High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints



Abstract
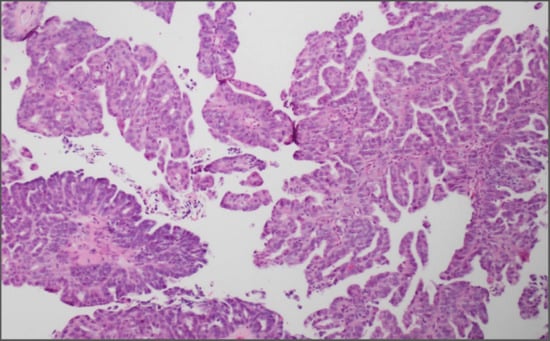
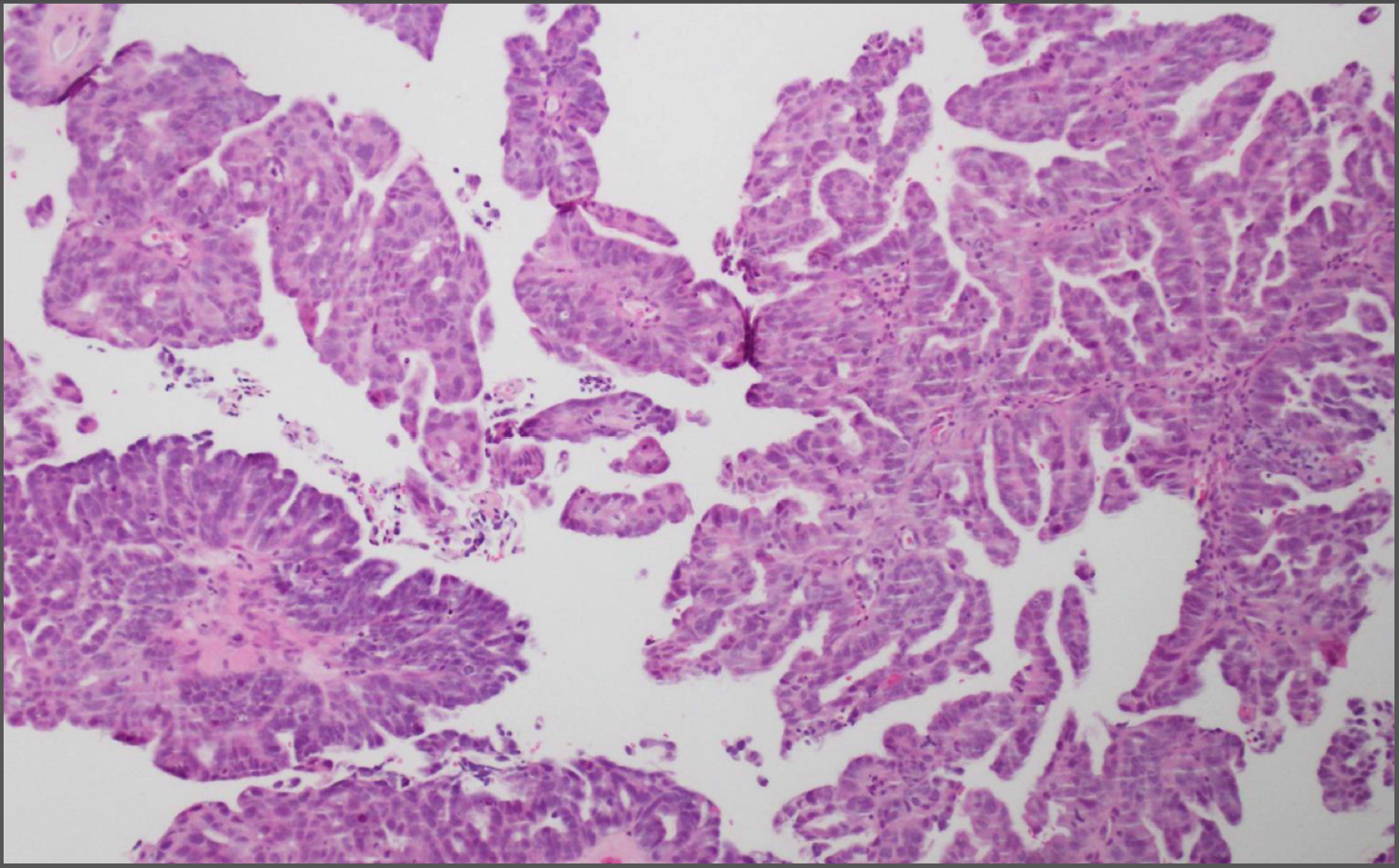{kind=link}
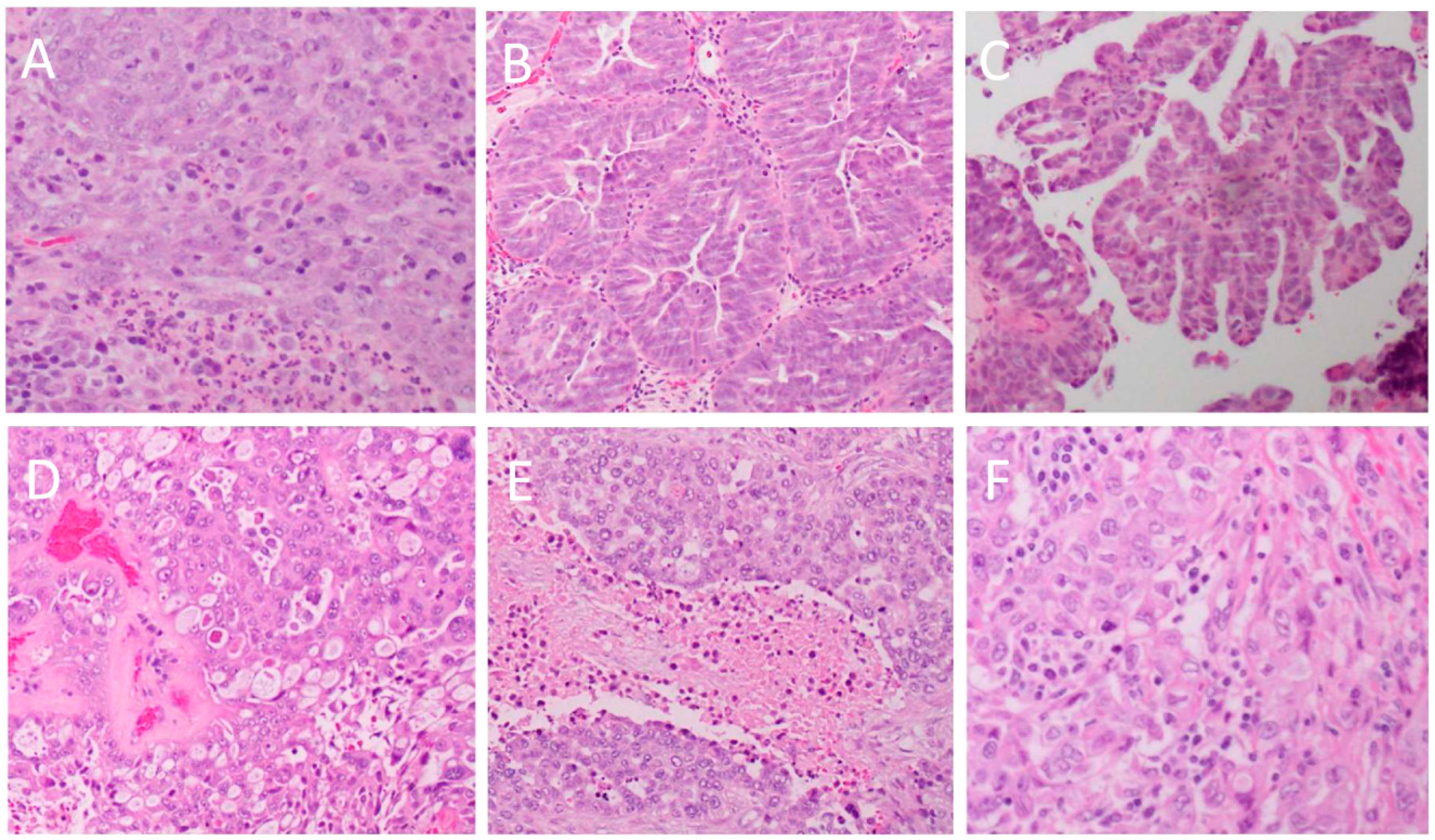{kind=link}
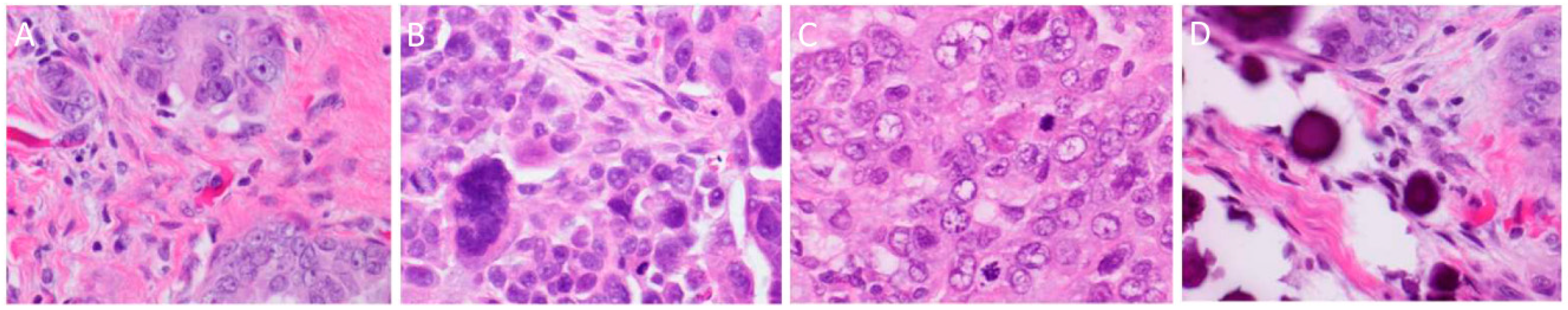{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Prevalence and Mortality
2. Subtype Classification
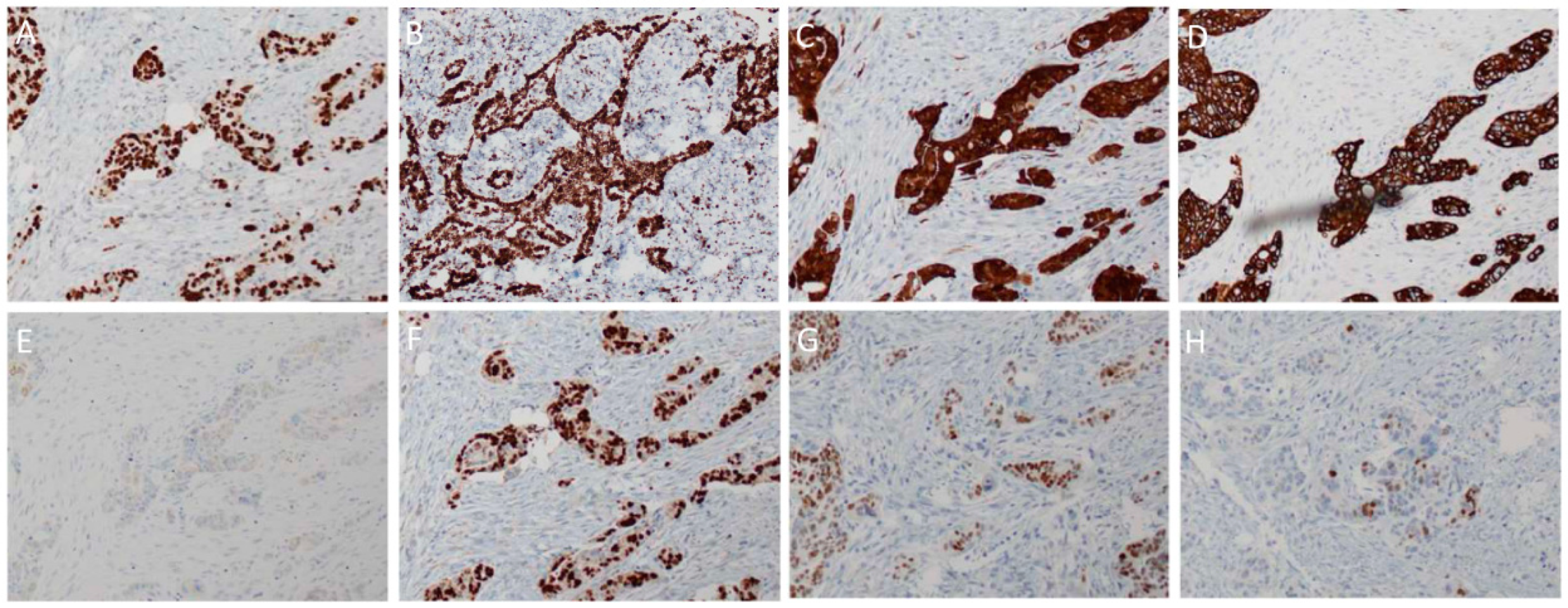
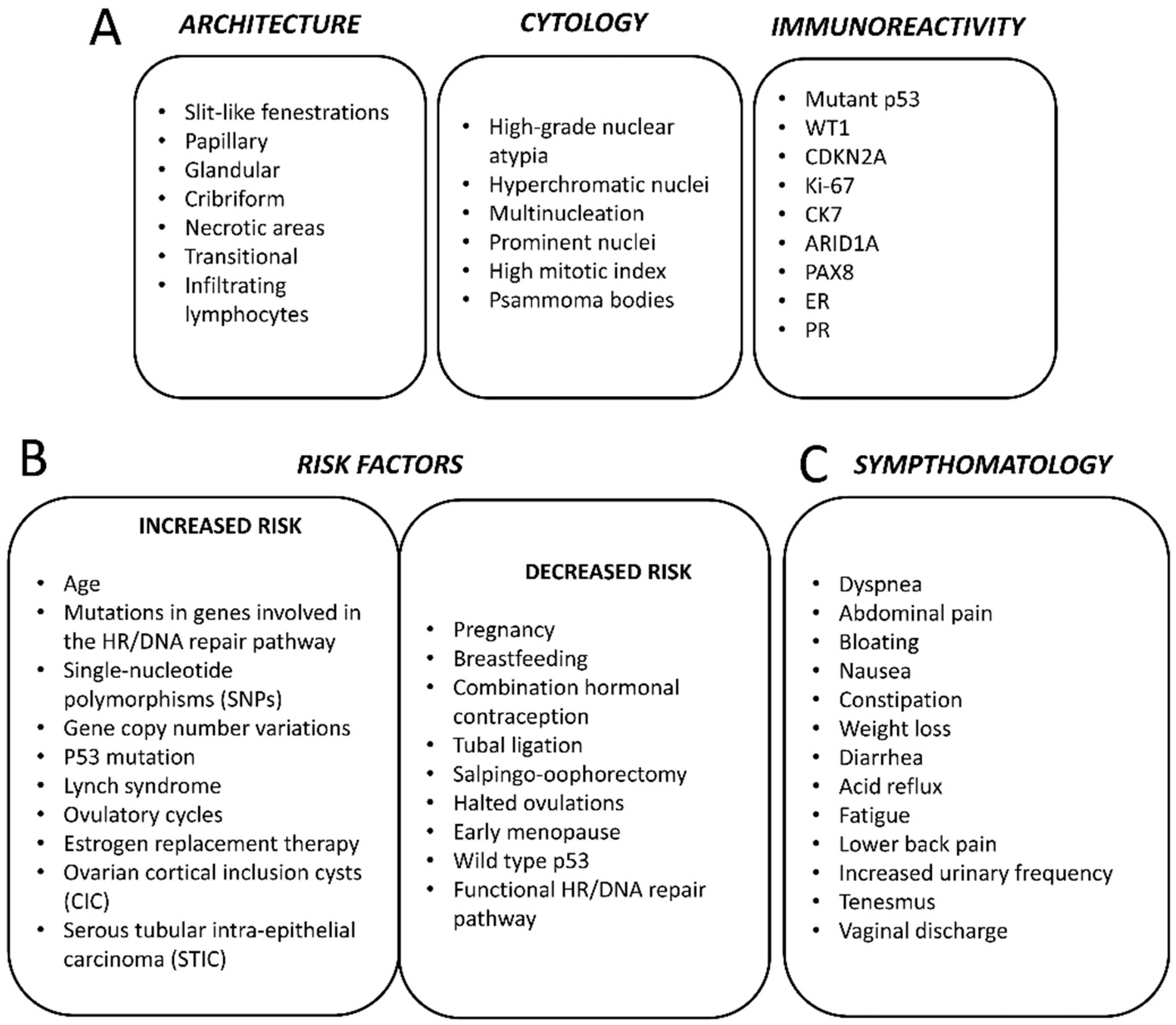
3. Histopathology
4. Epidemiology and Risk Factors
5. Origin
6. Dissemination
7. Symptomatology, Diagnosis and Staging
8. Genetics
9. Gene Expression Profiling and Molecular Subtypes
10. Surgery
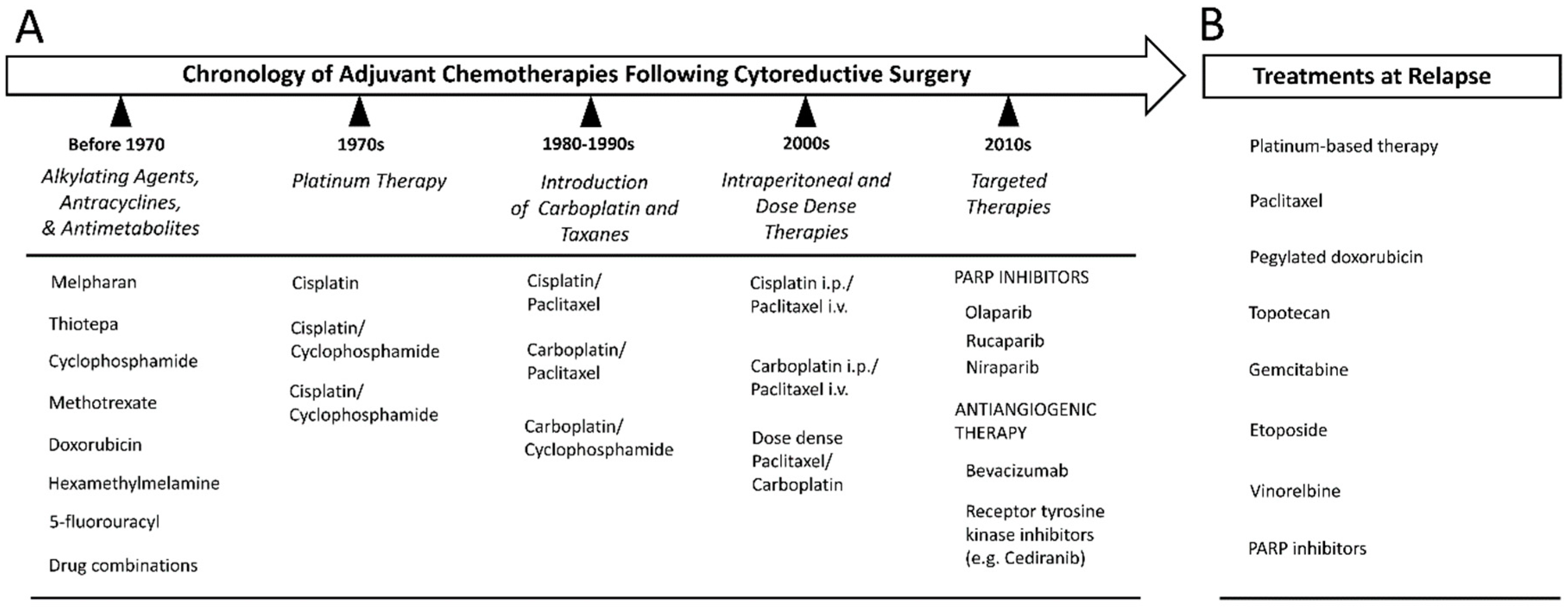
11. Cytotoxic Chemotherapy
12. Relapse and Treatment Resistance
13. Targeted Therapies
14. Preclinical Models of Study
14.1. In-Vitro Models
14.2. In-Vivo Models
15. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ARID1A | AT-Rich Interaction Domain 1A |
| ATM | ATM Serine/Threonine Kinase |
| AKT | AKT Serine/Threonine Kinase |
| ATR | ATR serine/threonine kinase |
| AUC | Area Under the Curve |
| BARD1 | BRCA1 Associated RING Domain 1 |
| BECN1 | Beclin 1 |
| BER | Base Excision Repair |
| BRAF | B-Raf Proto-Oncogene, Serine/Threonine Kinase |
| BRCA | BRCA, DNA Repair Associated |
| BRIP1 | BRCA1 Interacting Protein C-Terminal Helicase 1 |
| CA125 | Cancer Antigen 125 |
| CCNE1 | Cyclin E1 |
| CDKN2A | Cyclin Dependent Kinase Inhibitor 2A |
| CDKN2D | Cyclin Dependent Kinase Inhibitor 2D |
| CDK12 | Cyclin Dependent Kinase 12 |
| CHEK2 | Checkpoint Kinase 2 |
| CICs | Cortical Inclusion Cysts |
| CK7 | Cytokeratin 7 |
| CK20 | Cytokeratin 20 |
| CNAs | Copy Number Alterations |
| CT | Computer Tomography |
| CTNNB1 | Catenin Beta 1 |
| DICER | Dicer 1, Ribonuclease III |
| DNA | Deoxyribonucleic acid |
| EMA | European Medicines Agency |
| EOC | Epithelial Ovarian Cancer |
| ER | Estrogen Receptor |
| FDA | Food and Drug Administration |
| FIGO | International Federation of Gynecology and Obstetrics |
| FOXM1 | Forkhead Box M1 |
| FTSEC | Secretory Epithelial Cells of the Distal Fallopian Tube |
| GAB2 | GRB2 Associated Binding Protein 2 |
| GABRA6 | Gamma-Aminobutyric Acid Type A Receptor Alpha6 Subunit |
| g-H2AX | Gamma H2A histone family member X |
| GEMM | Genetically Engineered Mouse Models |
| GOG | Gynecologic Oncology Group |
| GWAS | Genome Wide Association Studies |
| HDM2 | HDM2 Proto-Oncogene |
| HGSOC | High Grade Serous Ovarian Cancer |
| HIF-1a | Hypoxia Inducible Factor 1 Subunit Alpha |
| HMGA2 | High Mobility Group AT-Hook 2 |
| HNF1b | HNF1 Homeobox B |
| HOXA9 | Homeobox A9 |
| IARC | International Agency for Research on Cancer |
| KRAS | KRAS Proto-Oncogene, GTPase |
| LC3 | Microtubule Associated Protein 1 Light Chain 3 Alpha |
| LOH | Loss of Heterozygosity |
| MAPK | Mitogen-Activated Protein Kinase 1 |
| MECOM | MDS1 And EVI1 Complex Locus |
| MRI | Magnetic Resonance Imaging |
| MRE11A | MRE11 Homolog, Double Strand Break Repair Nuclease |
| MYC | MYC Proto-Oncogene, BHLH Transcription Factor |
| NACT | Neoadjuvant Chemotherapy |
| NCI | American National Cancer Institute |
| NF1 | Neurofibromin 1 |
| NSG | NOD-SCID IL2R-gammanull |
| OSE | Ovarian Surface Epithelium |
| PALB2 | Partner and Localizer of BRCA2 |
| PARP | Poly (ADP-Ribose) Polymerase |
| PAX8 | Paired Box 8 |
| PDXs | Patient Derived Xerographs |
| PET | Positron-Emission Tomography |
| PI3K | Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit |
| PIK3CB | Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Beta |
| PIK3CA | Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha |
| PR | Progesterone Receptor |
| PTEN | Phosphatase and Tensin Homolog |
| P53 | Tumor Protein P53 |
| RAD50 | RAD50 Double Strand Break Repair Protein |
| RAD51C | RAD51 Paralog C |
| RAS | KRAS Proto-Oncogene, GTPase |
| RB | RB Transcriptional Corepressor |
| ROS | Radical Oxygen Species |
| RTK | Receptor Tyrosine Kinases |
| SCID | Severe Combined Immunodeficient |
| SEER | Surveillance Epidemiology and End Results |
| SET | Solid pseudo-Endometrioid and/or Transitional Cell Carcinoma-like |
| SNPs | Single Nucleotide Polymorphisms |
| STIC | Serous Tubal Intra Epithelial Carcinoma |
| TCGA | The Tumor Genome Atlas |
| UK | United Kingdom |
| VEGF-A | Vascular Endothelial Growth Factor A |
| WDFY2 | WD Repeat and FYVE Domain Containing 2 |
| WHO | World Health Organization |
| WNT | Wnt Family Member |
| WT1 | Wilms Tumor 1 |
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Berns, E.M.; Bowtell, D.D. The changing view of high-grade serous ovarian cancer. Cancer Res. 2012, 72, 2701–2704. [Google Scholar] [CrossRef] [PubMed]
- SEER Ovarian Cancer. Available online: https://seer.cancer.gov/statfacts/html/ovary.html (accessed on 26 May 2018).
- Canadian Cancer Statistics 2017. Available online: cancer.ca/Canadian-CancerStatistics-2017-EN.pdf (accessed on 26 May 2018).
- Vaughan, S.; Coward, J.I.; Bast, R.C., Jr.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih Ie, M. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer—Shifting the paradigm. Hum. Pathol. 2011, 42, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B.Y. Ovarian cancer. Nat. Rev. Dis. Primers 2016, 2, 16061. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Carcangiu, M.L.; Herrington, C.S.; Young, R.H. WHO Classification of Tumours of Female Reproductive Organs, 4th ed.; WHO: Geneva, Switzerland, 2014. [Google Scholar]
- Kurman, R.J.; Shih Ie, M. The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised and Expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef]
- Lengyel, E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef]
- Riopel, M.A.; Ronnett, B.M.; Kurman, R.J. Evaluation of diagnostic criteria and behaviour of ovarian intestinal-type mucinous tumours: Atypical proliferative (borderline) tumours and intraepithelial, microinvasive, invasive and metastatic carcinomas. Am. J. Surg. Pathol. 1999, 23, 617–635. [Google Scholar] [CrossRef]
