Characterization of a Novel Third-Generation Anti-CD24-CAR against Ovarian Cancer

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
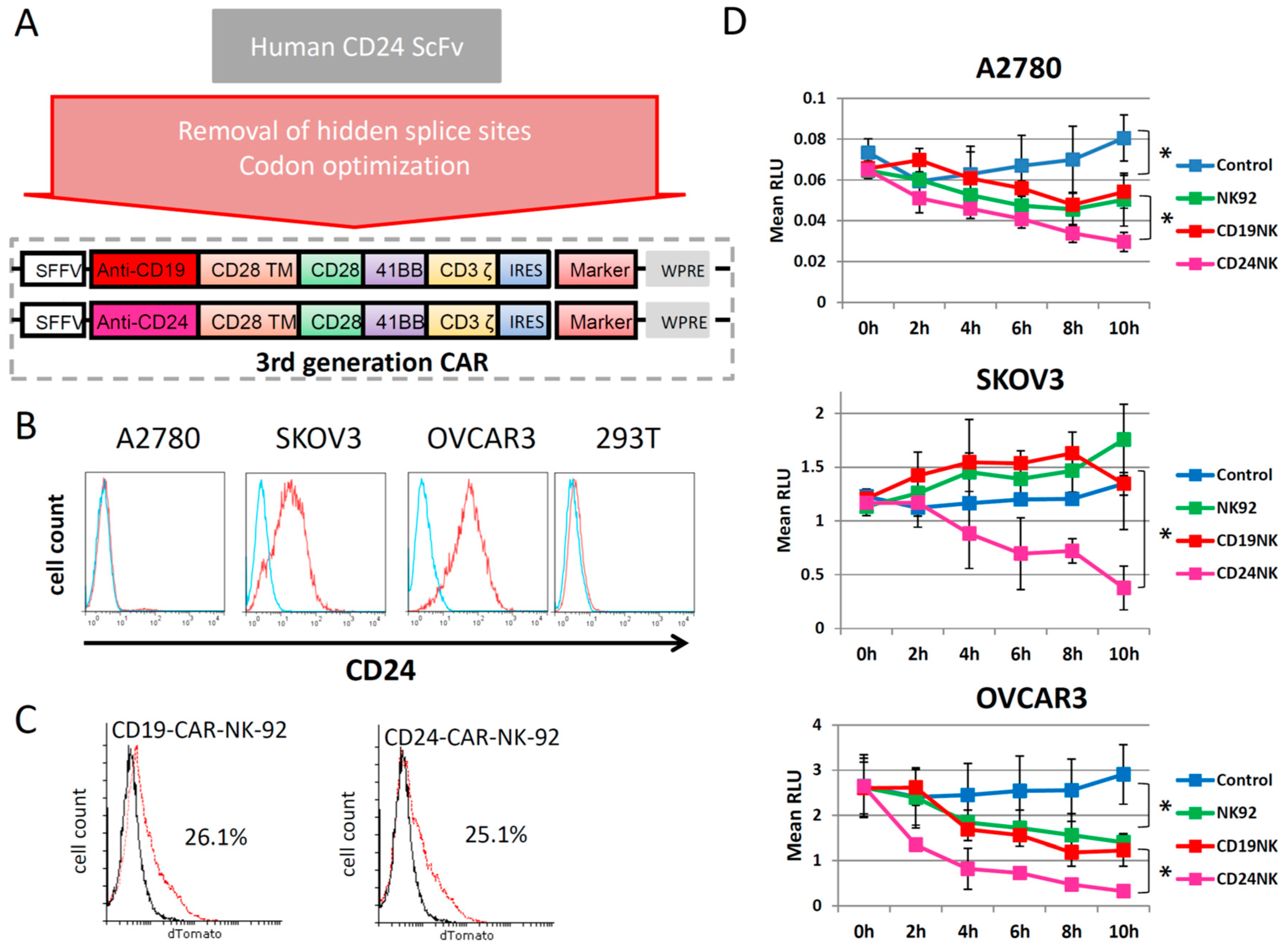
2.1. Generation of a New Third-Generation Anti-CD24 CAR
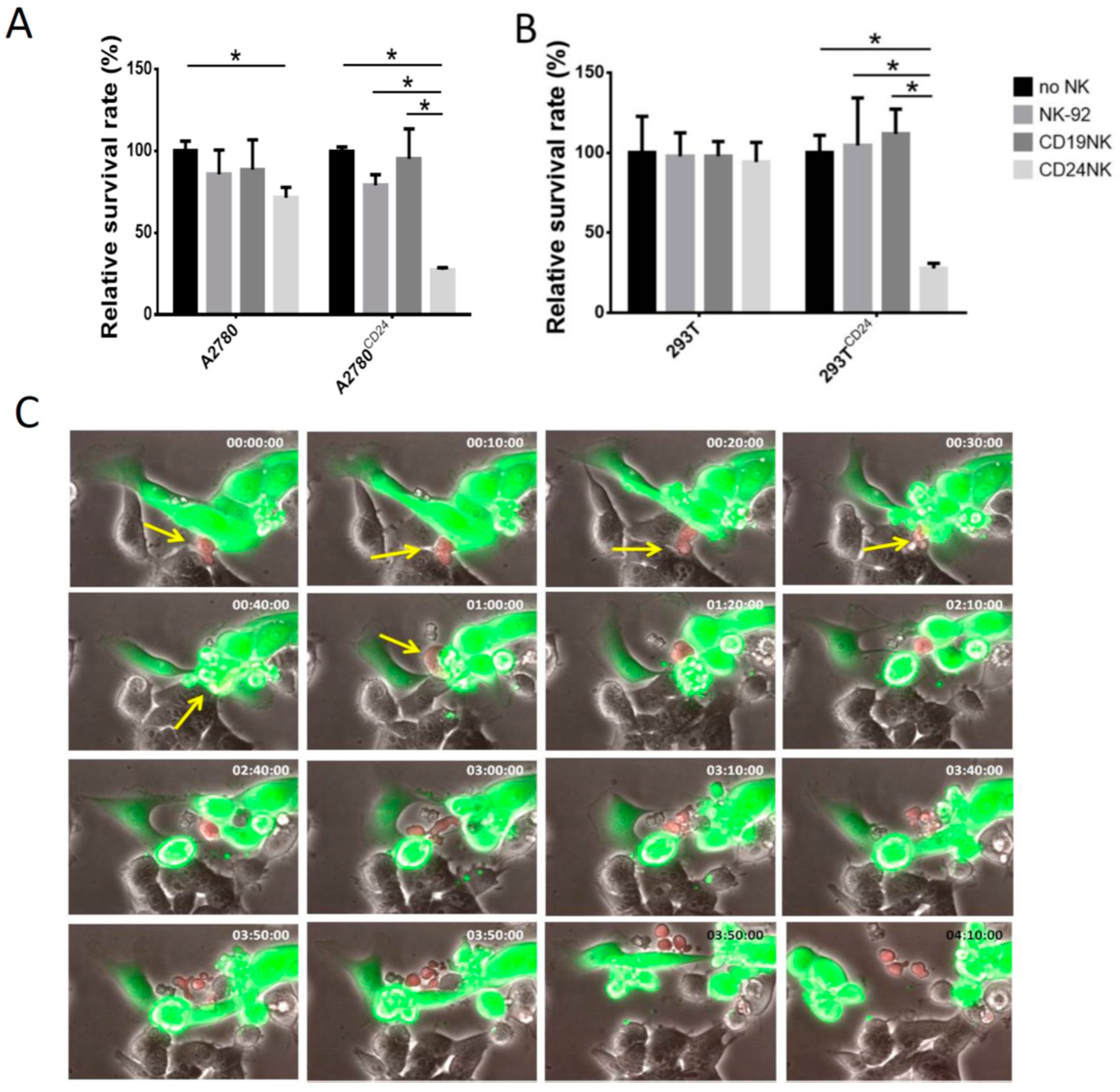
2.2. The novel CD24-CAR-NK-92 Cells Show Specific Killing of Ovarian Cancer Cell Lines
2.3. Specific Killing of Engineered NK Cells
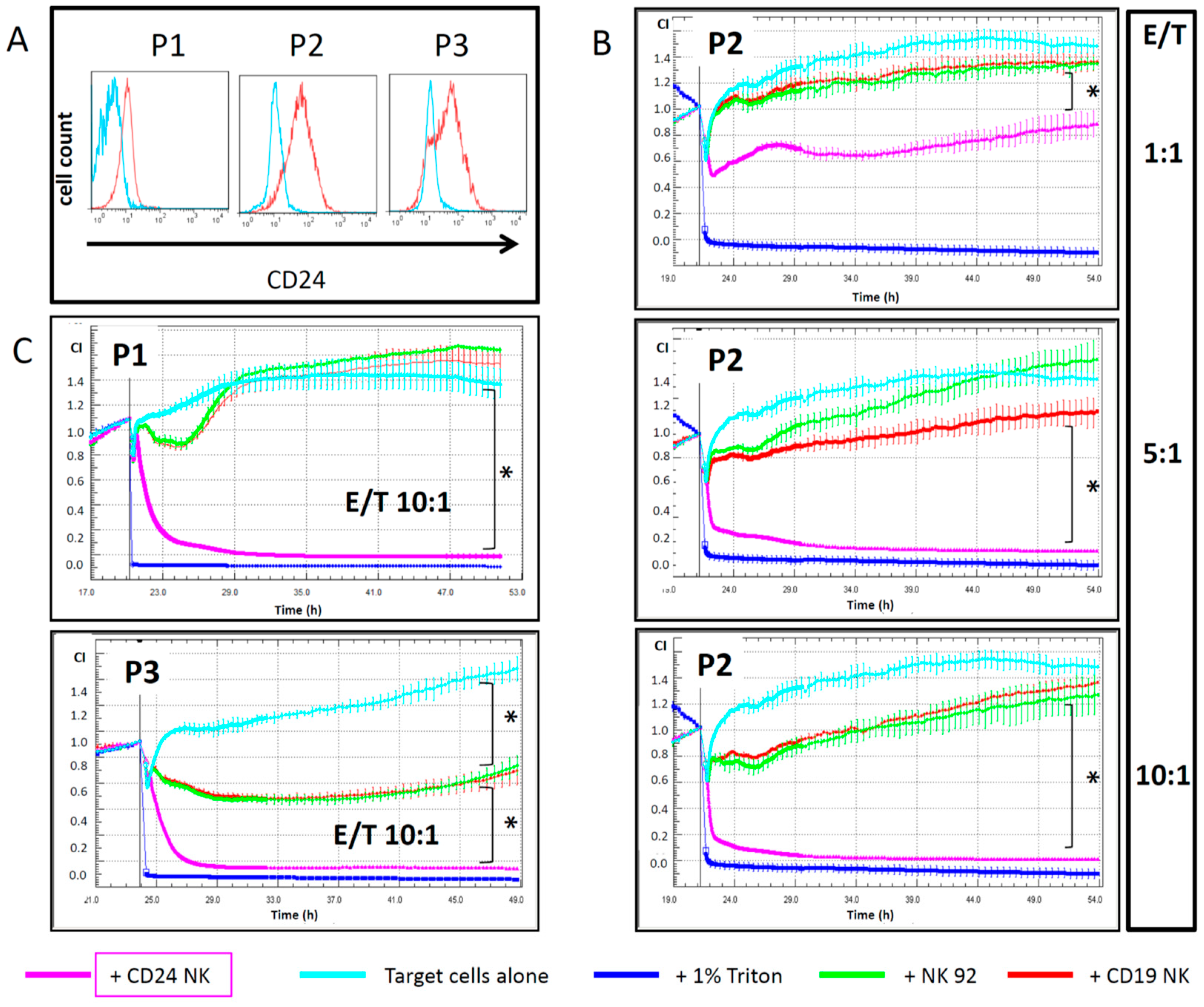
2.4. Engineered NK Cells Specifically Kill Primary Patient-Derived Ovarian Cancer Cells
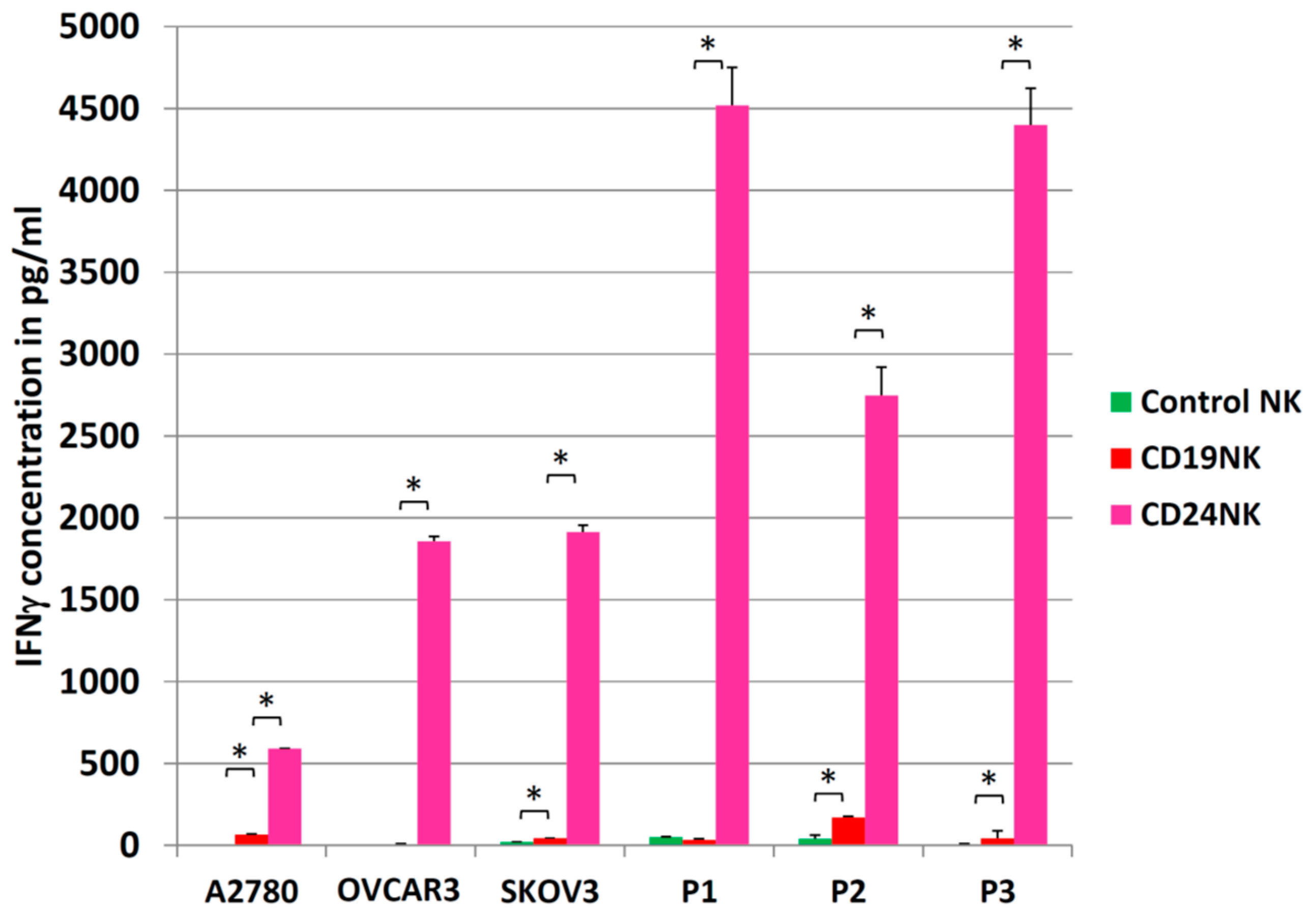
2.5. Activation of NK Cells
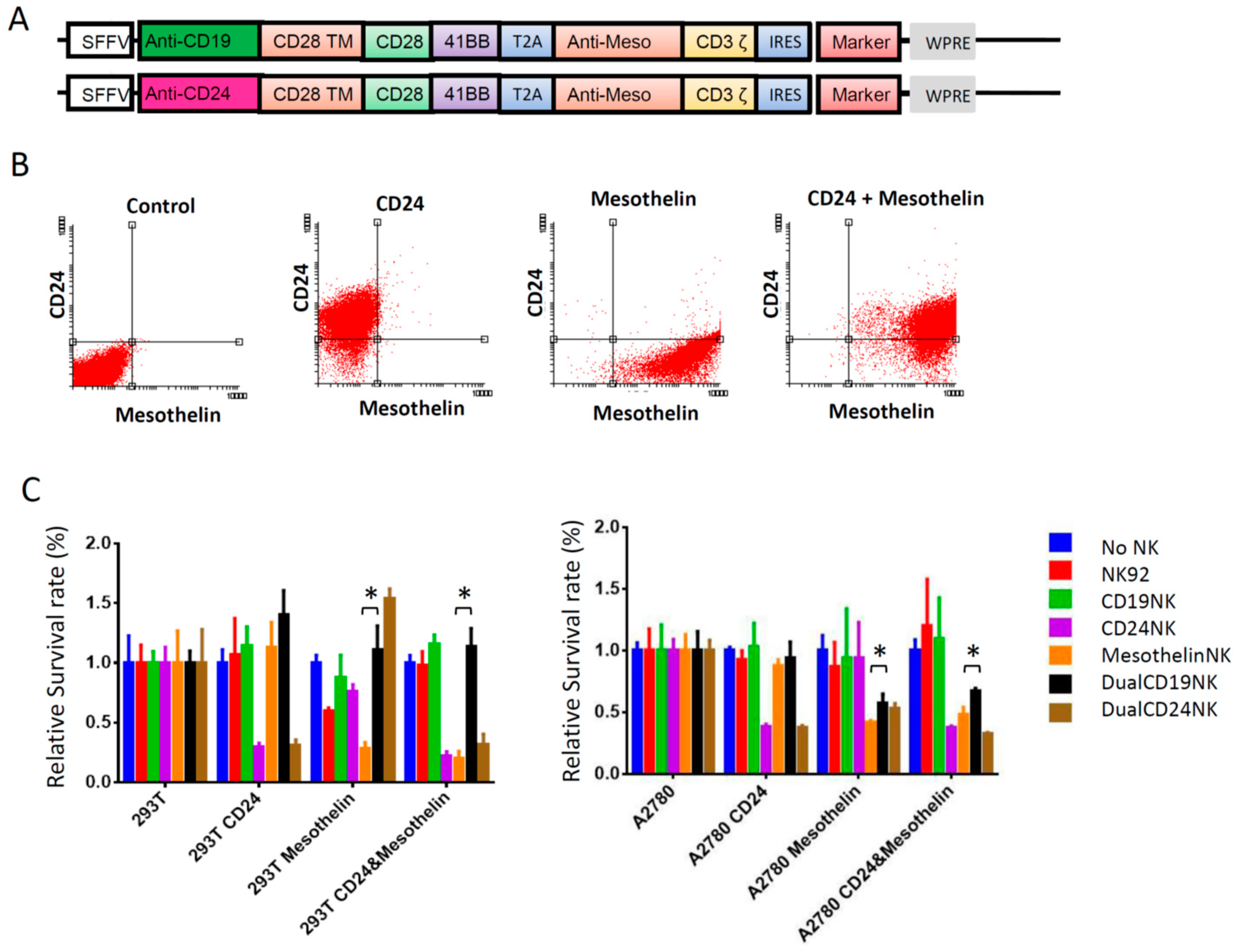
2.6. Dual-CAR
3. Discussion
4. Material and Methods
4.1. Cell lines
4.2. Cloning of Vectors and Virus Production
4.3. Transduction of NK Cells
4.4. Antibodies and ELISA
4.5. Cytotoxicity Assays
4.5.1. Fluoroskan Ascent™ FL
4.5.2. xCELLigence
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Burges, A.; Schmalfeldt, B. Ovarian cancer: Diagnosis and treatment. Deutsch. Arztebl. Int. 2011, 108, 635–641. [Google Scholar]
- Harter, P.; Sehouli, J.; Reuss, A.; Hasenburg, A.; Scambia, G.; Cibula, D.; Mahner, S.; Vergote, I.; Reinthaller, A.; Burges, A.; et al. Prospective validation study of a predictive score for operability of recurrent ovarian cancer: The Multicenter Intergroup Study DESKTOP II. A project of the AGO Kommission OVAR, AGO Study Group, NOGGO, AGO-Austria, and MITO. Int. J. Gynecol. Cancer 2011, 21, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.M.; Landen, C.N. Ovarian Cancer Stem Cells: Are They Real and Why are they Important? Gynecol. Oncol. 