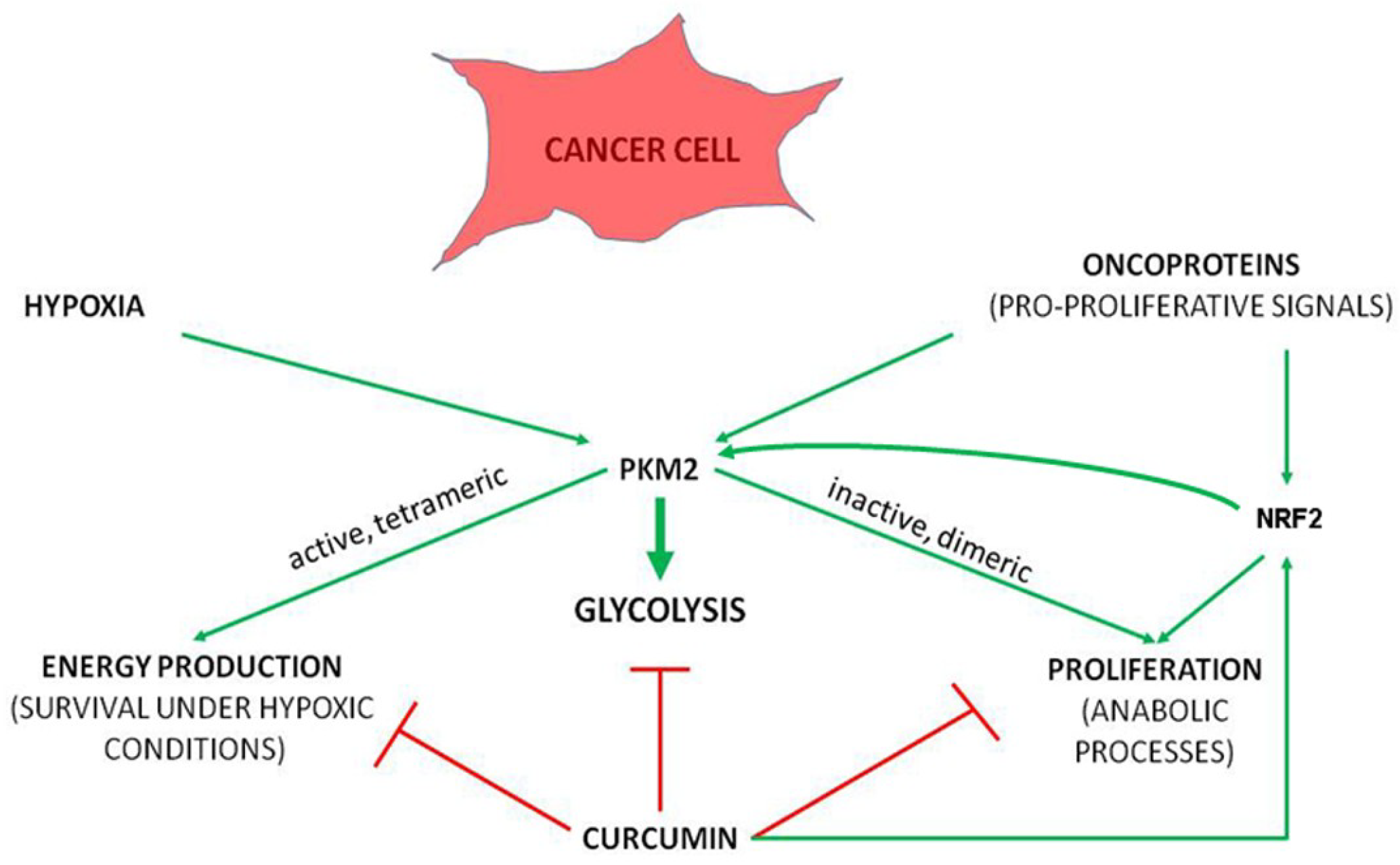{kind=link}
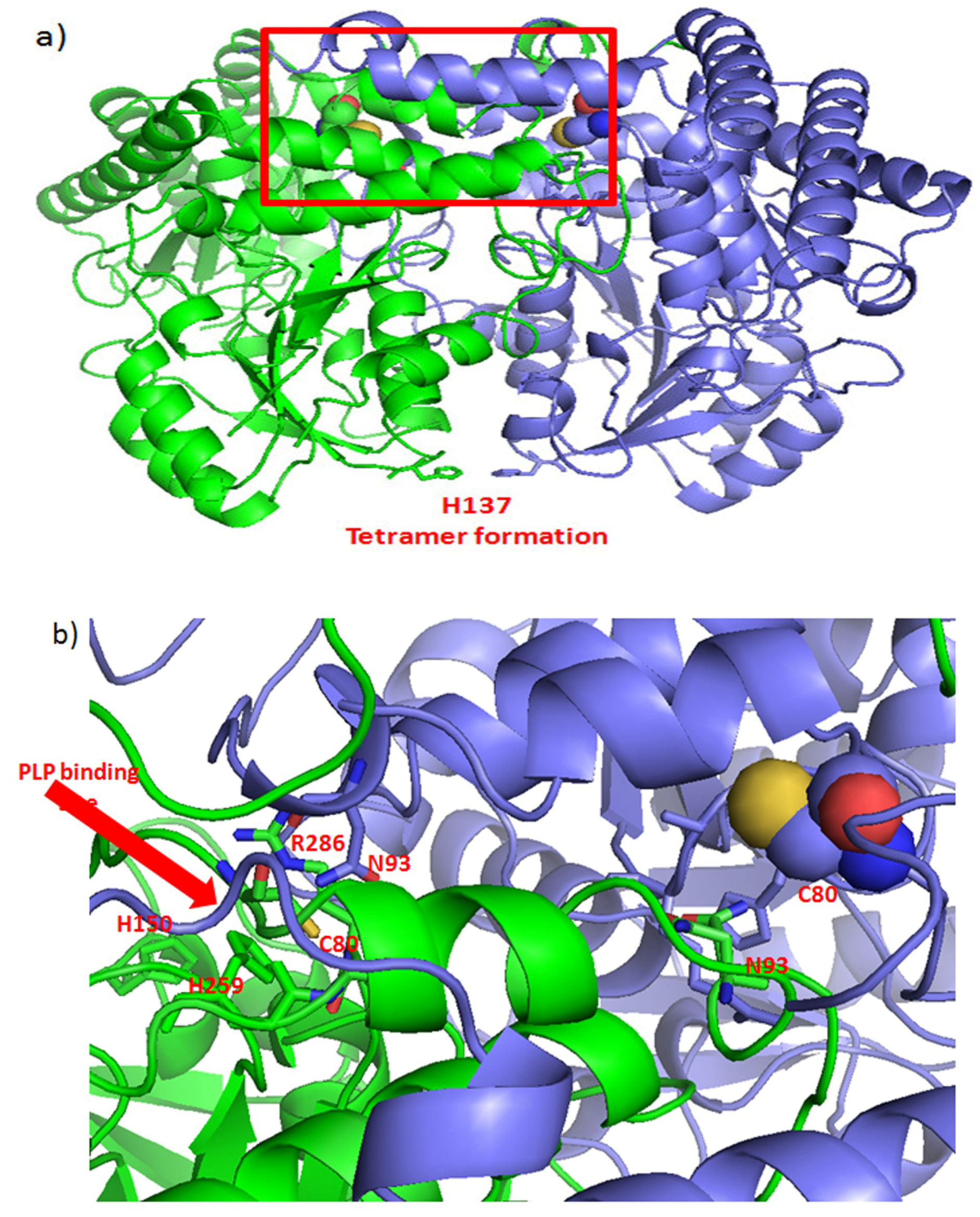{kind=link}
Abstract
1. Introduction
2. Curcumin and Inflamm-aging
2.1. Inflamm-Aging, Interleukin-17 and Curcumin
2.2. Breast Tissue Inflamm-Aging and Curcumin
2.3. Curcumin and the Concept of Network Medicine
3. Oxidative Metabolites of Curcumin
Curcumin and ROS-Producing Compounds
4. Curcumin, Cancer and Metabolic Reprogramming
4.1. Cellular Senescence (CS), Hypoxia and Cancer Metabolic Plasticity
4.2. Curcumin, STAT3, and Modes of Survival
4.3. Oxidative Stress, Curcumin and Gerometabolite NAD+
4.4. Curcumin in Regulation of Metabolic and Stress Response Pathways
4.5. Metabolic Enzymes as Targets for Curcumin Binding
Phosphoglycerate Dehydrogenase and Serine Hydroxymethyltransferase 2
5. Potential for Therapeutic Application?
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 1MNA | 1-Methylnicotinamide |
| 3-PG | 3-Phosphoglycerate |
| AD | Alzheimer’s Disease |
| AMPK | AMP-Activated Kinase |
| ATP | Adenosine Triphosphate |
| CBR1 | Carbonyl Reductase 1 |
| CRP | C-Reactive Protein |
| CS | Cellular Senescence |
| CSC | Cancer Stem Cells |
| ER | Estrogen Receptor |
| FADH2 | Flavin Adenine Dinucleotide |
| G6PD | Glucose-6-Phosphate Dehydrogenase |
| GAPDH | Glyceraldehyde 3-Phosphate Dehydrogenase |
| GM | Glioblastoma Multiforme |
| GLUT 1 | Glucose Transporter 1 |
| GSH | Glutathione |
| GSTP1 | Glutathione-S-Transferase Phi 1 |
| HCC | Hepatocellular Carcinoma |
| HIF-1α | Hypoxia-Inducible Factor 1α |
| HKII | Hexokinase II |
| HNC | Head and Neck Carcinoma |
| IKK b | Inhibitor of Nuclear Factor Kappa-B Kinase Subunit Beta |
| IL-17R | IL-17 Receptor |
| LDHA | L-Lactate Dehydrogenase A |
| MBN | MidBrain Neuron |
| MCP-1 | Monocyte Chemoattractant Protein-1 |
| MDH | Malate Dehydrogenase |
| MMP-2 | Matrix Metalloproteinase 2 |
| NADH/NAD+ | Nicotinamide Adenine Dinucleotide |
