The Role of Fibrinolytic Regulators in Vascular Dysfunction of Systemic Sclerosis


{kind=link}
{kind=link}
{kind=link}
Abstract
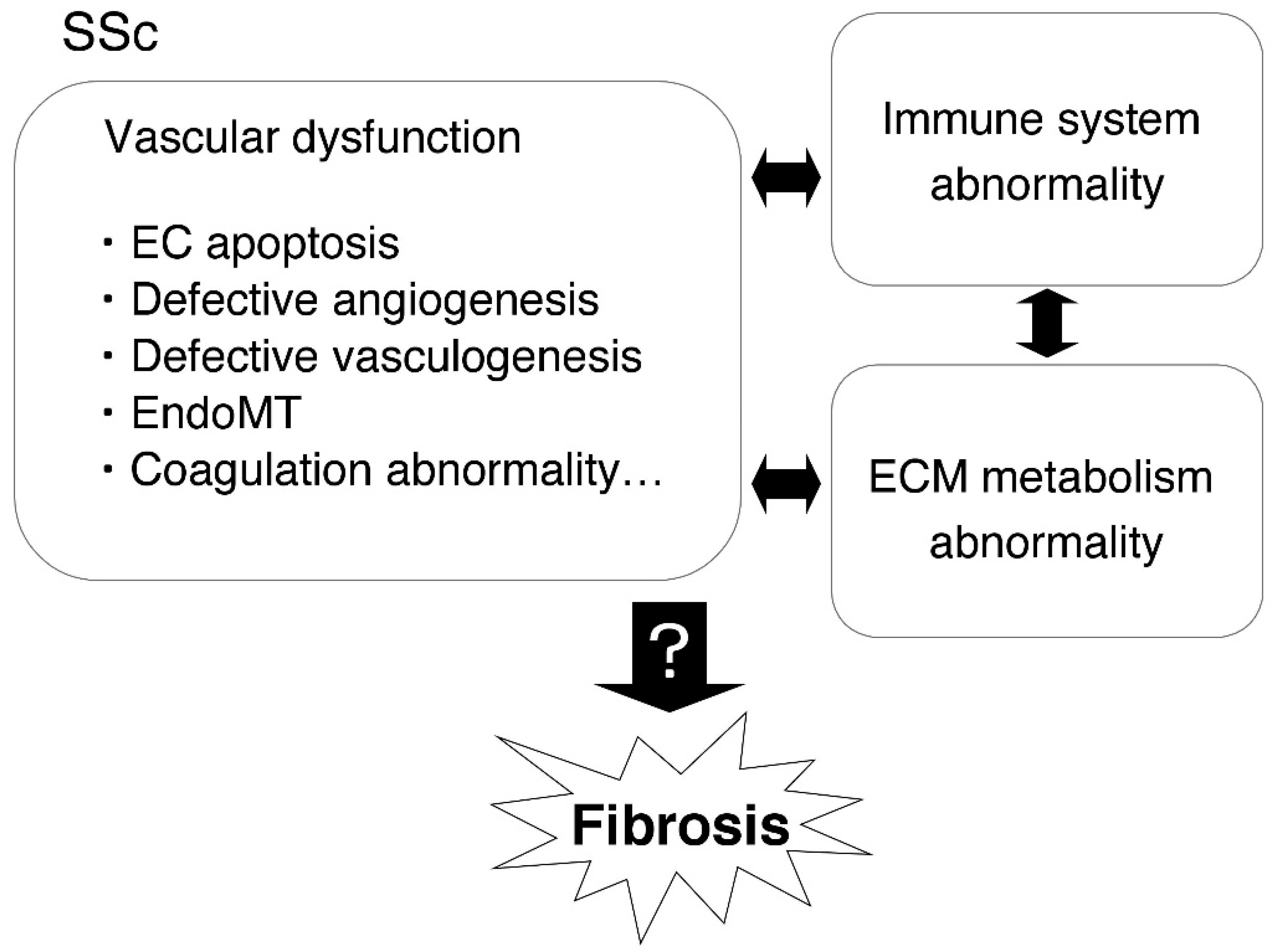
1. Introduction
2. The Various Functions of Fibrinolytic Regulators
2.1. Plasminogen (Plg) and Plasmin
2.2. α2-Antiplasmin (α2AP)
2.3. Urokinase-Type Plasminogen Activator (uPA) and Its Receptor (uPAR)
2.4. Tissue-Type Plasminogen Activator (tPA)
2.5. Plasminogen Activator Inhibitor-1 (PAI-1)
2.6. Angiostatin
3. The Role of Fibrinolytic Regulators in Vascular and EC Injury in SSc
4. The Role of Fibrinolytic Regulators in Defective Angiogenesis in SSc
5. The Role of Fibrinolytic Regulators on EPC Functions
6. The Role of Fibrinolytic Regulators in EndoMT in SSc
7. The Role of Fibrinolytic Regulators in Coagulation Abnormalities in SSc
8. The Role of Fibrinolytic Regulators in Vascular Tone Alteration in SSc
9. The Effect of Fibrinolytic Regulators on SSc-Associated PAH
10. Conclusion and Therapeutic Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Gilbane, A.J.; Denton, C.P.; Holmes, A.M. Scleroderma pathogenesis: A pivotal role for fibroblasts as effector cells. Arthritis Res. Ther. 2013, 15, 215. [Google Scholar] [CrossRef] [PubMed]
- Kavian, N.; Batteux, F. Macro- and microvascular disease in systemic sclerosis. Vascul. Pharmacol. 2015, 71, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Block, J.A.; Sequeira, W. Raynaud’s phenomenon. Lancet 2001, 357, 2042–2048. [Google Scholar] [CrossRef]
- Walker, J.G.; Stirling, J.; Beroukas, D.; Dharmapatni, K.; Haynes, D.R.; Smith, M.D.; Ahern, M.J.; Roberts-Thomson, P.J. Histopathological and ultrastructural features of dermal telangiectasias in systemic sclerosis. Pathology 2005, 37, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Bulkley, B.H.; Ridolfi, R.L.; Salyer, W.R.; Hutchins, G.M. Myocardial lesions of progressive systemic sclerosis. A cause of cardiac dysfunction. Circulation 1976, 53, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Akram, M.R.; Handler, C.E.; Williams, M.; Carulli, M.T.; Andron, M.; Black, C.M.; Denton, C.P.; Coghlan, J.G. Angiographically proven coronary artery disease in scleroderma. Rheumatology 2006, 45, 1395–1398. [Google Scholar] [CrossRef] [PubMed]
- Mostmans, Y.; Cutolo, M.; Giddelo, C.; Decuman, S.; Melsens, K.; Declercq, H.; Vandecasteele, E.; De Keyser, F.; Distler, O.; Gutermuth, J.; et al. The role of endothelial cells in the vasculopathy of systemic sclerosis: A systematic review. Autoimmun. Rev. 2017, 16, 774–786. [Google Scholar] [CrossRef] [PubMed]
- Manetti, M.; Rosa, I.; Milia, A.F.; Guiducci, S.; Carmeliet, P.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Inactivation of urokinase-type plasminogen activator receptor (uPAR) gene induces dermal and pulmonary fibrosis and peripheral microvasculopathy in mice: A new model of experimental scleroderma? Ann. Rheum. Dis. 2014, 73, 1700–1709. [Google Scholar] [CrossRef]
- Zhao, Y.Y.; Liu, Y.; Stan, R.V.; Fan, L.; Gu, Y.; Dalton, N.; Chu, P.H.; Peterson, K.; Ross, J., Jr.; Chien, K.R. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc. Natl. Acad. Sci. USA 2002, 99, 11375–11380. [Google Scholar] [CrossRef]
- Di Guglielmo, G.M.; Le Roy, C.; Goodfellow, A.F.; Wrana, J.L. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat. Cell Biol. 2003, 5, 410–421. [Google Scholar] [CrossRef]
- Asano, Y.; Stawski, L.; Hant, F.; Highland, K.; Silver, R.; Szalai, G.; Watson, D.K.; Trojanowska, M. Endothelial Fli1 deficiency impairs vascular homeostasis: A role in scleroderma vasculopathy. Am. J. Pathol. 2010, 176, 1983–1998. [Google Scholar] [CrossRef] [PubMed]
- Maurer, B.; Distler, J.H.; Distler, O. The Fra-2 transgenic mouse model of systemic sclerosis. Vascul. Pharmacol. 2013, 58, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Collen, D. Identification and some properties of a new fast-reacting plasmin inhibitor in human plasma. Eur. J. Biochem. 1976, 69, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Fay, W.P.; Eitzman, D.T.; Shapiro, A.D.; Madison, E.L.; Ginsburg, D. Platelets inhibit fibrinolysis in vitro by both plasminogen activator inhibitor-1-dependent and -independent mechanisms. Blood 1994, 83, 351–356. [Google Scholar] [PubMed]
- Draxler, D.F.; Sashindranath, M.; Medcalf, R.L. Plasmin: A Modulator of Immune Function. Semin. Thromb. Hemost. 2017, 43, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Kolev, K.; Machovich, R. Molecular and cellular modulation of fibrinolysis. Thromb. Haemost. 2003, 89, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Del Rosso, M.; Margheri, F.; Serratì, S.; Chillà, A.; Laurenzana, A.; Fibbi, G. The urokinase receptor system, a key regulator at the intersection between inflammation, immunity, and coagulation. Curr. Pharm. Des. 2011, 17, 1924–1943. [Google Scholar] [CrossRef] [PubMed]
- Booyse, F.M.; Aikens, M.L.; Grenett, H.E. Endothelial cell fibrinolysis: Transcriptional regulation of fibrinolytic protein gene expression (t-PA, u-PA, and PAI-1) by low alcohol. Alcohol. Clin. Exp. Res. 1999, 23, 1119–1124. [Google Scholar] [CrossRef]
- Shen, G.X. Impact and mechanism for oxidized and glycated lipoproteins on generation of fibrinolytic regulators from vascular endothelial cells. Mol. Cell. Biochem. 2003, 246, 69–74. [Google Scholar] [CrossRef]
- Mondino, A.; Blasi, F. uPA and uPAR in fibrinolysis, immunity and pathology. Trends Immunol. 2004, 25, 450–455. [Google Scholar] [CrossRef]