2014, 132, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Garson, K.; Vanderhyden, B.C. Epithelial ovarian cancer stem cells: Underlying complexity of a simple paradigm. Reproduction 2015, 149, R59–R70. [Google Scholar] [CrossRef] [PubMed]
- Burgos-Ojeda, D.; Rueda, B.R.; Buckanovich, R.J. Ovarian cancer stem cell markers: Prognostic and therapeutic implications. Cancer Lett. 2012, 322, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Zheng, P.; Tang, J.; Liu, Y. CD24: From A to Z. Cell. Mol. Immunol. 2010, 7, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Muinao, T.; Deka Boruah, H.P.; Pal, M. Diagnostic and Prognostic Biomarkers in ovarian cancer and the potential roles of cancer stem cells—An updated review. Exp. Cell Res. 2018, 362, 1–10. [Google Scholar] [CrossRef]
- Nakamura, K.; Terai, Y.; Tanabe, A.; Ono, Y.J.; Hayashi, M.; Maeda, K.; Fujiwara, S.; Ashihara, K.; Nakamura, M.; Tanaka, Y.; et al. CD24 expression is a marker for predicting clinical outcome and regulates the epithelial-mesenchymal transition in ovarian cancer via both the Akt and ERK pathways. Oncol. Rep. 2017, 37, 3189–3200. [Google Scholar] [CrossRef]
- Sun, F.; Wang, T.; Jiang, J.; Wang, Y.; Ma, Z.; Li, Z.; Han, Y.; Pan, M.; Cai, J.; Wang, M.; et al. Engineering a high-affinity humanized anti-CD24 antibody to target hepatocellular carcinoma by a novel CDR grafting design. Oncotarget 2017, 8, 51238–51252. [Google Scholar] [CrossRef] [PubMed]