| NADPH/NADP+ | Nicotinamide Adenine Dinucleotide Phosphate |
| NAM | Nicotinamide |
| NAMPT | Nicotinamide Phosphoribosyltransferase |
| NF-κB | Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells |
| NNMT | Nicotinamide N-Methyltransferase |
| NOROS | Nitric Oxide and Reactive Oxygen Species |
| NQO-1 | NAD(P)H Quinone Dehydrogenase 1 |
| NRF2 | Nuclear Factor (Erythroid-Derived 2)-Like 2 |
| OS | Overall Survival |
| OXPHOS | Oxidative Phosphorylation |
| PARP-1 | Poly (ADP-Ribose) Polymerase 1 |
| PD | Parkinson’s Disease |
| PEP | Phosphoenolpyruvate |
| PGAM1 | Phosphoglycerate Mutase |
| PKM | Pyruvate Kinase |
| PPP | Pentose Phosphate Pathway |
| PR | Progesterone Receptor |
| R5P | Ribulose 5-Phosphate |
| RFS | Relapse Free Survival |
| ROS | Reactive Oxygen Species |
| SAH | S-Adenosylhomocysteine |
| SAM | S-Adenosylmethionine |
| SEMF | Subepithelial Myofibroblast |
| SHMT2 | Serine Hidroxymethyltransferase 2 |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| TCA | Tricarboxylic Acid |
| TGF-b | Transforming Growth Factor Beta |
| TNF-a | Tumor Necrosis Factor-Alpha |
| VEGF | Vascular Endothelial Growth Factor |
References
- World Health Statistics 2018: Monitoring Health for the SDGs, Sustainable Development Goals; World Health Organization: Geneva, Switzerland, 2018; Licence: CC BY-NC-SA 3.0 IGO; Available online: https://apps.who.int/iris/bitstream/handle/10665/272596/9789241565585-eng.pdf?ua=1 (accessed on 17 January 2019).
- United Nations, Department of Economic and Social Affairs, Population Division. World Population Prospects: The 2017 Revision, Key Findings and Advance Tables; Working Paper No. ESA/P/WP/248; United Nations, Department of Economic and Social Affairs, Population Division, 2017; Available online: https://esa.un.org/unpd/wpp/publications/files/wpp2017_keyfindings.pdf (accessed on 22 January 2019).