- Jinnin, M.; Ihn, H.; Yamane, K.; Asano, Y.; Yazawa, N.; Tamaki, K. Plasma plasmin-alpha2-plasmin inhibitor complex levels are increased in systemic sclerosis patients with pulmonary hypertension. Rheumatology 2003, 42, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Saigusa, R.; Asano, Y.; Nakamura, K.; Yamashita, T.; Ichimura, Y.; Takahashi, T.; Toyama, T.; Taniguchi, T.; Yoshizaki, A.; Miyazaki, M.; et al. Plasma plasmin-α2-plasmin inhibitor complex levels may predict the effect of cyclophosphamide for systemic sclerosis-related interstitial lung disease. Mod. Rheumatol. 2017, 27, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Marie, I.; Borg, J.Y.; Hellot, M.F.; Levesque, H. Plasma D-dimer concentration in patients with systemic sclerosis. Br. J. Dermatol. 2008, 158, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Kanno, Y.; Shu, E.; Kanoh, H.; Seishima, M. The Antifibrotic Effect of α2AP Neutralization in Systemic Sclerosis Dermal Fibroblasts and Mouse Models of Systemic Sclerosis. J. Investig. Dermatol. 2016, 136, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Kanno, Y.; Kawashita, E.; Minamida, M.; Kaneiwa, A.; Okada, K.; Ueshima, S.; Matsuo, O.; Matsuno, H. alpha2-antiplasmin is associated with the progression of fibrosis. Am. J. Pathol. 2010, 176, 238–245. [Google Scholar] [CrossRef]
- Kanno, Y.; Kuroki, A.; Okada, K.; Tomogane, K.; Ueshima, S.; Matsuo, O.; Matsuno, H. alpha2-Antiplasmin is involved in the production of transforming growth factor beta1 and fibrosis. J. Thromb. Haemost. 2007, 5, 2266–2273. [Google Scholar] [CrossRef] [PubMed]
- Kanno, Y.; Kawashita, E.; Kokado, A.; Okada, K.; Ueshima, S.; Matsuo, O.; Matsuno, H. Alpha2-antiplasmin regulates the development of dermal fibrosis in mice by prostaglandin F2α synthesis through adipose triglyceride lipase/calcium-independent phospholipase A2. Arthritis Rheum. 2013, 65, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Kanno, Y.; Kaneiwa, A.; Minamida, M.; Kanno, M.; Tomogane, K.; Takeuchi, K.; Okada, K.; Ueshima, S.; Matsuo, O.; Matsuno, H. The absence of uPAR is associated with the progression of dermal fibrosis. J. Investig. Dermatol. 2008, 128, 2792–2797. [Google Scholar] [CrossRef] [PubMed]
- Bandinelli, F.; Bartoli, F.; Perfetto, E.; Del Rosso, A.; Moggi-Pignone, A.; Guiducci, S.; Cinelli, M.; Fatini, C.; Generini, S.; Gabrielli, A.; et al. The fibrinolytic system components are increased in systemic sclerosis and modulated by Alprostadil (alpha1 ciclodestryn). Clin. Exp. Rheumatol. 2005, 23, 671–677. [Google Scholar] [PubMed]
- Legány, N.; Toldi, G.; Distler, J.H.; Beyer, C.; Szalay, B.; Kovács, L.; Vásárhelyi, B.; Balog, A. Increased plasma soluble urokinase plasminogen activator receptor levels in systemic sclerosis: Possible association with microvascular abnormalities and extent of fibrosis. Clin. Chem. Lab. Med. 2015, 53, 1799–1805. [Google Scholar] [CrossRef]
- Lemaire, R.; Burwell, T.; Sun, H.; Delaney, T.; Bakken, J.; Cheng, L.; Rebelatto, M.C.; Czapiga, M.; de-Mendez, I.; Coyle, A.J.; et al. Resolution of Skin Fibrosis by Neutralization of the Antifibrinolytic Function of Plasminogen Activator Inhibitor 1. Arthritis Rheumatol. 2016, 68, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Almeida, I.; Oliveira Gomes, A.; Lima, M.; Silva, I.; Vasconcelos, C. Different contributions of angiostatin and endostatin in angiogenesis impairment in systemic sclerosis: A cohort study. Clin. Exp. Rheumatol. 2016, 100, 37–42. [Google Scholar]
- Kanno, Y.; Shu, E.; Kanoh, H.; Matsuda, A.; Seishima, M. α2AP regulates vascular alteration by inhibiting VEGF signaling in systemic sclerosis: The roles of α2AP in vascular dysfunction in systemic sclerosis. Arthritis Res. Ther. 2017, 19, 22. [Google Scholar] [CrossRef] [PubMed]
- Syrovets, T.; Lunov, O.; Simmet, T. Plasmin as a proinflammatory cell activator. J. Leukoc. Biol. 2012, 92, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Godier, A.; Hunt, B.J. Plasminogen receptors and their role in the pathogenesis of inflammatory, autoimmune and malignant disease. J. Thromb. Haemost. 2013, 11, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Surette, A.P.; Madureira, P.A.; Phipps, K.D.; Miller, V.A.; Svenningsson, P.; Waisman, D.M. Regulation of fibrinolysis by S100A10 in vivo. Blood 2011, 118, 3172–3181. [Google Scholar] [CrossRef] [PubMed]
- Cesarman-Maus, G.; Rios-Luna, N.P.; Deora, A.B.; Huang, B.; Villa, R.; Cravioto Mdel, C.; Alarcon-Segovia, D.; Sanchez-Guerrero, J.; Hajjar, K.A. Autoantibodies against the fibrinolytic receptor, annexin 2, in antiphospholipid syndrome. Blood 2006, 107, 4375–4382. [Google Scholar] [CrossRef]
- Herren, T.; Burke, T.A.; Das, R.; Plow, E.F. Identification of histone H2B as a regulated plasminogen receptor. Biochemistry 2006, 45, 9463–9474. [Google Scholar] [CrossRef]
- Das, R.; Plow, E.F. Phosphatidylserine as an anchor for plasminogen and its plasminogen receptor, histone H2B, to the macrophage surface. J. Thromb. Haemost. 2011, 9, 339–349. [Google Scholar] [CrossRef]
- Wygrecka, M.; Marsh, L.M.; Morty, R.E.; Henneke, I.; Guenther, A.; Lohmeyer, J.; Markart, P.; Preissner, K.T. Enolase-1 promotes plasminogen-mediated recruitment of monocytes to the acutely inflamed lung. Blood 2009, 113, 5588–5598. [Google Scholar] [CrossRef]
- Lighvani, S.; Baik, N.; Diggs, J.E.; Khaldoyanidi, S.; Parmer, R.J.; Miles, L.A. Regulation of macrophage migration by a novel plasminogen receptor Plg-R KT. Blood 2011, 118, 5622–5630. [Google Scholar] [CrossRef]
- Kanno, Y.; Ishisaki, A.; Kawashita, E.; Chosa, N.; Nakajima, K.; Nishihara, T.; Toyoshima, K.; Okada, K.; Ueshima, S.; Matsushita, K.; et al. Plasminogen/plasmin modulates bone metabolism by regulating the osteoblast and osteoclast function. J. Biol. Chem. 2011, 286, 8952–8960. [Google Scholar] [CrossRef]
- Kanno, Y.; Sakai, A.; Miyashita, M.; Tsuchida, K.; Matsuo, O. Plasminogen deficiency is associated with improved glucose tolerance, and lower DPP-4 activity. Diabetes Res. Clin. Pract. 2016, 120, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Law, R.H.; Abu-Ssaydeh, D.; Whisstock, J.C. New insights into the structure and function of the plasminogen/plasmin system. Curr. Opin. Struct. Biol. 2013, 23, 836–841. [Google Scholar] [CrossRef]