- Maliar, A.; Servais, C.; Waks, T.; Chmielewski, M.; Lavy, R.; Altevogt, P.; Abken, H.; Eshhar, Z. Redirected T cells that target pancreatic adenocarcinoma antigens eliminate tumors and metastases in mice. Gastroenterology 2012, 143, 1375–1384.e5. [Google Scholar] [CrossRef]
- Salnikov, A.V.; Bretz, N.P.; Perne, C.; Hazin, J.; Keller, S.; Fogel, M.; Herr, I.; Schlange, T.; Moldenhauer, G.; Altevogt, P. Antibody targeting of CD24 efficiently retards growth and influences cytokine milieu in experimental carcinomas. Br. J. Cancer 2013, 108, 1449–1459. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.; Waibel, R.; Weber, E.; Bell, J.; Stahel, R.A. CD24, a signal-transducing molecule expressed on human B cells, is a major surface antigen on small cell lung carcinomas. Cancer Res. 1992, 52, 5264–5270. [Google Scholar] [PubMed]
- Sadelain, M.; Brentjens, R.; Riviere, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Caruana, I.; Diaconu, I.; Dotti, G. From monoclonal antibodies to chimeric antigen receptors for the treatment of human malignancies. Semin. Oncol. 2014, 41, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Lanitis, E.; Poussin, M.; Hagemann, I.S.; Coukos, G.; Sandaltzopoulos, R.; Scholler, N.; Powell, D.J. Redirected antitumor activity of primary human lymphocytes transduced with a fully human anti-mesothelin chimeric receptor. Mol. Ther. 2012, 20, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Chekmasova, A.A.; Rao, T.D.; Nikhamin, Y.; Park, K.J.; Levine, D.A.; Spriggs, D.R.; Brentjens, R.J. Successful eradication of established peritoneal ovarian tumors in SCID-Beige mice following adoptive transfer of T cells genetically targeted to the MUC16 antigen. Clin. Cancer Res. 2010, 16, 3594–3606. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef]