- Virchow, R. Die Cellularpathologie in Ihrer Begründung auf Physiologische und Pathologische Gewebelehre; A. Hirschwald: Berlin, Germany, 1859; pp. 174, 441. [Google Scholar]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [PubMed]
- Schäfer, M.; Werner, S. Oxidative stress in normal and impaired wound repair. Pharmacol. Res. 2008, 58, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Kasuya, A.; Tokura, Y. Attempts to accelerate wound healing. J. Dermatol. Sci. 2014, 76, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 2007, 128, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and Age-Related Diseases: Role of inflammation triggers and cytokines. Front. Immunol. 2018, 9, 586. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5900450/ (accessed on 22 November 2018). [CrossRef] [PubMed]
- Michaud, M.; Balardy, L.; Moulis, G.; Gaudin, C.; Peyrot, C.; Vellas, B.; Cesari, M.; Nourhashemi, F. Proinflammatory cytokines, aging, and age-related diseases. J. Am. Med. Dir. Assoc. 2013, 14, 877–882. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Tomorrow; International Agency for Research on Cancer: Lyon, France, 2018; Available online: https://gco.iarc.fr/tomorrow (accessed on 12 January 2019).
- Surh, Y.J.; Lee, E.; Lee, J.M. Chemoprotective properties of some pungent ingredients present in red pepper and ginger. Mutat. Res. 1998, 402, 259–267. [Google Scholar] [CrossRef]
- Lee, W.-H.; Loo, C.-Y.; Bebawy, M.; Luk, F.; Mason, R.S.; Rohanizadeh, R. Curcumin and its derivatives: Their application in neuropharmacology and neuroscience in the 21st century. Curr. Neuropharmacol. 2013, 11, 338–378. [Google Scholar] [CrossRef]
- Esatbeyoglu, T.; Huebbe, P.; Ernst, I.M.A.; Chin, D.; Wagner, A.E.; Rimbach, G. Curcumin-From molecule to biological function. Angew. Chem. Int. Ed. 2012, 51, 5308–5332. [Google Scholar] [CrossRef] [PubMed]
- Global Market Insights. Report ID: GMI788. Available online: https://www.gminsights.com/industry-analysis/curcumin-market (accessed on 20 December 2018).
- Oppenheimer, A. Turmeric (curcumin) in biliary diseases. Lancet 1937, 229, 619–621. [Google Scholar] [CrossRef]
- Ammon, H.P.; Wahl, M.A. Pharmacology of curcuma longa. Planta Med. 1991, 57, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.D.; Smith, G.L.; Hurria, A.; Hortobagyi, G.N.; Buchholz, T.A. Future of cancer incidence in the United States: Burdens upon an aging, changing nation. J. Clin. Oncol. 2009, 27, 2758–2765. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Nolen, S.C.; Evans, M.A.; Fischer, A.; Corrada, M.M.; Kawas, C.H.; Bota, D.A. Cancer-Incidence, prevalence and mortality in the oldest-old. A comprehensive review. Mech. Ageing Dev. 2017, 164, 113–126. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Varkaris, A.; Katsiampoura, A.; Davis, J.S.; Shah, N.; Lam, M.; Frias, R.L.; Ivan, C.; Shimizu, M.; Morris, J.; Menter, D.; et al. Circulating inflammation signature predicts overall survival and relapse-free survival in metastatic colorectal cancer. Br. J. Cancer 2019, 120, 340–345. [Google Scholar] [CrossRef]
- Vainer, N.; Dehlendorff, C.; Johansen, J.S. Systematic literature review of IL-6 as a biomarker or treatment target in patients with gastric, bile duct, pancreatic and colorectal cancer. Oncotarget 2018, 9, 29820–29841. Available online: http://www.oncotarget.com/index.php?journal=oncotarget&page=article&op=view&path[]=25661&pubmed-linkout=1 (accessed on 28 December 2018 ). [CrossRef]
- Sirniö, P.; Väyrynen, J.P.; Klintrup, K.; Mäkelä, J.; Karhu, T.; Herzig, K.H.; Minkkinen, I.; Mäkinen, M.J.; Karttunen, T.J.; Tuomisto, A. Alterations in serum amino-acid profile in the progression of colorectal cancer: Associations with systemic inflammation, tumour stage and patient survival. Br. J. Cancer 2019, 120, 238–246. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation induces neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5260818/ (accessed on 11 January 2019). [PubMed]
- Giunta, B.; Fernandez, F.; Nikolic, W.V.; Obregon, D.; Rrapo, E.; Town, T.; Tan, J. Inflammaging as a prodrome to Alzheimer’s Disease. J. Neuroinflamm. 2008, 5, 51. Available online: https://jneuroinflammation.biomedcentral.com/articles/10.1186/1742-2094-5-51 (accessed on 13 November 2018). [CrossRef] [PubMed]
- Calabrese, V.; Santoro, A.; Monti, D.; Crupi, R.; Di Paola, R.; Latteri, S.; Cuzzocrea, S.; Zappia, M.; Giordano, J.; Calabrese, E.J.; et al. Aging and Parkinson’s disease: Inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic. Biol. Med. 2018, 115, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Boyko, A.A.; Troyanova, N.I.; Kovalenko, E.I.; Sapozhnikov, A.M. Similarity and differences in inflammation-related characteristics of the peripheral immune system of patients with Parkinson’s and Alzheimer’s diseases. Int. J. Mol. Sci. 2017, 18, 2633. Available online: https://www.mdpi.com/1422-0067/18/12/2633 (accessed on 13 November 2018). [CrossRef] [PubMed]
- Xia, Y.; Yeddula, N.; Leblanc, M.; Ke, E.; Zhang, Y.; Oldfield, E.; Shaw, R.J.; Verma, I.M. Reduced cell proliferation by IKK2 depletion in a mouse lung-cancer model. Nat. Cell Biol. 2012, 14, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhao, C.-H.; Sun, G.; Zhang, Z.-W.; Qian, B.-M.; Zhu, Y.-F.; Cai, M.-Y.; Pandey, S.; Zhao, D.; Wang, Y.-W.; et al. IL-17 induces the proliferation and migration of glioma cells through the activation of PI3K/Akt1/NF-κB-p65. Cancer Lett. 2019, 447, 93–104. [Google Scholar] [CrossRef]
- Hata, K.; Andoh, A.; Shimada, M.; Fujino, S.; Bamba, S.; Araki, Y.; Okuno, T.; Fujiyama, Y.; Bamba, T. IL-17 stimulates inflammatory responses via NF-kappaB and MAP kinase pathways in human colonic myofibroblasts. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G1035–G1044. [Google Scholar] [CrossRef]
- Sommer, A.; Maxreiter, F.; Krach, F.; Fadler, T.; Grosch, J.; Maroni, M.; Graef, D.; Eberhardt, E.; Riemenschneider, M.J.; Yeo, G.W.; et al. Th17 lymphocytes induce neuronal cell death in a human iPSC-based model of Parkinson’s disease. Cell Stem Cell 2018, 23, 123–131. [Google Scholar] [CrossRef]