- Veras, E.; Mao, T.L.; Ayhan, A.; Ueda, S.; Lai, H.; Hayran, M.; Shih Ie, M.; Kurman, R.J. Cystic and adenofibromatous clear cell carcinomas of the ovary: Distinctive tumours that differ in their pathogenesis and behaviour: A clinicopathologic analysis of 122 cases. Am. J. Surg. Pathol. 2009, 33, 844–853. [Google Scholar] [CrossRef]
- Shih Ie, M.; Kurman, R.J. Ovarian tumorigenesis: A proposed model based on morphological and molecular genetic analysis. Am. J. Pathol. 2004, 164, 1511–1518. [Google Scholar] [CrossRef]
- Bowtell, D.D.; Böhm, S.; Ahmed, A.A.; Aspuria, P.-J.; Bast, R.C., Jr.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A. Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Soslow, R.A.; Han, G.; Park, K.J.; Garg, K.; Olvera, N.; Spriggs, D.R.; Kauff, N.D.; Levine, D.A. Morphologic patterns associated with BRCA1 and BRCA2 genotype in ovarian carcinoma. Mod. Pathol. 2012, 25, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Kato, N.; Sasou, S.; Motoyama, T. Expression of hepatocyte nuclear factor-1beta (HNF-1beta) in clear cell tumours and endometriosis of the ovary. Mod. Pathol. 2006, 19, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Liliac, L.; Carcangiu, M.L.; Canevari, S.; Caruntu, I.D.; Ciobanu Apostol, D.G.; Danciu, M.; Onofriescu, M.; Amalinei, C. The value of PAX8 and WT1 molecules in ovarian cancer diagnosis. Rom. J. Morphol. Embryol. 2013, 54, 17–27. [Google Scholar]
- Webb, P.M.; Jordan, S.J. Epidemiology of epithelial ovarian cancer. Best Pract. Res. Clin. Obstet. Gynaecol. 2017, 41, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Stratton, J.F.; Pharoah, P.; Smith, S.K.; Easton, D.; Ponder, B.A. A systematic review and meta-analysis of family history and risk of ovarian cancer. Br. J. Obstet. Gynaecol. 1998, 105, 493–499. [Google Scholar] [CrossRef]
- Cunningham, J.M.; Cicek, M.S.; Larson, N.B.; Davila, J.; Wang, C.; Larson, M.C.; Song, H.; Dicks, E.M.; Harrington, P.; Wick, M.; et al. Clinical characteristics of ovarian cancer classified by BRCA1, BRCA2 and RAD51C status. Sci. Rep. 2014, 4, 4026. [Google Scholar] [CrossRef]
- Testa, U.; Petrucci, E.; Pasquini, L.; Castelli, G.; Pelosi, E. Ovarian Cancers: Genetic Abnormalities, Tumor Heterogeneity and Progression, Clonal Evolution and Cancer Stem Cells. Medicines 2018, 5, 16. [Google Scholar] [CrossRef]
- Ford, D.; Easton, D.F.; Stratton, M.; Narod, S.; Goldgar, D.; Devilee, P.; Bishop, D.T.; Weber, B.; Lenoir, G.; Chang-Claude, J.; et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am. J. Hum. Genet. 1998, 62, 676–689. [Google Scholar] [CrossRef]
- Song, H.; Dicks, E.; Ramus, S.J.; Tyrer, J.P.; Intermaggio, M.P.; Hayward, J.; Edlund, C.K.; Conti, D.; Harrington, P.; Fraser, L.; et al. Contribution of Germline Mutations in the RAD51B, RAD51C and RAD51D Genes to Ovarian Cancer in the Population. J. Clin. Oncol. 2015, 33, 2901–2907. [Google Scholar] [CrossRef] [PubMed]
- Ramus, S.J.; Song, H.; Dicks, E.; Tyrer, J.P.; Rosenthal, A.N.; Intermaggio, M.P.; Fraser, L.; Gentry-Maharaj, A.; Hayward, J.; Philpott, S.; et al. Germline Mutations in the BRIP1, BARD1, PALB2 and NBN Genes in Women with Ovarian Cancer. J. Natl. Cancer Inst. 2015, 107, djv214. [Google Scholar] [CrossRef] [PubMed]
- Norquist, B.M.; Harrell, M.I.; Brady, M.F.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Yi, Q.; Burger, R.A.; et al. Inherited Mutations in Women with Ovarian Carcinoma. JAMA Oncol. 2016, 2, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Ketabi, Z.; Bartuma, K.; Bernstein, I.; Malander, S.; Gronberg, H.; Bjorck, E.; Holck, S.; Nilbert, M. Ovarian cancer linked to Lynch syndrome typically presents as early-onset, non-serous epithelial tumors. Gynecol. Oncol. 2011, 121, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Phelan, C.M.; Kuchenbaecker, K.B.; Tyrer, J.P.; Kar, S.P.; Lawrenson, K.; Winham, S.J.; Dennis, J.; Pirie, A.; Riggan, M.J.; Chornokur, G.; et al. Identification of 12 new susceptibility loci for different histotypes of epithelial ovarian cancer. Nat. Genet. 2017, 49, 680–691. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.T.; Wu, Q.J.; Vogtmann, E.; Lin, B.; Wang, Y.L. Age at menarche and risk of ovarian cancer: A meta-analysis of epidemiological studies. Int. J. Cancer 2013, 132, 2894–2900. [Google Scholar] [CrossRef] [PubMed]
- Wentzensen, N.; Poole, E.M.; Trabert, B.; White, E.; Arslan, A.A.; Patel, A.V.; Setiawan, V.W.; Visvanathan, K.; Weiderpass, E.; Adami, H.O.; et al. Ovarian cancer risk factors by histologic subtype: An analysis from the Ovarian Cancer Cohort Consortium. J. Clin. Oncol. 2016, 34, 2888–2898. [Google Scholar] [CrossRef]
- Luan, N.N.; Wu, Q.J.; Gong, T.T.; Vogtmann, E.; Wang, Y.L.; Lin, B. Breastfeeding and ovarian cancer risk: A meta-analysis of epidemiologic studies. Am. J. Clin. Nutr. 2013, 98, 1020–1031. [Google Scholar] [CrossRef]
- Beral, V.; Doll, R.; Hermon, C.; Peto, R.; Reeves, G.; Brinton, L.; Green, A.C.; Marchbanks, P.; Negri, E.; Ness, R.; et al. Ovarian cancer and oral contraceptives: Collaborative reanalysis of data from 45 epidemiological studies including 23,257 women with ovarian cancer and 87,303 controls. Lancet 2008, 371, 303–314. [Google Scholar]
- Sieh, W.; Salvador, S.; McGuire, V.; Weber, R.P.; Terry, K.L.; Rossing, M.A.; Risch, H.; Wu, A.H.; Webb, P.M.; Moysich, K.; et al. Tubal ligation and risk of ovarian cancer subtypes: A pooled analysis of case-control studies. Int. J. Epidemiol. 2013, 42, 579–589. [Google Scholar] [CrossRef]
- Beral, V.; Gaitskell, K.; Hermon, C.; Moser, K.; Reeves, G.; Peto, R. Menopausal hormone use and ovarian cancer risk: Individual participant meta-analysis of 52 epidemiological studies. Lancet 2015, 385, 1835–1842. [Google Scholar] [PubMed]