- Trejo, J. Protease-activated receptors: New concepts in regulation of G protein-coupled receptor signaling and trafficking. J. Pharmacol. Exp. Ther. 2003, 307, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Barthel, D.; Schindler, S.; Zipfel, P.F. Plasminogen is a complement inhibitor. J. Biol. Chem. 2012, 287, 18831–18842. [Google Scholar] [CrossRef] [PubMed]
- Didiasova, M.; Wujak, L.; Wygrecka, M.; Zakrzewicz, D. From plasminogen to plasmin: Role of plasminogen receptors in human cancer. Int. J. Mol. Sci. 2014, 15, 21229–21252. [Google Scholar] [CrossRef]
- Kanno, Y.; Ishisaki, A.; Kawashita, E.; Kuretake, H.; Ikeda, K.; Matsuo, O. uPA Attenuated LPS-induced Inflammatory Osteoclastogenesis through the Plasmin/PAR-1/Ca2+/CaMKK/AMPK Axis. Int. J. Biol. Sci. 2016, 12, 63–71. [Google Scholar] [CrossRef]
- Lijnen, H.R.; De Cock, F.; Van Hoef, B.; Schlott, B.; Collen, D. Characterization of the interaction between plasminogen and staphylokinase. Eur. J. Biochem. 1994, 224, 143–149. [Google Scholar] [CrossRef]
- Lee, K.N.; Jackson, K.W.; Christiansen, V.J.; Lee, C.S.; Chun, J.G.; McKee, P.A. Antiplasmin-cleaving enzyme is a soluble form of fibroblast activation protein. Blood 2006, 107, 1397–1404. [Google Scholar] [CrossRef]
- Christiansen, V.J.; Jackson, K.W.; Lee, K.N.; McKee, P.A. Effect of fibroblast activation protein and α2-antiplasmin cleaving enzyme on collagen types I.; III, and IV. Arch. Biochem. Biophys. 2007, 457, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Menoud, P.A.; Sappino, N.; Boudal-Khoshbeen, M.; Vassalli, J.D.; Sappino, A.P. The kidney is a major site of alpha2-antiplasmin production. J. Clin. Investig. 1996, 97, 2478–2484. [Google Scholar] [CrossRef]
- Kanno, Y.; Hirade, K.; Ishisaki, A.; Nakajima, K.; Suga, H.; Into, T.; Matsushita, K.; Okada, K.; Matsuo, O.; Matsuno, H. Lack of alpha2-antiplasmin improves cutaneous wound healing via over-released vascular endothelial growth factor-induced angiogenesis in wound lesions. J. Thromb. Haemost. 2006, 4, 1602–1610. [Google Scholar] [CrossRef] [PubMed]
- Kanno, Y.; Ishisaki, A.; Kawashita, E.; Kuretake, H.; Ikeda, K.; Matsuo, O. α2-antiplasmin modulates bone formation by negatively regulating osteoblast differentiation and function. Int. J. Mol. Med. 2017, 40, 854–858. [Google Scholar] [CrossRef] [PubMed]
- Kawashita, E.; Kanno, Y.; Asayama, H.; Okada, K.; Ueshima, S.; Matsuo, O.; Matsuno, H. Involvement of α2-antiplasmin in dendritic growth of hippocampal neurons. J. Neurochem. 2013, 126, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Kawashita, E.; Kanno, Y.; Ikeda, K.; Kuretake, H.; Matsuo, O.; Matsuno, H. Altered behavior in mice with deletion of the alpha2-antiplasmin gene. PLoS ONE 2014, 9, e97947. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Okada, K.; Okamoto, C.; Ueshima, S.; Matsuo, O. Alpha2-antiplasmin is a critical regulator of angiotensin II-mediated vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1257–1262. [Google Scholar] [CrossRef] [PubMed]
- Kager, L.M.; Weehuizen, T.A.; Wiersinga, W.J.; Roelofs, J.J.; Meijers, J.C.; Dondorp, A.M.; van ’t Veer, C.; van der Poll, T. Endogenous α2-antiplasmin is protective during severe gram-negative sepsis (melioidosis). Am. J. Respir. Crit. Care Med. 2013, 188, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Law, R.H.; Sofian, T.; Kan, W.T.; Horvath, A.J.; Hitchen, C.R.; Langendorf, C.G.; Buckle, A.M.; Whisstock, J.C.; Coughlin, P.B. X-ray crystal structure of the fibrinolysis inhibitor alpha2-antiplasmin. Blood 2008, 111, 2049–2052. [Google Scholar] [CrossRef]
- Irving, J.A.; Pike, R.N.; Lesk, A.M.; Whisstock, J.C. Phylogeny of the serpin superfamily: Implications of patterns of amino acid conservation for structure and function. Genome Res. 2000, 10, 1845–1864. [Google Scholar] [CrossRef]
- Tombran-Tink, J.; Aparicio, S.; Xu, X.; Tink, A.R.; Lara, N.; Sawant, S.; Barnstable, C.J.; Zhang, S.S. PEDF and the serpins: Phylogeny, sequence conservation, and functional domains. J. Struct. Biol. 2005, 151, 130–150. [Google Scholar] [CrossRef] [PubMed]
- Kanno, Y.; Kawashita, E.; Kokado, A.; Kuretake, H.; Ikeda, K.; Okada, K.; Seishima, M.; Ueshima, S.; Matsuo, O.; Matsuno, H. α2AP mediated myofibroblast formation and the development of renal fibrosis in unilateral ureteral obstruction. Sci. Rep. 2014, 4, 5967. [Google Scholar] [CrossRef] [PubMed]
- Abdul, S.; Leebeek, F.W.; Rijken, D.C.; Uitte de Willige, S. Natural heterogeneity of α2-antiplasmin: Functional and clinical consequences. Blood 2016, 127, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Mondino, A.; Resnati, M.; Blasi, F. Structure and function of the urokinase receptor. Thromb. Haemost. 1999, 82, 19–22. [Google Scholar] [PubMed]
- Binder, B.R.; Mihaly, J.; Prager, G.W. uPAR-uPA-PAI-1 interactions and signaling: A vascular biologist’s view. Thromb. Haemost. 2007, 97, 336–342. [Google Scholar] [PubMed]
- Enocsson, H.; Sjöwall, C.; Wetterö, J. Soluble urokinase plasminogen activator receptor-a valuable biomarker in systemic lupus erythematosus? Clin. Chim. Acta 2015, 444, 234–241. [Google Scholar] [CrossRef]
- Duru, E.A.; Fu, Y.; Davies, M.G. Role of formic receptors in soluble urokinase receptor-induced human vascular smooth muscle migration. J. Surg. Res. 2015, 195, 396–405. [Google Scholar] [CrossRef]
- Pliyev, B.K. Activated human neutrophils rapidly release the chemotactically active D2D3 form of the urokinase-type plasminogen activator receptor (uPAR/CD87). Mol. Cell. Biochem. 2009, 321, 111–122. [Google Scholar] [CrossRef]