- Wilkie, S.; van Schalkwyk, M.; Hobbs, S.; Davies, M.; van der Stegen, S.; Pereira, A.P.; Burbridge, S.; Box, C.; Eccles, S.A.; Maher, J. Dual Targeting of ErbB2 and MUC1 in Breast Cancer Using Chimeric Antigen Receptors Engineered to Provide Complementary Signaling. J. Clin. Immunol. 2012, 32, 1059–1070. [Google Scholar] [CrossRef]
- Beatty, G.L.; O’Hara, M.H.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155, 29–32. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef]
- Li, H.; Zhao, Y. Increasing the safety and efficacy of chimeric antigen receptor T cell therapy. Protein Cell 2017, 8, 573–589. [Google Scholar] [PubMed]
- Engel, J.; Eckel, R.; Schubert-Fritschle, G.; Kerr, J.; Kuhn, W.; Diebold, J.; Kimmig, R.; Rehbock, J.; Holzel, D. Moderate progress for ovarian cancer in the last 20 years: Prolongation of survival, but no improvement in the cure rate. Eur. J. Cancer 2002, 38, 2435–2445. [Google Scholar] [CrossRef]
- Bapat, S.A.; Mali, A.M.; Koppikar, C.B.; Kurrey, N.K. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res. 2005, 65, 3025–3029. [Google Scholar] [CrossRef] [PubMed]
- Latifi, A.; Abubaker, K.; Castrechini, N.; Ward, A.C.; Liongue, C.; Dobill, F.; Kumar, J.; Thompson, E.W.; Quinn, M.A.; Findlay, J.K.; et al. Cisplatin treatment of primary and metastatic epithelial ovarian carcinomas generates residual cells with mesenchymal stem cell-like profile. J. Cell. Biochem. 2011, 112, 2850–2864. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Abubaker, K.; Findlay, J.; Quinn, M. Cancerous ovarian stem cells: Obscure targets for therapy but relevant to chemoresistance. J. Cell. Biochem. 2013, 114, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Steg, A.D.; Bevis, K.S.; Katre, A.A.; Ziebarth, A.; Dobbin, Z.C.; Alvarez, R.D.; Zhang, K.; Conner, M.; Landen, C.N. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin. Cancer Res. 2012, 18, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xu, T.; Cui, M. Are ovarian cancer stem cells the target for innovative immunotherapy? OncoTargets Ther. 2018, 11, 2615–2626. [Google Scholar] [CrossRef]
- Klapdor, R.; Wang, S.; Hacker, U.; Büning, H.; Morgan, M.; Dörk, T.; Hillemanns, P.; Schambach, A. Improved Killing of Ovarian Cancer Stem Cells by Combining a Novel Chimeric Antigen Receptor-Based Immunotherapy and Chemotherapy. Hum. Gene Ther. 2017, 28, 886–896. [Google Scholar] [CrossRef]
- Aigner, S.; Sthoeger, Z.M.; Fogel, M.; Weber, E.; Zarn, J.; Ruppert, M.; Zeller, Y.; Vestweber, D.; Stahel, R.; Sammar, M.; et al. CD24, a mucin-type glycoprotein, is a ligand for P-selectin on human tumor cells. Blood 1997, 89, 3385–3395. [Google Scholar]
- Baumann, P.; Cremers, N.; Kroese, F.; Orend, G.; Chiquet-Ehrismann, R.; Uede, T.; Yagita, H.; Sleeman, J.P. CD24 expression causes the acquisition of multiple cellular properties associated with tumor growth and metastasis. Cancer Res. 2005, 65, 10783–10793. [Google Scholar] [CrossRef]