- Zhang, J.; Ke, K.F.; Liu, Z.; Qiu, Y.H.; Peng, Y.P. Th17 cell-mediated neuroinflammation is involved in neurodegeneration of aβ1-42-induced Alzheimer’s disease model rats. PLoS ONE 2013, 8, e75786. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3790825/ (accessed on 3 December 2018). [CrossRef]
- Li, T.-J.; Jiang, Y.-M.; Hu, Y.-F.; Huang, L.; Yu, J.; Zhao, L.-Y.; Deng, H.-J.; Mou, T.-Y.; Liu, H.; Yang, Y.; et al. Interleukin-17-producing neutrophils link inflammatory stimuli to disease progression by promoting angiogenesis in gastric cancer. Clin. Cancer Res. 2017, 23, 1575–1585. [Google Scholar] [CrossRef]
- Lee, M.-H.; Chang, J.T.-C.; Liao, C.-T.; Chen, Y.-S.; Kuo, M.-L.; Shen, C.-R. Interleukin 17 and peripheral IL-17-expressing T cells are negatively correlated with the overall survival of head and neck cancer patients. Oncotarget 2018, 9, 9825–9837. Available online: http://www.oncotarget.com/index.php?journal=oncotarget&page=article&op=view&path[]=23934&pubmed-linkout=1 (accessed on 4 December 2018). [CrossRef] [PubMed]
- Latimer, B.; Ekshyyan, O.; Nathan, N.; Moore-Medlin, T.; Rong, X.; Ma, X.; Khandelwal, A.; Christy, H.T.; Abreo, F.; McClure, G.; et al. Enhanced systemic bioavailability of curcumin through transmucosal administration of a novel microgranular formulation. Anticancer Res. 2015, 35, 6411–6418. Available online: http://ar.iiarjournals.org/content/35/12/6411.long (accessed on 4 December 2018). [PubMed]
- Verma, S.P.; Goldin, B.R.; Lin, P.S. The inhibition of the estrogenic effects of pesticides and environmental chemicals by curcumin and isoflavonoids. Environ. Health Perspect. 1998, 106, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.M.; Shen, Z.Z.; Liu, C.H.; Sartippour, M.R.; Go, V.L.; Heber, D.; Nguyen, M. Curcumin exerts multiple suppressive effects on human breast carcinoma cells. Int. J. Cancer 2002, 98, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Leone, B.A.; Leone, J.; Leone, J.P. Breast cancer is a systemic disease rather than an anatomical process. Breast Cancer Res. Treat. 2017, 161, 619. [Google Scholar] [CrossRef] [PubMed]
- Morris, P.G.; Hudis, C.A.; Giri, D.; Morrow, M.; Falcone, D.J.; Zhou, X.K.; Du, B.; Brogi, E.; Crawford, C.B.; Kopelovich, L.; et al. Inflammation and increased aromatase expression occur in the breasttissue of obese women with breast cancer. Cancer Prev. Res. 2011, 4, 1021–1029. [Google Scholar] [CrossRef]
- Brown, K.A.; Iyengar, N.M.; Zhou, X.K.; Gucalp, A.; Subbaramaiah, K.; Wang, H.; Giri, D.D.; Morrow, M.; Falcone, D.J.; Wendel, N.K.; et al. Menopause is a determinant of breast aromatase expression and its associations with BMI, inflammation, and systemic markers. J. Clin. Endocrinol. Metab. 2017, 102, 1692–1701. [Google Scholar] [CrossRef]
- Key, T.J.; Appleby, P.N.; Reeves, G.K.; Roddam, A.; Dorgan, J.F.; Longcope, C.; Stanczyk, F.Z.; Stephenson, H.E., Jr.; Falk, R.T.; Miller, R.; et al. Body mass index, serum sex hormones, and breast cancer risk in postmenopausal women. J. Natl. Cancer Inst. 2003, 95, 1218–1226. [Google Scholar]
- Nicklas, B.J.; Rogus, E.M.; Colman, E.G.; Goldberg, A.P. Visceral adiposity, increased adipocyte lipolysis, and metabolic dysfunction in obese postmenopausal women. Am. J. Physiol. 1996, 270, E72–E78. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Favelyukis, S.; Nguyen, A.K.; Reichart, D.; Scott, P.A.; Jenn, A.; Liu-Bryan, R.; Glass, C.K.; Neels, J.G.; Olefsky, J.M. A subpopulation of macrophages infiltrates hypertrophic tissue and is activated by free fatty acids via Toll-likereceptors 2 and 4 and JNK-dependent pathways. J. Biol. Chem. 2007, 282, 35279–35292. [Google Scholar] [CrossRef]
- Subbaramaiah, K.; Sue, E.; Bhardwaj, P.; Du, B.; Hudis, C.A.; Giri, D.; Kopelovich, L.; Zhou, X.K.; Dannenberg, A.J. Dietary polyphenols suppress elevated levels of proinflammatory mediators and aromatase in the mammary gland of obese mice. Cancer Prev. Res. 2013, 6, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Devassy, J.G.; Nwachukwu, I.D.; Jones, P.J. Curcumin and cancer: Barriers to obtaining a health claim. Nutr. Rev. 2015, 73, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Long, J.; He, T.; Belshaw, R.; Scott, J. Integrated genomic approaches identify major pathways and upstream regulators in late onset Alzheimer’s disease. Sci. Rep. 2015, 5, 12393. Available online: https://www.nature.com/articles/srep12393 (accessed on 5 December 2018). [CrossRef] [PubMed]
- Ghosh, A.; Roy, A.; Liu, X.; Kordower, J.H.; Mufson, E.J.; Hartley, D.M.; Ghosh, S.; Mosley, R.L.; Gendelman, H.E.; Pahan, K. Selective inhibition of NF-kappaB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 18754–18759. [Google Scholar] [CrossRef]
- Barabási, A.L.; Gulbahce, N.; Loscalzo, J. Network Medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. Available online: https://www.nature.com/articles/nrg2918 (accessed on 10 January 2019). [CrossRef]
- Kunnumakkara, A.B.; Bordoloi, D.; Padmavathi, G.; Monisha, J.; Roy, N.K.; Prasad, S.; Aggarwal, B.B. Curcumin, the golden nutraceutical: Multitargeting for multiple chronic diseases. Br. J. Pharmacol. 2017, 174, 1325–1348. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5429333/ (accessed on 26 February 2019). [CrossRef] [PubMed]
- Abdolahi, M.; Tafakhori, A.; Togha, M.; Okhovat, A.A.; Siassi, F.; Eshraghian, M.R.; Sedighiyan, M.; Djalali, M.; Mohammadzadeh Honarvar, N.; Djalali, M. The synergistic effects of ω-3 fatty acids and nano-curcumin supplementation on tumor necrosis factor (TNF)-α gene expression and serum level in migraine patients. Immunogenetics 2017, 69, 371–378. [Google Scholar] [CrossRef]
- Panahi, Y.; Hosseini, M.S.; Khalili, N.; Naimi, E.; Simental-Mendía, L.E.; Majeed, M.; Sahebkar, A. Effects of curcumin on serum cytokine concentrations in subjects with metabolic syndrome: A post-hoc analysis of a randomized controlled trial. Biomed. Pharmacother. 2016, 82, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Hewlings, S.J.; Kalman, D.S. Curcumin: A review of its’ effects on human health. Foods 2017, 6, 92. Available online: https://www.mdpi.com/2304-8158/6/10/92 (accessed on 17 October 2018). [CrossRef] [PubMed]