- Sloand, E.M.; Pfannes, L.; Scheinberg, P.; More, K.; Wu, C.O.; Horne, M.; Young, N.S. Increased soluble urokinase plasminogen activator receptor (suPAR) is associated with thrombosis and inhibition of plasmin generation in paroxysmal nocturnal hemoglobinuria (PNH) patients. Exp. Hematol. 2008, 36, 1616–1624. [Google Scholar] [CrossRef]
- Blasi, F.; Carmeliet, P. uPAR: A versatile signalling orchestrator. Nat. Rev. Mol. Cell Biol. 2002, 3, 932–943. [Google Scholar] [CrossRef]
- Smith, H.W.; Marshall, C.J. Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell Biol. 2010, 11, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Kanno, Y.; Matsuno, H.; Kawashita, E.; Okada, K.; Suga, H.; Ueshima, S.; Matsuo, O. Urokinase-type plasminogen activator receptor is associated with the development of adipose tissue. Thromb. Haemost. 2010, 104, 1124–1132. [Google Scholar] [PubMed]
- Tomogane, K.; Kanno, Y.; Kawashita, E.; Okada, K.; Takeuchi, K.; Ueshima, S.; Matsuo, O.; Matsuno, H. The absence of urokinase-type plasminogen activator receptor plays a role in the insulin-independent glucose metabolism. J. Cardiovasc. Pharmacol. 2011, 57, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Navaratna, D.; Menicucci, G.; Maestas, J.; Srinivasan, R.; McGuire, P.; Das, A. A peptide inhibitor of the urokinase/urokinase receptor system inhibits alteration of the blood-retinal barrier in diabetes. FASEB J. 2008, 22, 3310–3317. [Google Scholar] [CrossRef] [PubMed]
- Chevilley, A.; Lesept, F.; Lenoir, S.; Ali, C.; Parcq, J.; Vivien, D. Impacts of tissue-type plasminogen activator (tPA) on neuronal survival. Front. Cell. Neurosci. 2015, 9, 415. [Google Scholar] [CrossRef] [PubMed]
- Adibhatla, R.M.; Hatcher, J.F. Tissue plasminogen activator (tPA) and matrix metalloproteinases in the pathogenesis of stroke: Therapeutic strategies. CNS Neurol. Disord. Drug Targets 2008, 7, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, D.E.; Rai, R.; Khan, S.S.; Eren, M.; Ghosh, A.K. Plasminogen Activator Inhibitor-1 Is a Marker and a Mediator of Senescence. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1446–1452. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Vaughan, D.E. PAI-1 in tissue fibrosis. J. Cell. Physiol. 2012, 227, 493–507. [Google Scholar] [CrossRef]
- Rabieian, R.; Boshtam, M.; Zareei, M.; Kouhpayeh, S.; Masoudifar, A.; Mirzaei, H. Plasminogen Activator Inhibitor Type-1 as a Regulator of Fibrosis. J. Cell. Biochem. 2018, 119, 17–27. [Google Scholar] [CrossRef]
- Yang, C.; Patel, K.; Harding, P.; Sorokin, A.; Glass, W.F. Regulation of TGFbeta1/MAPK-mediated PAI-1 gene expression by the actin cytoskeleton in human mesangial cells. Exp. Cell Res. 2007, 313, 1240–1250. [Google Scholar] [CrossRef]
- Paugh, B.S.; Paugh, S.W.; Bryan, L.; Kapitonov, D.; Wilczynska, K.M.; Gopalan, S.M.; Rokita, H.; Milstien, S.; Spiegel, S.; Kordula, T. EGF regulates plasminogen activator inhibitor-1 (PAI-1) by a pathway involving c-Src, PKCdelta, and sphingosine kinase 1 in glioblastoma cells. FASEB J. 2008, 22, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Crandall, D.L.; Groeling, T.M.; Busler, D.E.; Antrilli, T.M. Release of PAI-1 by human preadipocytes and adipocytes independent of insulin and IGF-1. Biochem. Biophys. Res. Commun. 2000, 279, 984–988. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Woodcock-Mitchell, J.; Mitchell, J.; Sakamoto, T.; Marutsuka, K.; Sobel, B.E.; Fujii, S. Induction of plasminogen activator inhibitor type 1 and type 1 collagen expression in rat cardiac microvascular endothelial cells by interleukin-1 and its dependence on oxygen-centered free radicals. Circulation 1998, 97, 2175–2182. [Google Scholar] [CrossRef] [PubMed]
- Farnoodian, M.; Wang, S.; Dietz, J.; Nickells, R.W.; Sorenson, C.M.; Sheibani, N. Negative regulators of angiogenesis: Important targets for treatment of exudative AMD. Clin. Sci. 2017, 131, 1763–1780. [Google Scholar] [CrossRef] [PubMed]
- Jurasz, P.; Santos-Martinez, M.J.; Radomska, A.; Radomski, M.W. Generation of platelet angiostatin mediated by urokinase plasminogen activator: Effects on angiogenesis. J. Thromb. Haemost. 2006, 4, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- van Tilborg, A.A.; Sweep, F.C.; Geurts-Moespot, A.J.; Wetzels, A.M.; de Waal, R.M.; Westphal, J.R.; Massuger, L.F. Plasminogen activators are involved in angiostatin generation in vivo in benign and malignant ovarian tumor cyst fluids. Int. J. Oncol. 2014, 44, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.D.; Moore, H.B.; Banerjee, A.; Silliman, C.C.; Moore, E.E.; Yaffe, M.B. Human neutrophil elastase mediates fibrinolysis shutdown through competitive degradation of plasminogen and generation of angiostatin. J. Trauma Acute Care Surg. 2017, 83, 1053–1061. [Google Scholar] [CrossRef]
- Xu, Z.; Shi, H.; Li, Q.; Mei, Q.; Bao, J.; Shen, Y.; Xu, J. Mouse macrophage metalloelastase generates angiostatin from plasminogen and suppresses tumor angiogenesis in murine colon cancer. Oncol. Rep. 2008, 20, 81–88. [Google Scholar] [CrossRef]
- Griscelli, F.; Li, H.; Bennaceur-Griscelli, A.; Soria, J.; Opolon, P.; Soria, C.; Perricaudet, M.; Yeh, P.; Lu, H. Angiostatin gene transfer: Inhibition of tumor growth in vivo by blockage of endothelial cell proliferation associated with a mitosis arrest. Proc. Natl. Acad. Sci. USA 1998, 95, 6367–6372. [Google Scholar] [CrossRef]
- Troyanovsky, B.; Levchenko, T.; Månsson, G.; Matvijenko, O.; Holmgren, L. Angiomotin: An angiostatin binding protein that regulates endothelial cell migration and tube formation. J. Cell Biol. 2001, 152, 1247–1254. [Google Scholar] [CrossRef]