- Whiteman, D.C.; Webb, P.M.; Green, A.C.; Neale, R.E.; Fritschi, L.; Bain, C.J.; Parkin, D.M.; Wilson, L.F.; Olsen, C.M.; Nagle, C.M.; et al. Cancers in Australia in 2010 attributable to modifiable factors: Summary and conclusions. Aust. New Zealand J. Public Health 2015, 39, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Klotz, D.M.; Wimberger, P. Cells of origin of ovarian cancer: Ovarian surface epithelium or fallopian tube? Arch. Gynecol. Obstet. 2017, 296, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Abubaker, K.; Findlay, J.; Quinn, M. Cancerous ovarian stem cells: Obscure targets for therapy but relevant to chemoresistance. J. Cell Biochem. 2013, 114, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Fathalla, M.F. Incessant ovulation—A factor in ovarian neoplasia? Lancet 1971, 2, 163. [Google Scholar] [CrossRef]
- Kuhn, E.; Kurman, R.J.; Shih, I.M. Ovarian Cancer Is an Imported Disease: Fact or Fiction? Curr. Obstet. Gynecol. Rep. 2012, 1, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih, I.-M. The Origin and Pathogenesis of Epithelial Ovarian Cancer—A Proposed Unifying Theory. Am. J. Surg. Pathol. 2010, 34, 433–443. [Google Scholar] [CrossRef]
- Cheng, W.; Liu, J.; Yoshida, H.; Rosen, D.; Naora, H. Lineage infidelity of epithelial ovarian cancers is controlled by HOX genes that specify regional identity in the reproductive tract. Nat. Med. 2005, 11, 531–537. [Google Scholar] [CrossRef]
- Karnezis, A.N.; Cho, K.R.; Gilks, C.B.; Pearce, C.L.; Huntsman, D.G. The disparate origins of ovarian cancers: Pathogenesis and prevention strategies. Nat. Rev. Cancer 2017, 17, 65–74. [Google Scholar] [CrossRef]
- Dubeau, L. The cell of origin of ovarian epithelial tumors and the ovarian surface epithelium dogma: Does the emperor have no clothes? Gynecol. Oncol. 1999, 72, 437–442. [Google Scholar] [CrossRef]
- Piek, J.M.; van Diest, P.J.; Zweemer, R.P.; Jansen, J.W.; Poort-Keesom, R.J.; Menko, F.H.; Gille, J.J.; Jongsma, A.P.; Pals, G.; Kenemans, P.; et al. Dysplastic changes in prophylactically removed Fallopian tubes of women predisposed to developing ovarian cancer. J. Pathol. 2001, 195, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, F.; Muto, M.G.; Lee, Y.; Elvin, J.A.; Callahan, M.J.; Feltmate, C.; Garber, J.E.; Cramer, D.W.; Crum, C.P. The tubal fimbria is a preferred site for early adenocarcinoma in women with familial ovarian cancer syndrome. Am. J. Surg. Pathol. 2006, 30, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Kindelberger, D.W.; Lee, Y.; Miron, A.; Hirsch, M.S.; Feltmate, C.; Medeiros, F.; Callahan, M.J.; Garner, E.O.; Gordon, R.W.; Birch, C.; et al. Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: Evidence for a causal relationship. Am. J. Surg. Pathol. 2007, 31, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, E.; Kurman, R.J.; Vang, R.; Sehdev, A.S.; Han, G.; Soslow, R.; Wang, T.L.; Shih Ie, M. TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma—Evidence supporting the clonal relationship of the two lesions. J. Pathol. 2012, 226, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, E.; Meeker, A.; Wang, T.L.; Sehdev, A.S.; Kurman, R.J.; Shih Ie, M. Shortened telomeres in serous tubal intraepithelial carcinoma: An early event in ovarian high-grade serous carcinogenesis. Am. J. Surg. Pathol. 2010, 34, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.C.; Blanco, L.Z., Jr.; Vang, R.; Ronnett, B.M. Incidental serous tubal intraepithelial carcinoma and early invasive serous carcinoma in the nonprophylactic setting: Analysis of a case series. Am. J. Surg. Pathol. 2015, 39, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, E.; Wang, T.L.; Doberstein, K.; Bahadirli-Talbott, A.; Ayhan, A.; Sehdev, A.S.; Drapkin, R.; Kurman, R.J.; Shih Ie, M. CCNE1 amplification and centrosome number abnormality in serous tubal intraepithelial carcinoma: Further evidence supporting its role as a precursor of ovarian high-grade serous carcinoma. Mod. Pathol. 2016, 29, 1254–1261. [Google Scholar] [CrossRef]
- Marquez, R.T.; Baggerly, K.A.; Patterson, A.P.; Liu, J.; Broaddus, R.; Frumovitz, M.; Atkinson, E.N.; Smith, D.I.; Hartmann, L.; Fishman, D.; et al. Patterns of gene expression in different histotypes of epithelial ovarian cancer correlate with those in normal fallopian tube, endometrium and colon. Clin. Cancer Res. 2005, 11, 6116–6126. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Ning, G.; Howitt, B.E.; Mehra, K.; Wu, L.; Wang, X.; Hong, Y.; Kern, F.; Wei, T.S.; Zhang, T.; et al. In vitro and in vivo correlates of physiological and neoplastic human Fallopian tube stem cells. J. Pathol. 2016, 238, 519–530. [Google Scholar] [CrossRef]
- Perets, R.; Wyant, G.A.; Muto, K.W.; Bijron, J.G.; Poole, B.B.; Chin, K.T.; Chen, J.Y.; Ohman, A.W.; Stepule, C.D.; Kwak, S.; et al. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer Cell 2013, 24, 751–765. [Google Scholar] [CrossRef]
- Auersperg, N. The stem-cell profile of ovarian surface epithelium is reproduced in the oviductal fimbriae, with increased stem-cell marker density in distal parts of the fimbriae. Int. J. Gynecol. Pathol. 2013, 32, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sacchetti, A.; van Dijk, M.R.; van der Zee, M.; van der Horst, P.H.; Joosten, R.; Burger, C.W.; Grootegoed, J.A.; Blok, L.J.; Fodde, R. Identification of quiescent, stem-like cells in the distal female reproductive tract. PLoS ONE 2012, 7, e40691. [Google Scholar] [CrossRef] [PubMed]
- Bast, R.C., Jr.; Hennessy, B.; Mills, G.B. The biology of ovarian cancer: New opportunities for translation. Nat. Rev. Cancer 2009, 9, 415–428. [Google Scholar] [CrossRef]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Naora, H.; Montell, D.J. Ovarian Cancer Metastasis: Integrating insights from disparate model organisms. Nat. Rev. Cancer 2005, 5, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Tarin, D.; Price, J.E.; Kettlewell, M.G.W.; Souter, R.G.; Vass, A.C.R.; Crossley, B. Mechanisms of Human Tumor Metastasis Studied in Patients with Peritoneovenous Shunts. Cancer Res. 1984, 44, 3584–3592. [Google Scholar]