- Philip, S.; Kundu, G.C. Osteopontin induces nuclear factor B-mediated promatrix metalloproteinase-2 activation through I kappa B alpha /IKK signaling pathways, and curcumin (diferulolylmethane) down-regulates these pathways. J. Biol. Chem. 2003, 278, 14487–14497. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Aggarwal, B.B. Activation of transcription factor NF-kappa B is suppressed by curcumin (diferuloylmethane) [corrected]. J. Biol. Chem. 1995, 270, 24995–25000. Available online: http://www.jbc.org/content/270/42/24995.long (accessed on 1 October 2018). [CrossRef] [PubMed]
- Edwards, R.L.; Luis, P.B.; Varuzza, P.V.; Joseph, A.I.; Presley, S.H.; Chaturvedi, R.; Schneider, C. The anti-inflammatory activity of curcumin is mediated by its oxidative metabolites. J. Biol. Chem. 2017, 292, 21243–21252. Available online: http://www.jbc.org/content/292/52/21243.long (accessed on 3 January 2019). [CrossRef] [PubMed]
- Liochev, S.I. Reactive oxygen species and the free radical theory of aging. Free Radic. Biol. Med. 2013, 60, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Larasati, Y.A.; Yoneda-Kato, N.; Nakamae, I.; Yokoyama, T.; Meiyanto, E.; Kato, J. Curcumin targets multiple enzymes involved in the ROS metabolic pathway to suppress tumor cell growth. Sci. Rep. 2018, 8, 2039. Available online: https://www.nature.com/articles/s41598-018-20179-6 (accessed on 7 January 2019). [CrossRef] [PubMed]
- Gall Troselj, K.; Novak Kujundzic, R. Curcumin in combined cancer therapy. Curr. Pharm. Des. 2014, 20, 6682–6696. [Google Scholar] [CrossRef]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS ONE 2013, 8, e81162. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3834214/ (accessed on 8 January 2019). [CrossRef] [PubMed]
- Khor, T.O.; Huang, Y.; Wu, T.Y.; Shu, L.M.; Lee, J.; Kong, A.N.T. Pharmacodynamics of curcumin as DNA hypomethylation agent in restoring the expression of Nrf2 via promoter CpGs demethylation. Biochem. Pharmacol. 2011, 82, 1073–1078. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, S.; Jaiswal, A.K. Functional characterization and role of INrf2 in antioxidant response element-mediated expression and antioxidant induction of NAD(P)H:quinone oxidoreductase1 gene. Oncogene 2001, 20, 3906–3917. [Google Scholar] [CrossRef]
- Singh, A.P.; Lange, T.S.; Kim, K.K.; Brard, L.; Horan, T.; Moore, R.G.; Vorsa, N.; Singh, R.K. Purified cranberry proanthocyanidines (PAC-1A) cause pro-apoptotic signaling, ROS generation, cyclophosphamide retention and cytotoxicity in high-risk neuroblastoma cells. Int. J. Oncol. 2012, 40, 99–108. [Google Scholar] [CrossRef]
- Weh, K.M.; Aiyer, H.S.; Howell, A.B.; Kresty, L.A. Cranberry proanthocyanidins modulate reactive oxygen species in Barrett’s and esophageal adenocarcinoma cell lines. J. Berry Res. 2016, 6, 125–136. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5002987/ (accessed on 7 January 2019). [CrossRef]
- Ravindranathan, P.; Pasham, D.; Balaji, U.; Cardenas, J.; Gu, J.; Toden, S.; Goel, A. A combination of curcumin and oligomeric proanthocyanidins offer superior anti-tumorigenic properties in colorectal cancer. Sci. Rep. 2018, 8, 1–12. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6138725/ (accessed on 10 January 2019). [CrossRef] [PubMed]
- Beaconsfield, P.; Rainsbury, R.; Kalton, G. Glucose-6-phosphate dehydrogenase deficiency and the incidence of cancer. Oncologia 1965, 19, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Foulds, L. The natural history of cancer. J. Chronic Dis. 1958, 8, 2–37. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Lecot, P.; Alimirah, F.; Desprez, P.Y.; Campisi, J.; Wiley, C. Context-dependent effects of cellular senescence in cancer development. Br. J. Cancer 2016, 114, 1180–1184. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4891501/ (accessed on 20 November 2018). [CrossRef] [PubMed]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. Available online: https://www.sciencedirect.com/science/article/pii/S009286740800620X?via%3Dihub (accessed on 18 December 2018). [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. Available online: http://science.sciencemag.org/content/324/5930/1029.long (accessed on 17 October 2018). [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Cell biology. Warburg effect and redox balance. Science 2011, 334, 1219–1220. [Google Scholar] [CrossRef] [PubMed]
- De Groof, A.J.C.; te Lindert, M.M.; van Dommelen, M.M.T.; Wu, M.; Willemse, M.; Smift, A.L.; Winer, M.; Oerlemans, F.; Pluk, H.; Fransen, J.A.M.; et al. Increased OXPHOS activity precedes rise in glycolytic rate in H-RasV12/E1A transformed fibroblasts that develop a Warburg phenotype. Mol. Cancer 2009, 8, 54. Available online: https://molecular-cancer.biomedcentral.com/articles/10.1186/1476-4598-8-54 (accessed on 20 September 2018). [CrossRef] [PubMed]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. Available online: https://www.pnas.org/content/95/20/11715.long (accessed on 8 October 2018). [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; David, C.A.; Donnelly, J.B.; Michaelides, M.; Chandel, N.S.; Huang, X.; Warrior, U.; Weinberg, F.; Tormos, K.V.; Fesik, S.W.; et al. A chemical genomics screen highlights the essential role of mitochondria in HIF-1 regulation. Proc. Natl. Acad. Sci. USA 2008, 105, 174–179. Available online: https://www.pnas.org/content/105/1/174.long (accessed on 9 October 2018). [CrossRef] [PubMed]
- Bae, M.-K.; Kim, S.-H.; Jeong, J.-W.; Lee, Y.M.; Kim, H.-S.; Kim, S.-R.; Yun, I.; Bae, S.-K.; Kim, K.-W. Curcumin inhibits hypoxia-induced angiogenesis via down-regulation of HIF-1. Oncol. Rep. 2006, 15, 1557–1562. [Google Scholar] [CrossRef]
- Khan, M.N.; Haggag, Y.A.; Lane, M.E.; McCarron, P.A.; Tambuwala, M.M. Polymeric nano-encapsulation of curcumin enhances its anti-cancer activity in breast (MDA-MB231) and lung (A549) cancer cells through reduction in expression of HIF-1α and nuclear p65 (REL A). Curr. Drug Deliv. 2018, 15, 286–295. [Google Scholar] [CrossRef]
- Wang, X.-P.; Wang, Q.-X.; Lin, H.-P.; Chang, N. Anti-tumor bioactivities of curcumin on mice loaded with gastric carcinoma. Food Funct. 2017, 8, 3319–3326. [Google Scholar] [CrossRef]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.-Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cell 2009, 15, 103–113. Available online: https://www.sciencedirect.com/science/article/pii/S1535610809000026?via%3Dihub (accessed on 25 October 2018).