- Hajitou, A.; Grignet, C.; Devy, L.; Berndt, S.; Blacher, S.; Deroanne, C.F.; Bajou, K.; Fong, T.; Chiang, Y.; Foidart, J.M.; et al. The antitumoral effect of endostatin and angiostatin is associated with a down-regulation of vascular endothelial growth factor expression in tumor cells. FASEB J. 2002, 16, 1802–1804. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.Y.; Muschal, S.; Pravda, E.A.; Folkman, J.; Abdollahi, A.; Javaherian, K. Angiostatin regulates the expression of antiangiogenic and proapoptotic pathways via targeted inhibition of mitochondrial proteins. Blood 2009, 114, 1987–1998. [Google Scholar] [CrossRef] [PubMed]
- Aulakh, G.K.; Balachandran, Y.; Liu, L.; Singh, B. Angiostatin inhibits activation and migration of neutrophils. Cell Tissue Res. 2014, 355, 375–396. [Google Scholar] [CrossRef] [PubMed]
- Perri, S.R.; Annabi, B.; Galipeau, J. Angiostatin inhibits monocyte/macrophage migration via disruption of actin cytoskeleton. FASEB J. 2007, 21, 3928–3936. [Google Scholar] [CrossRef]
- Chavakis, T.; Athanasopoulos, A.; Rhee, J.S.; Orlova, V.; Schmidt-Wöll, T.; Bierhaus, A.; May, A.E.; Celik, I.; Nawroth, P.P.; Preissner, K.T. Angiostatin is a novel anti-inflammatory factor by inhibiting leukocyte recruitment. Blood 2005, 105, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gronow, M.; Grenett, H.E.; Gawdi, G.; Pizzo, S.V. Angiostatin directly inhibits human prostate tumor cell invasion by blocking plasminogen binding to its cellular receptor, CD26. Exp. Cell Res. 2005, 303, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Radziwon-Balicka, A.; Ramer, C.; Moncada de la Rosa, C.; Zielnik-Drabik, B.; Jurasz, P. Angiostatin inhibits endothelial MMP-2 and MMP-14 expression: A hypoxia specific mechanism of action. Vascul. Pharmacol. 2013, 58, 280–291. [Google Scholar] [CrossRef]
- Manetti, M.; Guiducci, S.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Mechanisms in the loss of capillaries in systemic sclerosis: Angiogenesis versus vasculogenesis. J. Cell. Mol. Med. 2010, 14, 1241–1254. [Google Scholar] [CrossRef] [PubMed]
- Pattanaik, D.; Brown, M.; Postlethwaite, B.C.; Postlethwaite, A.E. Pathogenesis of Systemic Sclerosis. Front. Immunol. 2015, 6, 272. [Google Scholar] [CrossRef] [PubMed]
- Greeno, E.W.; Bach, R.R.; Moldow, C.F. Apoptosis is associated with increased cell surface tissue factor procoagulant activity. Lab. Investig. 1996, 75, 281–289. [Google Scholar] [PubMed]
- Tsuji, S.; Kaji, K.; Nagasawa, S. Activation of the alternative pathway of human complement by apoptotic human umbilical vein endothelial cells. J. Biochem. 1994, 116, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yao, Y.C.; Gu, X.Q.; Che, D.; Ma, C.Q.; Dai, Z.Y.; Li, C.; Zhou, T.; Cai, W.B.; Yang, Z.H.; et al. Plasminogen kringle 5 induces endothelial cell apoptosis by triggering a voltage-dependent anion channel 1 (VDAC1) positive feedback loop. J. Biol. Chem. 2014, 289, 32628–32638. [Google Scholar] [CrossRef] [PubMed]
- Okajima, K.; Abe, H.; Binder, B.R. Endothelial cell injury induced by plasmin in vitro. J. Lab. Clin. Med. 1995, 126, 377–384. [Google Scholar] [PubMed]
- Plow, E.F.; Hoover-Plow, J. The functions of plasminogen in cardiovascular disease. Trends Cardiovasc. Med. 2004, 14, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Lyons, R.M.; Gentry, L.E.; Purchio, A.F.; Moses, H.L. Mechanism of activation of latent recombinant transforming growth factor beta 1 by plasmin. J. Cell Biol. 1990, 110, 1361–1367. [Google Scholar] [CrossRef] [PubMed]
- Mosesson, M.W. Fibrinogen and fibrin structure and functions. J. Thromb. Haemost. 2005, 3, 1894–1904. [Google Scholar] [CrossRef] [PubMed]
- Rundhaug, J.E. Matrix metalloproteinases and angiogenesis. J. Cell. Mol. Med. 2005, 9, 267–285. [Google Scholar] [CrossRef]
- Yan, Q.; Sage, E.H. Transforming growth factor-beta1 induces apoptotic cell death in cultured retinal endothelial cells but not pericytes: Association with decreased expression of p21waf1/cip1. J. Cell. Biochem. 1998, 70, 70–83. [Google Scholar] [CrossRef]
- Prager, G.W.; Mihaly, J.; Brunner, P.M.; Koshelnick, Y.; Hoyer-Hansen, G.; Binder, B.R. Urokinase mediates endothelial cell survival via induction of the X-linked inhibitor of apoptosis protein. Blood 2009, 113, 1383–1390. [Google Scholar] [CrossRef]
- Cao, D.J.; Guo, Y.L.; Colman, R.W. Urokinase-type plasminogen activator receptor is involved in mediating the apoptotic effect of cleaved high molecular weight kininogen in human endothelial cells. Circ. Res. 2004, 94, 1227–1234. [Google Scholar] [CrossRef]
- Al-Fakhri, N.; Chavakis, T.; Schmidt-Wöll, T.; Huang, B.; Cherian, S.M.; Bobryshev, Y.V.; Lord, R.S.; Katz, N.; Preissner, K.T. Induction of apoptosis in vascular cells by plasminogen activator inhibitor-1 and high molecular weight kininogen correlates with their anti-adhesive properties. Biol. Chem. 2003, 384, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Bajou, K.; Peng, H.; Laug, W.E.; Maillard, C.; Noel, A.; Foidart, J.M.; Martial, J.A.; DeClerck, Y.A. Plasminogen activator inhibitor-1 protects endothelial cells from FasL-mediated apoptosis. Cancer Cell 2008, 14, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.; Holmgren, L.; Garcia, I.; Jimenez, B.; Mandriota, S.J.; Borlat, F.; Sim, B.K.; Wu, Z.; Grau, G.E.; Shing, Y.; et al. Multiple forms of angiostatin induce apoptosis in endothelial cells. Blood 1998, 92, 4730–4741. [Google Scholar] [PubMed]
- Guiducci, S.; Distler, O.; Distler, J.H.; Matucci-Cerinic, M. Mechanisms of vascular damage in SSc-implications for vascular treatment strategies. Rheumatology 2008, 47, v18–v20. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M.; Claesson-Welsh, L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp. Cell Res. 2006, 312, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Liakouli, V.; Cipriani, P.; Marrelli, A.; Alvaro, S.; Ruscitti, P.; Giacomelli, R. Angiogenic cytokines and growth factors in systemic sclerosis. Autoimmun. Rev. 2011, 10, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Trojanowska, M. Cellular and molecular aspects of vascular dysfunction in systemic sclerosis. Nat. Rev. Rheumatol. 2010, 6, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Houck, K.A.; Leung, D.W.; Rowland, A.M.; Winer, J.; Ferrara, N. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J. Biol. Chem. 1992, 267, 26031–26037. [Google Scholar]