- Kang, K.S.; Choi, Y.P.; Gao, M.-Q.; Kang, S.; Kim, B.G.; Lee, J.H.; Kwon, M.J.; Shin, Y.K.; Cho, N.H. CD24⁺ ovary cancer cells exhibit an invasive mesenchymal phenotype. Biochem. Biophys. Res. Commun. 2013, 432, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Klingemann, H. Are natural killer cells superior CAR drivers? Oncoimmunology 2014, 3, e28147. [Google Scholar] [CrossRef] [PubMed]
- Tonn, T.; Schwabe, D.; Klingemann, H.G.; Becker, S.; Esser, R.; Koehl, U.; Suttorp, M.; Seifried, E.; Ottmann, O.G.; Bug, G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 2013, 15, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- Arai, S.; Meagher, R.; Swearingen, M.; Myint, H.; Rich, E.; Martinson, J.; Klingemann, H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: A phase I trial. Cytotherapy 2008, 10, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Maki, G.; Klingemann, H.G.; Martinson, J.A.; Tam, Y.K. Factors regulating the cytotoxic activity of the human natural killer cell line, NK-92. J. Hematother. Stem Cell Res. 2001, 10, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.-S.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol. Ther. 2010, 18, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Hudecek, M.; Lupo-Stanghellini, M.-T.; Kosasih, P.L.; Sommermeyer, D.; Jensen, M.C.; Rader, C.; Riddell, S.R. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin. Cancer Res. 2013, 19, 3153–3164. [Google Scholar] [CrossRef]
- Liang, D.; Ma, Y.; Liu, J.; Trope, C.G.; Holm, R.; Nesland, J.M.; Suo, Z. The hypoxic microenvironment upgrades stem-like properties of ovarian cancer cells. BMC Cancer 2012, 12, 201. [Google Scholar] [CrossRef]
- Landskron, G.; De La Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef]