- Hahn, Y.-I.; Kim, S.-J.; Choi, B.-Y.; Cho, K.-C.; Bandu, R.; Kim, K.P.; Kim, D.-H.; Kim, W.; Park, J.S.; Han, B.W.; et al. Curcumin interacts directly with the Cysteine 259 residue of STAT3 and induces apoptosis in H-Ras transformed human mammary epithelial cells. Sci. Rep. 2018, 8, 6409. Available online: https://www.nature.com/articles/s41598-018-23840-2 (accessed on 16 January 2019). [CrossRef]
- Grivennikov, S.I.; Karin, M. Inflammation and oncogenesis: A vicious connection. Curr. Opin. Genet. Dev. 2010, 20, 65–71. Available online: https://www.sciencedirect.com/science/article/pii/S0959437X09001919?via%3Dihub (accessed on 25 October 2018). [CrossRef] [PubMed]
- Deschênes-Simard, X.; Parisotto, M.; Rowell, M.-C.; Le Calvé, B.; Igelmann, S.; Moineau-Vallée, K.; Saint-Germain, E.; Kalegari, P.; Bourdeau, V.; Kottakis, F.; et al. Circumventing senescence is associated with stem cell properties and metformin sensitivity. Aging Cell 2019, e12889. [Google Scholar] [CrossRef] [PubMed]
- Deschênes-Simard, X.; Gaumont-Leclerc, M.-F.; Bourdeau, V.; Lessard, F.; Moiseeva, O.; Forest, V.; Igelmann, S.; Mallette, F.A.; Saba-El-Leil, M.K.; Meloche, S.; et al. Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation. Genes Dev. 2013, 27, 900–915. Available online: http://genesdev.cshlp.org/content/27/8/900.long (accessed on 3 December 2018). [CrossRef] [PubMed]
- Locken, H.; Clamor, C.; Muller, K. Napabucasin and related heterocycle-fused naphthoquinones as STAT3 inhibitors with antiproliferative activity against cancer cells. J. Nat. Prod. 2018, 81, 1636–1644. [Google Scholar] [CrossRef] [PubMed]
- Kanska, J.; Aspuria, P.-J.P.; Taylor-Harding, B.; Spurka, L.; Funari, V.; Orsulic, S.; Karlan, B.Y.; Wiedemeyer, W.R. Glucose deprivation elicits phenotypic plasticity via ZEB1-mediated expression of NNMT. Oncotarget 2017, 8, 26200–26220. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5432250/ (accessed on 10 October 2018). [CrossRef] [PubMed]
- Pastò, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305–4319. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4147325/ (accessed on 22 October 2018). [CrossRef] [PubMed]
- Pissios, P. Nicotinamide N-Methyltransferase: More than a vitamin B3 clearance enzyme. Trends Endocrinol. Metab. 2017, 28, 340–353. [Google Scholar] [CrossRef]
- Tomida, M.; Ohtake, H.; Yokota, T.; Kobayashi, Y.; Kurosumi, M. Stat3 up-regulates expression of nicotinamide N-methyltransferase in human cancer cells. J. Cancer Res. Clin. Oncol. 2008, 134, 551–559. [Google Scholar] [CrossRef]
- Ulanovskaya, O.A.; Zuhl, A.M.; Cravatt, B.F. NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat. Chem. Biol. 2013, 9, 300–306. Available online: https://www.nature.com/articles/nchembio.1204 (accessed on 25 September 2018). [CrossRef]
- Sperber, H.; Mathieu, J.; Wang, Y.; Ferreccio, A.; Hesson, J.; Xu, Z.; Fischer, K.A.; Devi, A.; Detraux, D.; Gu, H.; et al. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat. Cell Biol. 2015, 17, 1523–1535. Available online: https://www.nature.com/articles/ncb3264 (accessed on 5 December 2018). [CrossRef]
- Xu, X.; Zhu, Y. Curcumin inhibits human non-small cell lung cancer xenografts by targeting STAT3 pathway. Am. J. Transl. Res. 2017, 9, 3633–3641. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5575177/ (accessed on 21 November 2018). [PubMed]
- Menendez, J.A.; Alarcón, T.; Joven, J. Gerometabolites: The pseudohypoxic aging side of cancer oncometabolites. Cell Cycle 2014, 13, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Chini, C.C.S.; Tarragó, M.G.; Chini, E.N. NAD and the aging process: Role in life, death and everything in between. Mol. Cell. Endocrinol. 2017, 455, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.P.; Price, N.L.; Ling, A.J.Y.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. Available online: https://www.sciencedirect.com/science/article/pii/S0092867413015213?via%3Dihub (accessed on 8 October 2018). [CrossRef] [PubMed]
- Yaku, K.; Okabe, K.; Hikosaka, K.; Nakagawa, T. NAD metabolism in cancer therapeutics. Front. Oncol. 2018, 8, 622. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Bamezai, R.N. Human pyruvate kinase M2: A multifunctional protein. Protein Sci. 2010, 19, 2031–2044. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Chen, M.; Assanah, M.; Canoll, P.; Manley, J.L. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 2010, 463, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Clower, C.V.; Chatterjee, D.; Wang, Z.; Cantley, L.C.; Vander Heiden, M.G.; Krainer, A.R. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc. Natl. Acad. Sci. USA 2010, 107, 1894–1899. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.; Ojo, D.; Yan, J.; Tang, D. PKM2 contributes to cancer metabolism. Cancer Lett. 2015, 356 Pt A, 184–191. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Locasale, J.W.; Swanson, K.D.; Sharfi, H.; Heffron, G.J.; Amador-Noguez, D.; Christofk, H.R.; Wagner, G.; Rabinowitz, J.D.; Asara, J.M.; et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science 2010, 329, 1492–1499. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, R.A.; Garcia-Smith, R.; Dorsey, J.; Griffith, J.K.; Bisoffi, M.; Trujillo, K.A. Tumor necrosis factor alpha induces Warburg-like metabolism and is reversed by anti-inflammatory curcumin in breast epithelial cells. Int. J. Cancer 2013, 133, 2504–2510. [Google Scholar] [CrossRef] [PubMed]