- Narod, S. Can advanced-stage ovarian cancer be cured? Nat. Rev. Clin. Oncol. 2016, 13, 255–261. [Google Scholar] [CrossRef]
- Menon, U.; Ryan, A.; Kalsi, J.; Gentry-Maharaj, A.; Dawnay, A.; Habib, M.; Apostolidou, S.; Singh, N.; Benjamin, E.; Burnell, M.; et al. Risk Algorithm Using Serial Biomarker Measurements Doubles the Number of Screen-Detected Cancers Compared with a Single-Threshold Rule in the United Kingdom Collaborative Trial of Ovarian Cancer Screening. J. Clin. Oncol. 2015, 33, 2062–2071. [Google Scholar] [CrossRef]
- Jacobs, I.J.; Menon, U.; Ryan, A.; Gentry-Maharaj, A.; Burnell, M.; Kalsi, J.K.; Amso, N.N.; Apostolidou, S.; Benjamin, E.; Cruickshank, D.; et al. Ovarian cancer screening and mortality in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): A randomised controlled trial. Lancet 2016, 387, 945–956. [Google Scholar] [CrossRef]
- Walker, J.L.; Powell, C.B.; Chen, L.M.; Carter, J.; Bae Jump, V.L.; Parker, L.P.; Borowsky, M.E.; Gibb, R.K. Society of Gynecologic Oncology recommendations for the prevention of ovarian cancer. Cancer 2015, 121, 2108–2120. [Google Scholar] [CrossRef]
- Wang, Y.; Li, L.; Douville, C.; Cohen, J.D.; Yen, T.T.; Kinde, I.; Sundfelt, K.; Kjaer, S.K.; Hruban, R.H.; Shih, I.M.; et al. Evaluation of liquid from the Papanicolaou test and other liquid biopsies for the detection of endometrial and ovarian cancers. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.; Basso, O.; Sampalis, J.; Karp, I.; Martins, C.; Feng, J.; Piedimonte, S.; Quintal, L.; Ramanakumar, A.V.; Takefman, J.; et al. Assessment of symptomatic women for early diagnosis of ovarian cancer: Results from the prospective DOvE pilot project. Lancet Oncol. 2012, 13, 285–291. [Google Scholar] [CrossRef]
- Zivanovic, O.; Sima, C.S.; Iasonos, A.; Bell-McGuinn, K.M.; Sabbatini, P.J.; Leitao, M.M.; Levine, D.A.; Gardner, G.J.; Barakat, R.R.; Chi, D.S. Exploratory analysis of serum CA-125 response to surgery and the risk of relapse in patients with FIGO stage IIIC ovarian cancer. Gynecol. Oncol. 2009, 115, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Mutch, D.G.; Prat, J. 2014 FIGO staging for ovarian, fallopian tube and peritoneal cancer. Gynecol. Oncol. 2014, 133, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Network, C.G.A.R. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Vang, R.; Levine, D.A.; Soslow, R.A.; Zaloudek, C.; Shih Ie, M.; Kurman, R.J. Molecular Alterations of TP53 are a Defining Feature of Ovarian High-Grade Serous Carcinoma: A Rereview of Cases Lacking TP53 Mutations in The Cancer Genome Atlas Ovarian Study. Int. J. Gynecol. Pathol. 2016, 35, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, L.; Nguyen, D.; Lu, H. TP53 mutations in epithelial ovarian cancer. Transl. Cancer Res. 2016, 5, 650–663. [Google Scholar] [CrossRef]
- Seagle, B.L.; Eng, K.H.; Dandapani, M.; Yeh, J.Y.; Odunsi, K.; Shahabi, S. Survival of patients with structurally-grouped TP53 mutations in ovarian and breast cancers. Oncotarget 2015, 6, 18641–18652. [Google Scholar] [CrossRef]
- Brachova, P.; Mueting, S.R.; Carlson, M.J.; Goodheart, M.J.; Button, A.M.; Mott, S.L.; Dai, D.; Thiel, K.W.; Devor, E.J.; Leslie, K.K. TP53 oncomorphic mutations predict resistance to platinum and taxane based standard chemotherapy in patients diagnosed with advanced serous ovarian carcinoma. Int. J. Oncol. 2015, 46, 607–618. [Google Scholar] [CrossRef]
- Rzepecka, I.K.; Szafron, L.; Stys, A.; Bujko, M.; Plisiecka-Halasa, J.; Madry, R.; Osuch, B.; Markowska, J.; Bidzinski, M.; Kupryjanczyk, J. High frequency of allelic loss at the BRCA1 locus in ovarian cancers: Clinicopathologic and molecular associations. Cancer Genet. 2012, 205, 94–100. [Google Scholar] [CrossRef]
- Kanchi, K.L.; Johnson, K.J.; Lu, C.; McLellan, M.D.; Leiserson, M.D.; Wendl, M.C.; Zhang, Q.; Koboldt, D.C.; Xie, M.; Kandoth, C.; et al. Integrated analysis of germline and somatic variants in ovarian cancer. Nat. Commun. 2014, 5, 3156. [Google Scholar] [CrossRef] [PubMed]
- Labidi-Galy, S.I.; Olivier, T.; Rodrigues, M.; Ferraioli, D.; Derbel, O.; Bodmer, A.; Petignat, P.; Rak, B.; Chopin, N.; Tredan, O.; et al. Location of Mutation in BRCA2 Gene and Survival in Patients with Ovarian Cancer. Clin. Cancer Res. 2018, 24, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Candido-dos-Reis, F.J.; Song, H.; Goode, E.L.; Cunningham, J.M.; Fridley, B.L.; Larson, M.C.; Alsop, K.; Dicks, E.; Harrington, P.; Ramus, S.J.; et al. Germline mutation in BRCA1 or BRCA2 and ten-year survival for women diagnosed with epithelial ovarian cancer. Clin. Cancer Res. 2015, 21, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, L.; Greshock, J.; Colligon, T.A.; Wang, Y.; Ward, R.; Katsaros, D.; Lassus, H.; Butzow, R.; Godwin, A.K.; et al. Frequent genetic abnormalities of the PI3K/AKT pathway in primary ovarian cancer predict patient outcome. Genes Chromosomes Cancer 2011, 50, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.C.; Santiago, I.; Trinh, A.; Xian, J.; Guo, A.; Sayal, K.; Jimenez-Linan, M.; Deen, S.; Driver, K.; Mack, M.; et al. Combined image and genomic analysis of high-grade serous ovarian cancer reveals PTEN loss as a common driver event and prognostic classifier. Genome Biol. 2014, 15, 526. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Cheung, H.W.; Agarwalla, P.K.; Thomas, S.; Zektser, Y.; Karst, A.M.; Boehm, J.S.; Weir, B.A.; Berlin, A.M.; Zou, L.; et al. In vivo multiplexed interrogation of amplified genes identifies GAB2 as an ovarian cancer oncogene. Proc. Natl. Acad. Sci. USA 2014, 111, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Delaney, J.R.; Patel, C.B.; Willis, K.M.; Haghighiabyaneh, M.; Axelrod, J.; Tancioni, I.; Lu, D.; Bapat, J.; Young, S.; Cadassou, O.; et al. Haploinsufficiency networks identify targetable patterns of allelic deficiency in low mutation ovarian cancer. Nat. Commun. 2017, 8, 14423. [Google Scholar] [CrossRef] [PubMed]