- Lamers, C.H.J.; Sleijfer, S.; Vulto, A.G.; Kruit, W.H.J.; Kliffen, M.; Debets, R.; Gratama, J.W.; Stoter, G.; Oosterwijk, E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: First clinical experience. J. Clin. Oncol. 2006, 24, e20–e22. [Google Scholar] [CrossRef]
- Scarfò, I.; Maus, M.V. Current approaches to increase CAR T cell potency in solid tumors: Targeting the tumor microenvironment. J. Immunother. Cancer 2017, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Klingemann, H.; Boissel, L.; Toneguzzo, F. Natural Killer Cells for Immunotherapy–advantages of the NK-92 Cell Line over Blood NK Cells. Front. Immunol. 2016, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Lanitis, E.; Poussin, M.; Klattenhoff, A.W.; Song, D.; Sandaltzopoulos, R.; June, C.H.; Powell, D.J., Jr. Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol. Res. 2013, 1, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision Tumor Recognition by T Cells with Combinatorial Antigen-Sensing Circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.; Lee, S.-B.; Park, H.; Lee, H.J.; Cho, N.H.; Kim, J. Susceptibility of CD24(+) ovarian cancer cells to anti-cancer drugs and natural killer cells. Biochem. Biophys. Res. Commun. 2012, 427, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Feldman, S.A.; Zhao, Y.; Xu, H.; Black, M.A.; Morgan, R.A.; Wilson, W.H.; Rosenberg, S.A. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J. Immunother. (Hagerstown, Md.: 1997) 2009, 32, 689–702. [Google Scholar] [CrossRef]