- Das, L.; Vinayak, M. Long term effect of curcumin in regulation of glycolytic pathway and angiogenesis via modulation of stress activated genes in prevention of cancer. PLoS ONE 2014, 9, e99583. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4059662/ (accessed on 4 October 2018). [CrossRef] [PubMed]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. Available online: https://www.sciencedirect.com/science/article/pii/S1550413107002951?via%3Dihub (accessed on 28 September 2018). [CrossRef] [PubMed]
- Sun, Q.; Chen, X.; Ma, J.; Peng, H.; Wang, F.; Zha, X.; Wang, Y.; Jing, Y.; Yang, H.; Chen, R.; et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc. Natl. Acad. Sci. USA 2011, 108, 4129–4134. [Google Scholar] [CrossRef] [PubMed]
- Pusapati, R.V.; Daemen, A.; Wilson, C.; Sandoval, W.; Gao, M.; Haley, B.; Baudy, A.R.; Hatzivassiliou, G.; Evangelista, M.; Settleman, J. mTORC1-dependent metabolic reprogramming underlies escape from glycolysis addiction in cancer cells. Cancer Cell 2016, 29, 548–562. Available online: https://www.sciencedirect.com/science/article/pii/S1535610816300526?via%3Dihub (accessed on 30 October 2018). [CrossRef] [PubMed]
- Beevers, C.S.; Zhou, H.; Huang, S. Hitting the golden TORget: Curcumin’s effects on mTOR signaling. Anticancer Agents Med. Chem. 2013, 13, 988–994. Available online: http://www.eurekaselect.com/112990/article (accessed on 5 November 2018). [CrossRef] [PubMed]
- Siddiqui, F.A.; Prakasam, G.; Chattopadhyay, S.; Rehman, A.U.; Padder, R.A.; Ansari, M.A.; Irshad, R.; Mangalhara, K.; Bamezai, R.N.K.; Husain, M.; et al. Curcumin decreases Warburg effect in cancer cells by down-regulating pyruvate kinase M2 via mTOR-HIF-1αinhibition. Sci. Rep. 2018, 8, 8323. Available online: https://www.nature.com/articles/s41598-018-25524-3 (accessed on 20 December 2018). [CrossRef] [PubMed]
- Wang, K.; Fan, H.; Chen, Q.; Ma, G.; Zhu, M.; Zhang, X.; Zhang, Y.; Yu, J. Curcumin inhibits aerobic glycolysis and induces mitochondrial-mediated apoptosis through hexokinase II in human colorectal cancer cells in vitro. Anticancer Drugs 2015, 26, 15–24. [Google Scholar] [CrossRef]
- Zhang, F.-J.; Zhang, H.-S.; Liu, Y.; Huang, Y.-H. Curcumin inhibits Ec109 cell growth via an AMPK-mediated metabolic switch. Life Sci. 2015, 134, 49–55. [Google Scholar] [CrossRef]
- Angelo, L.S.; Maxwell, D.S.; Wu, J.Y.; Sun, D.; Hawke, D.H.; McCutcheon, I.E.; Slopis, J.M.; Peng, Z.; Bornmann, W.G.; Kurzrock, R. Binding partners for curcumin in human schwannoma cells: Biologic implications. Bioorg. Med. Chem. 2013, 21, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Abegg, D.; Frei, R.; Cerato, L.; Prasad Hari, D.; Wang, C.; Waser, J.; Adibekian, A. Proteome-wide profiling of targets of cysteine reactive small molecules by using ethynyl benziodoxolone reagents. Angew. Chem. Int. Ed. Engl. 2015, 54, 10852–10857. [Google Scholar] [CrossRef] [PubMed]
- McBean, G.J.; Aslan, M.; Griffiths, H.R.; Torrão, R.C. Thiol redox homeostasis in neurodegenerative disease. Redox Biol. 2015, 5, 186–194. Available online: https://www.sciencedirect.com/science/article/pii/S221323171500035X?via%3Dihub (accessed on 20 December 2018). [CrossRef] [PubMed]
- Wang, J.; Zhang, J.; Zhang, C.J.; Wong, Y.K.; Lim, T.K.; Hua, Z.C.; Liu, B.; Tannenbaum, S.R.; Shen, H.M.; Lin, Q. In situ proteomic profiling of curcumin targets in hct116 colon cancer cell line. Sci. Rep. 2016, 6, 22146. Available online: https://www.nature.com/articles/srep22146 (accessed on 3 January 2019). [CrossRef] [PubMed]
- Feng, D.; Cheng, Y.; Meng, Y.; Zou, L.; Huang, S.; Xie, J. Multiple effects of curcumin on promoting expression of the exon 7-containing SMN2 transcript. Genes Nutr. 2015, 10, 40. Available online: https://link.springer.com/article/10.1007%2Fs12263-015-0486-y (accessed on 18 January 2019). [CrossRef] [PubMed]
- Stine, Z.E.; Dang, C. Stress eating and tuning out: Cancer cells re-wire metabolism to counter stress. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.-K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.P.; Giacomantonio, M.A.; Paulo, J.A.; Everley, R.A.; Kennedy, B.E.; Pathak, G.P.; Clements, D.R.; Kim, Y.; Dai, C.; Sharif, T.; et al. The NAD+ salvage pathway supports PHGDH-driven serine biosynthesis. Cell Rep. 2018, 24, 2381–2391. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Park, H.J.; Bae, Y.H.; Ahn, S.C.; Wee, H.J.; Yun, I.; Jang, H.O.; Bae, M.K.; Bae, S.K. Curcumin down-regulates visfatin expression and inhibits breast cancer cell invasion. Endocrinology 2012, 153, 554–563. [Google Scholar] [CrossRef]