- Shell, S.; Park, S.-M.; Radjabi, A.R.; Schickel, R.; Kistner, E.O.; Jewell, D.A.; Feig, C.; Lengyel, E.; Peter, M.E. Let-7 expression defines two differentiation stages of cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 11400–11405. [Google Scholar] [CrossRef]
- Mahajan, A.; Liu, Z.; Gellert, L.; Zou, X.; Yang, G.; Lee, P.; Yang, X.; Wei, J.-J. HMGA2: A biomarker significantly overexpressed in high-grade ovarian serous carcinoma. Mod. Pathol. 2010, 23, 673–681. [Google Scholar] [CrossRef]
- Kannan, K.; Coarfa, C.; Rajapakshe, K.; Hawkins, S.M.; Matzuk, M.M.; Milosavljevic, A.; Yen, L. CDKN2D-WDFY2 is a cancer-specific fusion gene recurrent in high-grade serous ovarian carcinoma. PLoS Genet. 2014, 10, e1004216. [Google Scholar] [CrossRef]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Tamayo, P.; Yang, J.Y.; Hubbard, D.; Zhang, H.; Creighton, C.J.; Fereday, S.; Lawrence, M.; Carter, S.L.; Mermel, C.H.; et al. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J. Clin. Investig. 2013, 123, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Konecny, G.E.; Wang, C.; Hamidi, H.; Winterhoff, B.; Kalli, K.R.; Dering, J.; Ginther, C.; Chen, H.W.; Dowdy, S.; Cliby, W.; et al. Prognostic and therapeutic relevance of molecular subtypes in high-grade serous ovarian cancer. J. Natl. Cancer Inst. 2014, 106, dju249. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Armasu, S.M.; Kalli, K.R.; Maurer, M.J.; Heinzen, E.P.; Keeney, G.L.; Cliby, W.A.; Oberg, A.L.; Kaufmann, S.H.; Goode, E.L. Pooled Clustering of High-Grade Serous Ovarian Cancer Gene Expression Leads to Novel Consensus Subtypes Associated with Survival and Surgical Outcomes. Clin. Cancer Res. 2017, 23, 4077–4085. [Google Scholar] [CrossRef] [PubMed]
- Murakami, R.; Matsumura, N.; Mandai, M.; Yoshihara, K.; Tanabe, H.; Nakai, H.; Yamanoi, K.; Abiko, K.; Yoshioka, Y.; Hamanishi, J.; et al. Establishment of a Novel Histopathological Classification of High-Grade Serous Ovarian Carcinoma Correlated with Prognostically Distinct Gene Expression Subtypes. Am. J. Pathol. 2016, 186, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; D’Andrea, A.D.; Kozono, D. A DNA repair pathway-focused score for prediction of outcomes in ovarian cancer treated with platinum-based chemotherapy. J. Natl. Cancer Inst. 2012, 104, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Matondo, A.; Jo, Y.H.; Shahid, M.; Choi, T.G.; Nguyen, M.N.; Nguyen, N.N.Y.; Akter, S.; Kang, I.; Ha, J.; Maeng, C.H.; et al. The Prognostic 97 Chemoresponse Gene Signature in Ovarian Cancer. Sci. Rep. 2017, 7, 9689. [Google Scholar] [CrossRef]
- Cheon, D.J.; Tong, Y.; Sim, M.S.; Dering, J.; Berel, D.; Cui, X.; Lester, J.; Beach, J.A.; Tighiouart, M.; Walts, A.E.; et al. A collagen-remodeling gene signature regulated by TGF-beta signaling is associated with metastasis and poor survival in serous ovarian cancer. Clin. Cancer Res. 2014, 20, 711–723. [Google Scholar] [CrossRef]
- Riester, M.; Wei, W.; Waldron, L.; Culhane, A.C.; Trippa, L.; Oliva, E.; Kim, S.-H.; Michor, F.; Huttenhower, C.; Parmigiani, G.; et al. Risk Prediction for Late-Stage Ovarian Cancer by Meta-analysis of 1525 Patient Samples. J. Natl. Cancer Inst. 2014, 106, dju048. [Google Scholar] [CrossRef]
- McGrail, D.J.; Lin, C.C.-J.; Garnett, J.; Liu, Q.; Mo, W.; Dai, H.; Lu, Y.; Yu, Q.; Ju, Z.; Yin, J.; et al. Improved prediction of PARP inhibitor response and identification of synergizing agents through use of a novel gene expression signature generation algorithm. NPJ Syst. Biol. Appl. 2017, 3, 8. [Google Scholar] [CrossRef]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- Chang, S.-J.; Bristow, R.E.; Ryu, H.-S. Impact of complete cytoreduction leaving no gross residual disease associated with radical cytoreductive surgical procedures on survival in advanced ovarian cancer. Ann. Surg. Oncol. 2012, 19, 4059–4067. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-J.; Hodeib, M.; Chang, J.; Bristow, R.E. Survival impact of complete cytoreduction to no gross residual disease for advanced-stage ovarian cancer: A meta-analysis. Gynecol. Oncol. 2013, 130, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, N.S.; Miller, A.; Rungruang, B.; Richard, S.D.; Rodriguez, N.; Bookman, M.A.; Hamilton, C.A.; Krivak, T.C.; Maxwell, G.L. Does aggressive surgery improve outcomes? Interaction between preoperative disease burden and complex surgery in patients with advanced-stage ovarian cancer: An analysis of GOG 182. J. Clin. Oncol. 2015, 33, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Martin, A.; Sanchez-Lorenzo, L.; Bratos, R.; Marquez, R.; Chiva, L. First-line and maintenance therapy for ovarian cancer: Current status and future directions. Drugs 2014, 74, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Markman, M. Antineoplastic agents in the management of ovarian cancer: Current status and emerging therapeutic strategies. Trends Pharmacol. Sci. 2008, 29, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Markman, M. Optimizing primary chemotherapy in ovarian cancer. Hematol. Oncol. Clin. N. Am. 2003, 17, 957–968. [Google Scholar] [CrossRef]
- Thigpen, T.; Vance, R.; Lambuth, B.; Balducci, L.; Khansur, T.; Blessing, J.; McGehee, R. Chemotherapy for advanced or recurrent gynecologic cancer. Cancer 1987, 60, 2104–2116. [Google Scholar] [CrossRef]
- Alberts, D.S.; Green, S.; Hannigan, E.V.; O’Toole, R.; Stock-Novack, D.; Anderson, P.; Surwit, E.A.; Malvlya, V.K.; Nahhas, W.A.; Jolies, C.J. Improved therapeutic index of carboplatin plus cyclophosphamide versus cisplatin plus cyclophosphamide: Final report by the Southwest Oncology Group of a phase III randomized trial in Stages III and IV ovarian cancer. J. Clin. Oncol. 1992, 10, 706–717. [Google Scholar] [CrossRef]