- Park, J.E.; Keller, G.-A.; Ferrara, N. The vascular endothelial growth factor (VEGF) isoforms: Differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol. Biol. Cell 1993, 4, 1317–1326. [Google Scholar] [CrossRef]
- Serratì, S.; Cinelli, M.; Margheri, F.; Guiducci, S.; Del Rosso, A.; Pucci, M.; Fibbi, G.; Bazzichi, L.; Bombardieri, S.; Matucci-Cerinic, M.; et al. Systemic sclerosis fibroblasts inhibit in vitro angiogenesis by MMP-12-dependent cleavage of the endothelial cell urokinase receptor. J. Pathol. 2006, 210, 240–248. [Google Scholar] [CrossRef]
- Montuori, N.; Ragno, P. Role of uPA/uPAR in the modulation of angiogenesis. Chem. Immunol. Allergy 2014, 99, 105–122. [Google Scholar] [PubMed]
- Uhrin, P.; Breuss, JM. uPAR: A modulator of VEGF-induced angiogenesis. Cell Adh. Migr. 2013, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, S.; Fibbi, G.; Cinelli, M.; Guiducci, S.; Del Rosso, A.; Margheri, F.; Serrati, S.; Pucci, M.; Kahaleh, B.; Fan, P.; et al. Matrix metalloproteinase 12-dependent cleavage of urokinase receptor in systemic sclerosis microvascular endothelial cells results in impaired angiogenesis. Arthritis Rheum. 2004, 50, 3275–3285. [Google Scholar] [CrossRef]
- Margheri, F.; Manetti, M.; Serrati, S.; Nosi, D.; Pucci, M.; Matucci-Cerinic, M.; Kahaleh, B.; Bazzichi, L.; Fibbi, G.; Ibba-Manneschi, L.; et al. Domain 1 of the urokinase-type plasminogen activator receptor is required for its morphologic and functional, beta2 integrin-mediated connection with actin cytoskeleton in human microvascular endothelial cells: Failure of association in systemic sclerosis endothelial cells. Arthritis Rheum. 2006, 54, 3926–3938. [Google Scholar] [PubMed]
- Bagnato, G.L.; Irrera, N.; Pizzino, G.; Santoro, D.; Roberts, W.N.; Bagnato, G.; Pallio, G.; Vaccaro, M.; Squadrito, F.; Saitta, A.; et al. Dual αvβ3 and αvβ5 blockade attenuates fibrotic and vascular alterations in a murine model of systemic sclerosis. Clin. Sci. 2018, 132, 231–242. [Google Scholar] [CrossRef]
- Giusti, B.; Margheri, F.; Rossi, L.; Lapini, I.; Magi, A.; Serratì, S.; Chillà, A.; Laurenzana, A.; Magnelli, L.; Calorini, L.; et al. Desmoglein-2-integrin Beta-8 interaction regulates actin assembly in endothelial cells: Deregulation in systemic sclerosis. PLoS ONE 2013, 8, e68117. [Google Scholar] [CrossRef]
- Kanno, Y.; Kuroki, A.; Minamida, M.; Kaneiwa, A.; Okada, K.; Tomogane, K.; Takeuchi, K.; Ueshima, S.; Matsuo, O.; Matsuno, H. The absence of uPAR attenuates insulin-induced vascular smooth muscle cell migration and proliferation. Thromb. Res. 2008, 123, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Kiyan, J.; Kiyan, R.; Haller, H.; Dumler, I. Urokinase-induced signaling in human vascular smooth muscle cells is mediated by PDGFR-beta. EMBO J. 2005, 24, 1787–1797. [Google Scholar] [CrossRef]
- Wu, J.; Strawn, T.L.; Luo, M.; Wang, L.; Li, R.; Ren, M.; Xia, J.; Zhang, Z.; Ma, W.; Luo, T.; et al. Plasminogen activator inhibitor-1 inhibits angiogenic signaling by uncoupling vascular endothelial growth factor receptor-2-αVβ3 integrin cross talk. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 111–120. [Google Scholar] [CrossRef]
- Bajou, K.; Herkenne, S.; Thijssen, V.L.; D’Amico, S.; Nguyen, N.Q.; Bouché, A.; Tabruyn, S.; Srahna, M.; Carabin, J.Y.; Nivelles, O.; et al. PAI-1 mediates the antiangiogenic and profibrinolytic effects of 16K prolactin. Nat. Med. 2014, 20, 741–747. [Google Scholar] [CrossRef]
- Duan, P.; Ni, C. t-PA stimulates VEGF expression in endothelial cells via ERK2/p38 signaling pathways. Pharmazie 2014, 69, 70–75. [Google Scholar] [PubMed]
- Fukuhara, S.; Sako, K.; Noda, K.; Zhang, J.; Minami, M.; Mochizuki, N. Angiopoietin-1/Tie2 receptor signaling in vascular quiescence and angiogenesis. Histol. Histopathol. 2010, 25, 387–396. [Google Scholar] [PubMed]
- Augustin, H.G.; Koh, G.Y.; Thurston, G.; Alitalo, K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat. Rev. Mol. Cell Biol. 2009, 10, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Thurston, G.; Rudge, J.S.; Ioffe, E.; Zhou, H.; Ross, L.; Croll, S.D.; Glazer, N.; Holash, J.; McDonald, D.M.; Yancopoulos, G.D. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat. Med. 2000, 6, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Maisonpierre, P.C.; Suri, C.; Jones, P.F.; Bartunkova, S.; Wiegand, S.J.; Radziejewski, C.; Compton, D.; McClain, J.; Aldrich, T.H.; Papadopoulos, N.; et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997, 277, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Michalska-Jakubus, M.; Kowal-Bielecka, O.; Chodorowska, G.; Bielecki, M.; Krasowska, D. Angiopoietins-1 and -2 are differentially expressed in the sera of patients with systemic sclerosis: High angiopoietin-2 levels are associated with greater severity and higher activity of the disease. Rheumatology 2011, 50, 746–755. [Google Scholar] [CrossRef] [PubMed]
- Mishiro, K.; Ishiguro, M.; Suzuki, Y.; Tsuruma, K.; Shimazawa, M.; Hara, H. Tissue plasminogen activator prevents restoration of tight junction proteins through upregulation of angiopoietin-2. Curr. Neurovasc. Res. 2013, 10, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Kim, H.G.; Moon, S.O.; Chae, S.W.; So, J.N.; Koh, K.N.; Ahn, B.C.; Koh, G.Y. Angiopoietin-1 induces endothelial cell sprouting through the activation of focal adhesion kinase and plasmin secretion. Circ. Res. 2000, 86, 952–959. [Google Scholar] [CrossRef]