- Jing, Z.; Heng, W.; Xia, L.; Ning, W.; Yafei, Q.; Yao, Z.; Shulan, Z. Downregulation of phosphoglycerate dehydrogenase inhibits proliferation and enhances cisplatin sensitivity in cervical adenocarcinoma cells by regulating Bcl-2 and caspase-3. Cancer Biol. Ther. 2015, 16, 541–548. [Google Scholar] [CrossRef]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Fiske, B.P.; Birsoy, K.; Freinkman, E.; Kami, K.; Possemato, R.L.; Chudnovsky, Y.; Pacold, M.E.; Chen, W.W.; Cantor, J.R.; et al. SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature 2015, 520, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Giardina, G.; Brunotti, P.; Fiascarelli, A.; Cicalini, A.; Costa, M.G.; Buckle, A.M.; di Salvo, M.L.; Giorgi, A.; Marani, M.; Paone, A.; et al. How pyridoxal 5′-phosphate differentially regulates human cytosolic and mitochondrial serine hydroxymethyltransferase oligomeric state. FEBS J. 2015, 282, 1225–1241. [Google Scholar] [CrossRef] [PubMed]
- Chaneton, B.; Hillmann, P.; Zheng, L.; Martin, A.C.L.; Maddocks, O.D.K.; Chokkathukalam, A.; Coyle, J.E.; Jankevics, A.; Holding, F.P.; Vousden, K.H.; et al. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature 2012, 491, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fan, J.; Venneti, S.; Wan, Y.W.; Pawel, B.R.; Zhang, J.; Finley, L.W.; Lu, C.; Lindsten, T.; Cross, J.R.; et al. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov. 2014, 4, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.Y.; Haverty, P.M.; Li, L.; Kljavin, N.M.; Bourgon, R.; Lee, J.; Stern, H.; Modrusan, Z.; Seshagiri, S.; Zhang, Z.; et al. Comparative oncogenomics identifies PSMB4 and SHMT2 as potential cancer driver genes. Cancer Res. 2014, 74, 3114–3126. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Unterlass, J.E.; Baslé, A.; Blackburn, T.J.; Tucker, J.; Cano, C.; Noble, M.E.M.; Curtin, N.J. Validating and enabling phosphoglycerate dehydrogenase (PHGDH) as a target for fragment-based drug discovery in PHGDH-amplified breast cancer. Oncotarget 2016, 9, 13139–13153. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5862567/ (accessed on 17 October 2018). [CrossRef]
- Yoshino, H.; Nohata, N.; Miyamoto, K.; Yonemori, M.; Sakaguchi, T.; Sugita, S.; Itesako, T.; Kofuji, S.; Nakagawa, M.; Dahiya, R.; et al. PHGDH as a key enzyme for serine biosynthesis in HIF2α-targeting therapy for renal cell carcinoma. Cancer Res. 2017, 77, 6321–6329. [Google Scholar] [CrossRef]
- Antonov, A.; Agostini, M.; Morello, M.; Minieri, M.; Melino, G.; Amelio, I. Bioinformatics analysis of the serine and glycine pathway in cancer cells. Oncotarget 2014, 5, 11004–11013. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4294344/ (accessed on 17 October 2018). [CrossRef]
- DeNicola, G.M.; Chen, P.-H.; Mullarky, E.; Sudderth, J.A.; Hu, Z.; Wu, D.; Tang, H.; Xie, Y.; Asara, J.M.; Huffman, K.E.; et al. NRF2 regulates serine biosynthesis in non–small cell lung cancer. Nat. Genet. 2015, 47, 1475–1481. [Google Scholar] [CrossRef] [PubMed]
- Yin, K. Positive correlation between expression level of mitochondrial serine hydroxymethyltransferase and breast cancer grade. Onco Targets Ther. 2015, 8, 1069–1074. Available online: https://www.dovepress.com/positive-correlation-between-expression-level-of-mitochondrial-serine--peer-reviewed-article-OTT (accessed on 24 October 2018). [CrossRef] [PubMed]
- Wu, X.; Deng, L.; Tang, D.; Ying, G.; Yao, X.; Liu, F.; Liang, G. miR-615-5p prevents proliferation and migration through negatively regulating serine hydromethyltransferase 2 (SHMT2) in hepatocellular carcinoma. Tumor Biol. 2016, 37, 6813–6821. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, Z.; Li, X.; Liu, B.; Liu, M.; Liu, L.; Chen, S.; Ren, M.; Wang, Y.; Yu, M. SHMT2 desuccinylation by SIRT5 drives cancer cell proliferation. Cancer Res. 2018, 78, 372–386. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kujundžić, R.N.; Stepanić, V.; Milković, L.; Gašparović, A.Č.; Tomljanović, M.; Trošelj, K.G. Curcumin and its Potential for Systemic Targeting of Inflamm-Aging and Metabolic Reprogramming in Cancer. Int. J. Mol. Sci. 2019, 20, 1180. https://doi.org/10.3390/ijms20051180
Kujundžić RN, Stepanić V, Milković L, Gašparović AČ, Tomljanović M, Trošelj KG. Curcumin and its Potential for Systemic Targeting of Inflamm-Aging and Metabolic Reprogramming in Cancer. International Journal of Molecular Sciences. 2019; 20(5):1180. https://doi.org/10.3390/ijms20051180
Chicago/Turabian StyleKujundžić, Renata Novak, Višnja Stepanić, Lidija Milković, Ana Čipak Gašparović, Marko Tomljanović, and Koraljka Gall Trošelj. 2019. "Curcumin and its Potential for Systemic Targeting of Inflamm-Aging and Metabolic Reprogramming in Cancer" International Journal of Molecular Sciences 20, no. 5: 1180. https://doi.org/10.3390/ijms20051180
APA StyleKujundžić, R. N., Stepanić, V., Milković, L., Gašparović, A. Č., Tomljanović, M., & Trošelj, K. G. (2019). Curcumin and its Potential for Systemic Targeting of Inflamm-Aging and Metabolic Reprogramming in Cancer. International Journal of Molecular Sciences, 20(5), 1180. https://doi.org/10.3390/ijms20051180