- Swenerton, K.; Jeffrey, J.; Stuart, G.; Roy, M.; Krepart, G.; Carmichael, J.; Drouin, P.; Stanimir, R.; O’Connell, G.; MacLean, G.; et al. Cisplatin-cyclophosphamide versus carboplatin-cyclophosphamide in advanced ovarian cancer: A randomized phase III study of the national cancer institute of Canada clinical trials group. J. Clin. Oncol. 1992, 10, 718–726. [Google Scholar] [CrossRef]
- Bukowska, B.; Gajek, A.; Marczak, A. Two drugs are better than one. A short history of combined therapy of ovarian cancer. Contemp. Oncol. 2015, 19, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Thigpen, J.T.; Blessing, J.A.; Ball, H.; Hummel, S.J.; Barrett, R.J. Phase II trial of paclitaxel in patients with progressive ovarian carcinoma after platinum-based chemotherapy: A Gynecologic Oncology Group study. J. Clin. Oncol. 1994, 12, 1748–1753. [Google Scholar] [CrossRef] [PubMed]
- McGuire, W.P.; Rowinsky, E.K.; Rosenhein, N.B.; Grumbine, F.C.; Ettinger, D.S.; Armstrong, D.K.; Donehower, R.C. Taxol: A unique antineoplastic agent with significant activity in advanced ovarian epithelial neoplasms. Ann. Intern. Med. 1989, 111, 273–279. [Google Scholar] [CrossRef] [PubMed]
- McGuire, W.P.; Hoskins, W.J.; Brady, M.F.; Kucera, P.R.; Partridge, E.E.; Look, K.Y.; Clarke-Pearson, D.L.; Davidson, M. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N. Engl. J. Med. 1996, 334, 1–6. [Google Scholar] [CrossRef] [PubMed]
- International Collaborative Ovarian Neoplasm Group. Paclitaxel plus carboplatin versus standard chemotherapy with either single-agent carboplatin or cyclophosphamide, doxorubicin and cisplatin in women with ovarian cancer: The ICON3 randomised trial. Lancet 2002, 360, 505–515. [Google Scholar] [CrossRef]
- Bookman, M.A.; Greer, B.E.; Ozols, R.F. Optimal therapy of advanced ovarian cancer: Carboplatin and paclitaxel versus cisplatin and paclitaxel (GOG158) and an update on GOG0182-ICON5. Int. J. Gynecol. Cancer 2003, 13 (Suppl. 2), 149–155. [Google Scholar] [CrossRef]
- Neijt, J.P.; Engelholm, S.A.; Tuxen, M.K.; Sorensen, P.G.; Hansen, M.; Sessa, C.; de Swart, C.A.; Hirsch, F.R.; Lund, B.; van Houwelingen, H.C. Exploratory phase III study of paclitaxel and cisplatin versus paclitaxel and carboplatin in advanced ovarian cancer. J. Clin. Oncol. 2000, 18, 3084–3092. [Google Scholar] [CrossRef]
- du Bois, A.; Luck, H.J.; Meier, W.; Adams, H.P.; Mobus, V.; Costa, S.; Bauknecht, T.; Richter, B.; Warm, M.; Schroder, W.; et al. A randomized clinical trial of cisplatin/paclitaxel versus carboplatin/paclitaxel as first-line treatment of ovarian cancer. J. Natl. Cancer Inst. 2003, 95, 1320–1329. [Google Scholar] [CrossRef]
- Katsumata, N.; Yasuda, M.; Isonishi, S.; Michimae, H.; Kimura, E.; Aoki, D.; Jobo, T.; Kodama, S.; Terauchi, F.; Tsuda, H. Long-Term Follow-up of a Randomized Trial Comparing Conventional Paclitaxel and Carboplatin with Dose-Dense Weekly Paclitaxel and Carboplatin in Women with Advanced Epithelial Ovarian, Fallopian Tube or Primary Peritoneal Cancer: JGOG 3016 Trial; American Society of Clinical Oncology: Alexandria, VA, USA, 2012. [Google Scholar]
- Pignata, S.; Scambia, G.; Katsaros, D.; Gallo, C.; Pujade-Lauraine, E.; De Placido, S.; Bologna, A.; Weber, B.; Raspagliesi, F.; Panici, P.B.; et al. Carboplatin plus paclitaxel once a week versus every 3 weeks in patients with advanced ovarian cancer (MITO-7): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 396–405. [Google Scholar] [CrossRef]
- Chan, J.K.; Brady, M.F.; Penson, R.T.; Huang, H.; Birrer, M.J.; Walker, J.L.; DiSilvestro, P.A.; Rubin, S.C.; Martin, L.P.; Davidson, S.A.; et al. Weekly vs. Every-3-Week Paclitaxel and Carboplatin for Ovarian Cancer. N. Engl. J. Med. 2016, 374, 738–748. [Google Scholar] [CrossRef]
- Lopez, J.A.; Krikorian, J.G.; Reich, S.D.; Smyth, R.D.; Lee, F.H.; Issell, B.F. Clinical pharmacology of intraperitoneal cisplatin. Gynecol. Oncol. 1985, 20, 1–9. [Google Scholar] [CrossRef]
- Francis, P.; Rowinsky, E.; Schneider, J.; Hakes, T.; Hoskins, W.; Markman, M. Phase I feasibility and pharmacologic study of weekly intraperitoneal paclitaxel: A Gynecologic Oncology Group pilot Study. J. Clin. Oncol. 1995, 13, 2961–2967. [Google Scholar] [CrossRef]
- Alberts, D.S.; Liu, P.; Hannigan, E.V.; O’toole, R.; Williams, S.D.; Young, J.A.; Franklin, E.W.; Clarke-Pearson, D.L.; Malviya, V.K.; DuBeshter, B. Intraperitoneal cisplatin plus intravenous cyclophosphamide versus intravenous cisplatin plus intravenous cyclophosphamide for stage III ovarian cancer. N. Engl. J. Med. 1996, 335, 1950–1955. [Google Scholar] [CrossRef] [PubMed]
- Markman, M.; Bundy, B.N.; Alberts, D.S.; Fowler, J.M.; Clark-Pearson, D.L.; Carson, L.F.; Wadler, S.; Sickel, J. Phase III trial of standard-dose intravenous cisplatin plus paclitaxel versus moderately high-dose carboplatin followed by intravenous paclitaxel and intraperitoneal cisplatin in small-volume stage III ovarian carcinoma: An intergroup study of the Gynecologic Oncology Group, Southwestern Oncology Group and Eastern Cooperative Oncology Group. J. Clin. Oncol. 2001, 19, 1001–1007. [Google Scholar] [PubMed]
- Armstrong, D.K.; Bundy, B.; Wenzel, L.; Huang, H.Q.; Baergen, R.; Lele, S.; Copeland, L.J.; Walker, J.L.; Burger, R.A. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N. Engl. J. Med. 2006, 354, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.J.; Chan, J.K. Is intraperitoneal chemotherapy still an acceptable option in primary adjuvant chemotherapy for advanced ovarian cancer? Ann. Oncol. 2017, 28 (Suppl. 8), viii40–viii45. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.; Brady, M.; DiSilvestro, P.; Fujiwara, K.; Alberts, D.; Zheng, W.; Tewari, K.; Cohn, D.; Powell, M.; Van Le, L. A phase III trial of bevacizumab with IV versus IP chemotherapy for ovarian, fallopian tube and peritoneal carcinoma: An NRG Oncology Study. Gynecol. Oncol. 2016, 141, 208. [Google Scholar] [CrossRef]
- Walker, J.L.; Wenzel, L.; Huang, H.; Brady, M.F. Patient-reported outcomes in GOG 252: NRG Oncology Study of IV vs IP chemotherapy for ovarian, fallopian or peritoneal carcinoma. Gynecol. Oncol. 2016, 141, 208. [Google Scholar] [CrossRef]