- Suerth, J.D.; Morgan, M.A.; Kloess, S.; Heckl, D.; Neudörfl, C.; Falk, C.S.; Koehl, U.; Schambach, A. Efficient generation of gene-modified human natural killer cells via alpharetroviral vectors. J. Mol. Med. 2016, 94, 83–93. [Google Scholar] [CrossRef]
- Schambach, A.; Bohne, J.; Chandra, S.; Will, E.; Margison, G.P.; Williams, D.A.; Baum, C. Equal potency of gammaretroviral and lentiviral SIN vectors for expression of O6-methylguanine-DNA methyltransferase in hematopoietic cells. Mol. Ther. 2006, 13, 391–400. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klapdor, R.; Wang, S.; Morgan, M.; Dörk, T.; Hacker, U.; Hillemanns, P.; Büning, H.; Schambach, A. Characterization of a Novel Third-Generation Anti-CD24-CAR against Ovarian Cancer. Int. J. Mol. Sci. 2019, 20, 660. https://doi.org/10.3390/ijms20030660
Klapdor R, Wang S, Morgan M, Dörk T, Hacker U, Hillemanns P, Büning H, Schambach A. Characterization of a Novel Third-Generation Anti-CD24-CAR against Ovarian Cancer. International Journal of Molecular Sciences. 2019; 20(3):660. https://doi.org/10.3390/ijms20030660
Chicago/Turabian StyleKlapdor, Rüdiger, Shuo Wang, Michael Morgan, Thilo Dörk, Ulrich Hacker, Peter Hillemanns, Hildegard Büning, and Axel Schambach. 2019. "Characterization of a Novel Third-Generation Anti-CD24-CAR against Ovarian Cancer" International Journal of Molecular Sciences 20, no. 3: 660. https://doi.org/10.3390/ijms20030660
APA StyleKlapdor, R., Wang, S., Morgan, M., Dörk, T., Hacker, U., Hillemanns, P., Büning, H., & Schambach, A. (2019). Characterization of a Novel Third-Generation Anti-CD24-CAR against Ovarian Cancer. International Journal of Molecular Sciences, 20(3), 660. https://doi.org/10.3390/ijms20030660