- Raghu, H.; Lakka, S.S.; Gondi, C.S.; Mohanam, S.; Dinh, D.H.; Gujrati, M.; Rao, J.S. Suppression of uPA and uPAR attenuates angiogenin mediated angiogenesis in endothelial and glioblastoma cell lines. PLoS ONE 2010, 5, e12458. [Google Scholar] [CrossRef]
- Dallabrida, S.M.; Ismail, N.S.; Pravda, E.A.; Parodi, E.M.; Dickie, R.; Durand, E.M.; Lai, J.; Cassiola, F.; Rogers, R.A.; Rupnick, M.A. Integrin binding angiopoietin-1 monomers reduce cardiac hypertrophy. FASEB J. 2008, 22, 3010–3023. [Google Scholar] [CrossRef]
- Cascone, I.; Napione, L.; Maniero, F.; Serini, G.; Bussolino, F. Stable interaction between alpha5beta1 integrin and Tie2 tyrosine kinase receptor regulates endothelial cell response to Ang-1. J. Cell Biol. 2005, 170, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Del Papa, N.; Pignataro, F. The Role of Endothelial Progenitors in the Repair of Vascular Damage in Systemic Sclerosis. Front. Immunol. 2018, 9, 1383. [Google Scholar] [CrossRef] [PubMed]
- Del Papa, N.; Quirici, N.; Soligo, D.; Scavullo, C.; Cortiana, M.; Borsotti, C.; Maglione, W.; Comina, D.P.; Vitali, C.; Fraticelli, P.; et al. Bone marrow endothelial progenitors are defective in systemic sclerosis. Arthritis Rheum. 2006, 54, 2605–2615. [Google Scholar] [CrossRef] [PubMed]
- Laurenzana, A.; Fibbi, G.; Margheri, F.; Biagioni, A.; Luciani, C.; Del Rosso, M.; Chillà, A. Endothelial Progenitor Cells in Sprouting Angiogenesis: Proteases Pave the Way. Curr. Mol. Med. 2015, 15, 606–620. [Google Scholar] [CrossRef] [PubMed]
- Yip, H.K.; Sun, C.K.; Tsai, T.H.; Sheu, J.J.; Kao, Y.H.; Lin, Y.C.; Shiue, Y.L.; Chen, Y.L.; Chai, H.T.; Chua, S.; et al. Tissue plasminogen activator enhances mobilization of endothelial progenitor cells and angiogenesis in murine limb ischemia. Int. J. Cardiol. 2013, 168, 226–236. [Google Scholar] [CrossRef]
- Leu, S.; Day, Y.J.; Sun, C.K.; Yip, H.K. tPA-MMP-9 Axis Plays a Pivotal Role in Mobilization of Endothelial Progenitor Cells from Bone Marrow to Circulation and Ischemic Region for Angiogenesis. Stem Cells Int. 2016, 2016, 5417565. [Google Scholar] [CrossRef] [PubMed]
- Li, W.D.; Hu, N.; Lei, F.R.; Wei, S.; Rong, J.J.; Zhuang, H.; Li, X.Q. Autophagy inhibits endothelial progenitor cells migration via the regulation of MMP2, MMP9 and uPA under normoxia condition. Biochem. Biophys. Res. Commun. 2015, 466, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Margheri, F.; Papucci, L.; Schiavone, N.; D’Agostino, R.; Trigari, S.; Serratì, S.; Laurenzana, A.; Biagioni, A.; Luciani, C.; Chillà, A.; et al. Differential uPAR recruitment in caveolar-lipid rafts by GM1 and GM3 gangliosides regulates endothelial progenitor cells angiogenesis. J. Cell. Mol. Med. 2015, 19, 113–123. [Google Scholar] [CrossRef]
- Margheri, F.; Chillà, A.; Laurenzana, A.; Serratì, S.; Mazzanti, B.; Saccardi, R.; Santosuosso, M.; Danza, G.; Sturli, N.; Rosati, F.; et al. Endothelial progenitor cell-dependent angiogenesis requires localization of the full-length form of uPAR in caveolae. Blood 2011, 118, 3743–3755. [Google Scholar] [CrossRef]
- Ito, H.; Rovira, I.I.; Bloom, M.L.; Takeda, K.; Ferrans, V.J.; Quyyumi, A.A.; Finkel, T. Endothelial progenitor cells as putative targets for angiostatin. Cancer Res. 1999, 59, 5875–5877. [Google Scholar]
- Jimenez, S.A. Role of endothelial to mesenchymal transition in the pathogenesis of the vascular alterations in systemic sclerosis. ISRN Rheumatol. 2013, 23, 835948. [Google Scholar] [CrossRef]
- Manetti, M.; Romano, E.; Rosa, I.; Guiducci, S.; Bellando-Randone, S.; De Paulis, A.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Endothelial-to-mesenchymal transition contributes to endothelial dysfunction and dermal fibrosis in systemic sclerosis. Ann. Rheum. Dis. 2017, 76, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, S.A.; Piera-Velazquez, S. Endothelial to mesenchymal transition (EndoMT) in the pathogenesis of Systemic Sclerosis-associated pulmonary fibrosis and pulmonary arterial hypertension. Myth or reality? Matrix Biol. 2016, 51, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.G.; Lee, A.; Chang, W.; Lee, M.S.; Kim, J. Endothelial to Mesenchymal Transition Represents a Key Link in the Interaction between Inflammation and Endothelial Dysfunction. Front. Immunol. 2018, 9, 294. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Mendoza, F.A.; Jimenez, S.A. Endothelial to Mesenchymal Transition (EndoMT) in the Pathogenesis of Human Fibrotic Diseases. J. Clin Med. 2016, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Shiomi, A.; Kawao, N.; Yano, M.; Okada, K.; Tamura, Y.; Okumoto, K.; Matsuo, O.; Akagi, M.; Kaji, H. α2-Antiplasmin is involved in bone loss induced by ovariectomy in mice. Bone 2015, 79, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Lester, R.D.; Jo, M.; Montel, V.; Takimoto, S.; Gonias, S.L. uPAR induces epithelial-mesenchymal transition in hypoxic breast cancer cells. J. Cell Biol. 2007, 178, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.; Lester, R.D.; Montel, V.; Eastman, B.; Takimoto, S.; Gonias, S.L. Reversibility of epithelial-mesenchymal transition (EMT) induced in breast cancer cells by activation of urokinase receptor-dependent cell signaling. J. Biol. Chem. 2009, 284, 22825–22833. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, A.M.; Twining, S.S.; Warejcka, D.J.; Tall, E.; Masur, S.K. Urokinase receptor cleavage: A crucial step in fibroblast-to-myofibroblast differentiation. Mol. Biol. Cell 2007, 18, 2716–2727. [Google Scholar] [CrossRef] [PubMed]
- Kanno, Y.; Ishisaki, A.; Miyashita, M.; Matsuo, O. The blocking of uPAR suppresses lipopolysaccharide-induced inflammatory osteoclastogenesis and the resultant bone loss through attenuation of integrin β3/Akt pathway. Immun. Inflamm. Dis. 2016, 4, 338–349. [Google Scholar] [CrossRef]