- Vergote, I.; Tropé, C.G.; Amant, F.; Kristensen, G.B.; Ehlen, T.; Johnson, N.; Verheijen, R.H.; van der Burg, M.E.; Lacave, A.J.; Panici, P.B. Neoadjuvant chemotherapy or primary surgery in stage IIIC or IV ovarian cancer. N. Engl. J. Med. 2010, 363, 943–953. [Google Scholar] [CrossRef]
- Kehoe, S.; Hook, J.; Nankivell, M.; Jayson, G.C.; Kitchener, H.; Lopes, T.; Luesley, D.; Perren, T.; Bannoo, S.; Mascarenhas, M. Primary chemotherapy versus primary surgery for newly diagnosed advanced ovarian cancer (CHORUS): An open-label, randomised, controlled, non-inferiority trial. Lancet 2015, 386, 249–257. [Google Scholar] [CrossRef]
- Rustin, G.J.; Van Der Burg, M.E.; Griffin, C.L.; Guthrie, D.; Lamont, A.; Jayson, G.C.; Kristensen, G.; Mediola, C.; Coens, C.; Qian, W. Early versus delayed treatment of relapsed ovarian cancer (MRC OV05/EORTC 55955): A randomised trial. Lancet 2010, 376, 1155–1163. [Google Scholar] [CrossRef]
- Al Rawahi, T.; Lopes, A.; Bristow, R.; Bryant, A.; Elattar, A.; Chattopadhyay, S.; Galaal, K. Surgical cytoreduction for recurrent epithelial ovarian cancer. Cochrane Database Syst. Rev. 2013, 2, CD008765. [Google Scholar] [CrossRef] [PubMed]
- Papa, A.; Caruso, D.; Strudel, M.; Tomao, S.; Tomao, F. Update on Poly-ADP-ribose polymerase inhibition for ovarian cancer treatment. J. Transl. Med. 2016, 14, 267. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. The Concept of Synthetic Lethality in the Context of Anticancer Therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; De Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly(ADP)-ribose polymerase inhibition: Frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol. 2010, 28, 2512–2519. [Google Scholar] [CrossRef] [PubMed]
- Kaye, S.B.; Lubinski, J.; Matulonis, U.; Ang, J.E.; Gourley, C.; Karlan, B.Y.; Amnon, A.; Bell-McGuinn, K.M.; Chen, L.M.; Friedlander, M.; et al. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J. Clin. Oncol. 2012, 30, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Gelmon, K.A.; Tischkowitz, M.; Mackay, H.; Swenerton, K.; Robidoux, A.; Tonkin, K.; Hirte, H.; Huntsman, D.; Clemons, M.; Gilks, B.; et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: A phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011, 12, 852–861. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Oza, A.M.; Cibula, D.; Benzaquen, A.O.; Poole, C.; Mathijssen, R.H.; Sonke, G.S.; Colombo, N.; Spacek, J.; Vuylsteke, P.; Hirte, H.; et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: A randomised phase 2 trial. Lancet Oncol. 2015, 16, 87–97. [Google Scholar] [CrossRef]
- Pignata, S.; Cecere, S.; Du Bois, A.; Harter, P.; Heitz, F. Treatment of recurrent ovarian cancer. Ann. Oncol. 2017, 28 (Suppl. 8), viii51–viii56. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]
- Oza, A.M.; Cook, A.D.; Pfisterer, J.; Embleton, A.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A. Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): Overall survival results of a phase 3 randomised trial. Lancet Oncol. 2015, 16, 928–936. [Google Scholar] [CrossRef]
- Burger, R.A.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Huang, H.; Mannel, R.S.; Homesley, H.D.; Fowler, J.; Greer, B.E. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N. Engl. J. Med. 2011, 365, 2473–2483. [Google Scholar] [CrossRef] [PubMed]
- Aghajanian, C.; Blank, S.V.; Goff, B.A.; Judson, P.L.; Teneriello, M.G.; Husain, A.; Sovak, M.A.; Yi, J.; Nycum, L.R. OCEANS: A randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal or fallopian tube cancer. J. Clin. Oncol. 2012, 30, 2039–2045. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Hilpert, F.; Weber, B.; Reuss, A.; Poveda, A.; Kristensen, G.; Sorio, R.; Vergote, I.; Witteveen, P.; Bamias, A.; et al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: The AURELIA open-label randomized phase III trial. J. Clin. Oncol. 2014, 32, 1302–1308. [Google Scholar] [CrossRef]
- Du Bois, A.; Floquet, A.; Kim, J.-W.; Rau, J.; Del Campo, J.M.; Friedlander, M.; Pignata, S.; Fujiwara, K.; Vergote, I.; Colombo, N. Incorporation of pazopanib in maintenance therapy of ovarian cancer. J. Clin. Oncol. 2014, 32, 3374–3382. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Berlin, S.; Ivy, P.; Tyburski, K.; Krasner, C.; Zarwan, C.; Berkenblit, A.; Campos, S.; Horowitz, N.; Cannistra, S.A. Cediranib, an oral inhibitor of vascular endothelial growth factor receptor kinases, is an active drug in recurrent epithelial ovarian, fallopian tube and peritoneal cancer. J. Clin. Oncol. 2009, 27, 5601–5606. [Google Scholar] [CrossRef]
- Hirte, H.; Lheureux, S.; Fleming, G.F.; Sugimoto, A.; Morgan, R.; Biagi, J.; Wang, L.; McGill, S.; Ivy, S.P.; Oza, A.M. A phase 2 study of cediranib in recurrent or persistent ovarian, peritoneal or fallopian tube cancer: A trial of the Princess Margaret, Chicago and California Phase II Consortia. Gynecol. Oncol. 2015, 138, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.A.; Embleton, A.C.; Raja, F.; Perren, T.J.; Jayson, G.C.; Rustin, G.J.; Kaye, S.B.; Hirte, H.; Eisenhauer, E.; Vaughan, M. Cediranib in patients with relapsed platinum-sensitive ovarian cancer (ICON6): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2016, 387, 1066–1074. [Google Scholar] [CrossRef]
- Stark, D.P.; Cook, A.; Brown, J.M.; Brundage, M.D.; Embleton, A.C.; Kaplan, R.S.; Raja, F.A.; Swart, A.M.W.; Velikova, G.; Qian, W.; et al. Quality of life with cediranib in relapsed ovarian cancer: The ICON6 phase 3 randomized clinical trial. Cancer 2017, 123, 2752–2761. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Barry, W.T.; Birrer, M.; Lee, J.M.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.; Buss, M.K.; Nattam, S.; Hurteau, J.; et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: A randomised phase 2 study. Lancet Oncol. 2014, 15, 1207–1214. [Google Scholar] [CrossRef]
- Liu, J.F.; Barry, W.T.; Birrer, M.J.; Lee, J.-M.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.; Buss, M.K.; Nattam, S.R.; Hurteau, J. Overall Survival and Updated Progression-Free Survival Results from a Randomized Phase 2 Trial Comparing the Combination of Olaparib and Cediranib against Olaparib Alone in Recurrent Platinum-Sensitive Ovarian Cancer; American Society of Clinical Oncology: Alexandria, VA, USA, 2017. [Google Scholar]
- Blagden, S.; Hamilton, A.; Mileshkin, L.; Hall, M.; Meniawy, T.; Wong, S.; Anandra, S.; Buck, M.; McAleer, D.; Reedy, B. 3 Afuresertib (GSK2110183), an oral AKT kinase inhibitor, in combination with carboplatin and paclitaxel in recurrent ovarian cancer. Eur. J. Cancer 2014, 50, 7. [Google Scholar] [CrossRef]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef] [PubMed]
- Elias, K.M.; Emori, M.M.; Papp, E.; MacDuffie, E.; Konecny, G.E.; Velculescu, V.E.; Drapkin, R. Beyond genomics: Critical evaluation of cell line utility for ovarian cancer research. Gynecol. Oncol. 2015, 139, 97–103. [Google Scholar] [CrossRef]
- Coscia, F.; Watters, K.M.; Curtis, M.; Eckert, M.A.; Chiang, C.Y.; Tyanova, S.; Montag, A.; Lastra, R.R.; Lengyel, E.; Mann, M. Integrative proteomic profiling of ovarian cancer cell lines reveals precursor cell associated proteins and functional status. Nat. Commun. 2016, 7, 12645. [Google Scholar] [CrossRef]
- Langdon, S.P.; Lawrie, S.S.; Hay, F.G.; Hawkes, M.M.; McDonald, A.; Hayward, I.P.; Schol, D.J.; Hilgers, J.; Leonard, R.C.; Smyth, J.F. Characterization and properties of nine human ovarian adenocarcinoma cell lines. Cancer Res. 1988, 48, 6166–6172. [Google Scholar]
- Beaufort, C.M.; Helmijr, J.C.A.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van Ijcken, W.F.J.; Heine, A.A.J.; Smid, M.; et al. Ovarian Cancer Cell Line Panel (OCCP): Clinical Importance of In Vitro Morphological Subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef]
- Letourneau, I.J.; Quinn, M.C.; Wang, L.L.; Portelance, L.; Caceres, K.Y.; Cyr, L.; Delvoye, N.; Meunier, L.; de Ladurantaye, M.; Shen, Z.; et al. Derivation and characterization of matched cell lines from primary and recurrent serous ovarian cancer. BMC Cancer 2012, 12, 379. [Google Scholar] [CrossRef] [PubMed]
- Kenny, H.A.; Krausz, T.; Yamada, S.D.; Lengyel, E. Use of a novel 3D culture model to elucidate the role of mesothelial cells, fibroblasts and extra-cellular matrices on adhesion and invasion of ovarian cancer cells to the omentum. Int. J. Cancer 2007, 121, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E.; Burdette, J.E.; Kenny, H.A.; Matei, D.; Pilrose, J.; Haluska, P.; Nephew, K.P.; Hales, D.B.; Stack, M.S. Epithelial Ovarian Cancer Experimental Models. Oncogene 2014, 33, 3619–3633. [Google Scholar] [CrossRef] [PubMed]
- Goyeneche, A.A.; Telleria, C.M. Ovarian Cancer Research in the Post Genomic Era—Challenges and Opportunities. In Gynecologic Cancers-Basic Sciences, Clinical and Therapeutic Perspectives; InTech: London, UK, 2016. [Google Scholar]
- Kim, J.; Coffey, D.M.; Creighton, C.J.; Yu, Z.; Hawkins, S.M.; Matzuk, M.M. High-grade serous ovarian cancer arises from fallopian tube in a mouse model. Proc. Natl. Acad. Sci. USA 2012, 109, 3921–3926. [Google Scholar] [CrossRef] [PubMed]
- Sherman-Baust, C.A.; Kuhn, E.; Valle, B.L.; Shih, I.-M.; Kurman, R.J.; Wang, T.-L.; Amano, T.; Ko, M.S.H.; Miyoshi, I.; Araki, Y.; et al. A genetically engineered ovarian cancer mouse model based on fallopian tube transformation mimics human high-grade serous carcinoma development. J. Pathol. 2014, 233, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Wu, R.; Kuick, R.; Sessine, M.S.; Schulman, S.; Green, M.; Fearon, E.R.; Cho, K.R. High-grade serous carcinomas arise in the mouse oviduct via defects linked to the human disease. J. Pathol. 2017, 243, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Shaw, T.J.; Senterman, M.K.; Dawson, K.; Crane, C.A.; Vanderhyden, B.C. Characterization of intraperitoneal, orthotopic, and metastatic xenograft models of human ovarian cancer. Mol. Ther. 2004, 10, 1032–1042. [Google Scholar] [CrossRef]
- Mitra, A.K.; Davis, D.A.; Tomar, S.; Roy, L.; Gurler, H.; Xie, J.; Lantvit, D.D.; Cardenas, H.; Fang, F.; Liu, Y. In vivo tumor growth of high-grade serous ovarian cancer cell lines. Gynecol. Oncol. 2015, 138, 372–377. [Google Scholar] [CrossRef]
- Dobbin, Z.C.; Katre, A.A.; Steg, A.D.; Erickson, B.K.; Shah, M.M.; Alvarez, R.D.; Conner, M.G.; Schneider, D.; Chen, D.; Landen, C.N. Using heterogeneity of the patient-derived xenograft model to identify the chemoresistant population in ovarian cancer. Oncotarget 2014, 5, 8750–8764. [Google Scholar] [CrossRef]
- Eoh, K.J.; Chung, Y.S.; Lee, S.H.; Park, S.A.; Kim, H.J.; Yang, W.; Lee, I.O.; Lee, J.Y.; Cho, H.; Chay, D.B.; et al. Comparison of Clinical Features and Outcomes in Epithelial Ovarian Cancer according to Tumorigenicity in Patient-Derived Xenograft Models. Cancer Res. Treat. 2018, 50, 956–963. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lisio, M.-A.; Fu, L.; Goyeneche, A.; Gao, Z.-h.; Telleria, C. High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints. Int. J. Mol. Sci. 2019, 20, 952. https://doi.org/10.3390/ijms20040952
Lisio M-A, Fu L, Goyeneche A, Gao Z-h, Telleria C. High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints. International Journal of Molecular Sciences. 2019; 20(4):952. https://doi.org/10.3390/ijms20040952
Chicago/Turabian StyleLisio, Michael-Antony, Lili Fu, Alicia Goyeneche, Zu-hua Gao, and Carlos Telleria. 2019. "High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints" International Journal of Molecular Sciences 20, no. 4: 952. https://doi.org/10.3390/ijms20040952
APA StyleLisio, M.-A., Fu, L., Goyeneche, A., Gao, Z.-h., & Telleria, C. (2019). High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints. International Journal of Molecular Sciences, 20(4), 952. https://doi.org/10.3390/ijms20040952