- Kanno, Y.; Maruyama, C.; Matsuda, A.; Ishisaki, A. uPA-derived peptide, Å6 is involved in the suppression of lipopolysaccaride-promoted inflammatory osteoclastogenesis and the resultant bone loss. Immun. Inflamm. Dis. 2017, 5, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wermuth, P.J.; Benn, B.S.; Lisanti, M.P.; Jimenez, S.A. Caveolin-1 deficiency induces spontaneous endothelial-to-mesenchymal transition in murine pulmonary endothelial cells in vitro. Am. J. Pathol. 2013, 182, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Monaghan-Benson, E.; Mastick, C.C.; McKeown-Longo, P.J. A dual role for caveolin-1 in the regulation of fibronectin matrix assembly by uPAR. J. Cell Sci. 2008, 121, 3693–3703. [Google Scholar] [CrossRef] [PubMed]
- Cavallo-Medved, D.; Mai, J.; Dosescu, J.; Sameni, M.; Sloane, B.F. Caveolin-1 mediates the expression and localization of cathepsin B.; pro-urokinase plasminogen activator and their cell-surface receptors in human colorectal carcinoma cells. J. Cell Sci. 2005, 118, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Zidovetzki, R.; Wang, J.L.; Kim, J.A.; Chen, P.; Fisher, M.; Hofman, F.M. Endothelin-1 enhances plasminogen activator inhibitor-1 production by human brain endothelial cells via protein kinase C.-dependent pathway. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1768–1775. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Tan, R.; Dai, C.; Li, Y.; Wang, D.; Hao, S.; Kahn, M.; Liu, Y. Plasminogen activator inhibitor-1 is a transcriptional target of the canonical pathway of Wnt/beta-catenin signaling. J. Biol. Chem. 2010, 285, 24665–24675. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Bradham, W.S.; Gleaves, L.A.; De Taeye, B.; Murphy, S.B.; Covington, J.W.; Vaughan, D.E. Genetic deficiency of plasminogen activator inhibitor-1 promotes cardiac fibrosis in aged mice: Involvement of constitutive transforming growth factor-beta signaling and endothelial-to-mesenchymal transition. Circulation 2010, 122, 1200–1209. [Google Scholar] [CrossRef] [PubMed]
- Matucci Cerinic, M.M.; Valentini, G.; Sorano, G.G.; D’Angelo, S.; Cuomo, G.; Fenu, L.; Generini, S.; Cinotti, S.; Morfini, M.; Pignone, A.; et al. Blood coagulation, fibrinolysis, and markers of endothelial dysfunction in systemic sclerosis. Semin. Arthritis Rheum. 2003, 32, 285–295. [Google Scholar] [CrossRef]
- Chiang, T.M.; Takayama, H.; Postlethwaite, A.E. Increase in platelet non-integrin type I collagen receptor in patients with systemic sclerosis. Thromb. Res. 2006, 117, 299–306. [Google Scholar] [CrossRef]
- Ramirez, G.A.; Franchini, S.; Rovere-Querini, P.; Sabbadini, M.G.; Manfredi, A.A.; Maugeri, N. The role of platelets in the pathogenesis of systemic sclerosis. Front. Immunol. 2012, 3, 160. [Google Scholar] [CrossRef]
- Quinton, T.M.; Kim, S.; Derian, C.K.; Jin, J.; Kunapuli, S.P. Plasmin-mediated activation of platelets occurs by cleavage of protease-activated receptor 4. J. Biol. Chem. 2004, 279, 18434–18439. [Google Scholar] [CrossRef] [PubMed]
- Watabe, A.; Ohta, M.; Matsuyama, N.; Mizuno, K.; el Borai, N.; Tanimoto, T.; Kawanishi, T.; Hayakawa, T. Characterization of plasmin-induced platelet aggregation. Res. Commun. Mol. Pathol. Pharmacol. 1997, 96, 341–352. [Google Scholar] [PubMed]
- Niewiarowski, S.; Senyi, A.F.; Gillies, P. Plasmin-induced platelet aggregation and platelet release reaction. Effects on hemostasis. J. Clin. Investig. 1973, 52, 1647–1659. [Google Scholar] [CrossRef] [PubMed]
- Brogren, H.; Karlsson, L.; Andersson, M.; Wang, L.; Erlinge, D.; Jern, S. Platelets synthesize large amounts of active plasminogen activator inhibitor 1. Blood 2004, 104, 3943–3948. [Google Scholar] [CrossRef] [PubMed]
- Plow, E.F.; Collen, D. The presence and release of alpha 2-antiplasmin from human platelets. Blood 1981, 58, 1069–1074. [Google Scholar] [PubMed]
- Podor, T.J.; Peterson, C.B.; Lawrence, D.A.; Stefansson, S.; Shaughnessy, S.G.; Foulon, D.M.; Butcher, M.; Weitz, J.I. Type 1 plasminogen activator inhibitor binds to fibrin via vitronectin. J. Biol. Chem. 2000, 275, 19788–19794. [Google Scholar] [CrossRef]
- Heyman, S.N.; Hanna, Z.; Nassar, T.; Shina, A.; Akkawi, S.; Goldfarb, M.; Rosen, S.; Higazi, A.A. The fibrinolytic system attenuates vascular tone: Effects of tissue plasminogen activator (tPA) and aminocaproic acid on renal microcirculation. Br. J. Pharmacol. 2004, 141, 971–978. [Google Scholar] [CrossRef]
- Nassar, T.; Akkawi, S.; Shina, A.; Haj-Yehia, A.; Bdeir, K.; Tarshis, M.; Heyman, S.N.; Higazi, AA. In vitro and in vivo effects of tPA and PAI-1 on blood vessel tone. Blood 2004, 103, 897–902. [Google Scholar] [CrossRef]
- Kaikita, K.; Fogo, A.B.; Ma, L.; Schoenhard, J.A.; Brown, N.J.; Vaughan, D.E. Plasminogen activator inhibitor-1 deficiency prevents hypertension and vascular fibrosis in response to long-term nitric oxide synthase inhibition. Circulation 2001, 104, 839–844. [Google Scholar] [CrossRef]
- Makarova, A.M.; Lebedeva, T.V.; Nassar, T.; Higazi, A.A.; Xue, J.; Carinato, M.E.; Bdeir, K.; Cines, D.B.; Stepanova, V. Urokinase-type plasminogen activator (uPA) induces pulmonary microvascular endothelial permeability through low density lipoprotein receptor-related protein (LRP)-dependent activation of endothelial nitric-oxide synthase. J. Biol. Chem. 2011, 286, 23044–23053. [Google Scholar] [CrossRef]
- Takahashi, S.; Shinya, T.; Sugiyama, A. Angiostatin inhibition of vascular endothelial growth factor-stimulated nitric oxide production in endothelial cells. J. Pharmacol. Sci. 2010, 112, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Koshida, R.; Ou, J.; Matsunaga, T.; Chilian, W.M.; Oldham, K.T.; Ackerman, A.W.; Pritchard, K.A., Jr. Angiostatin: A negative regulator of endothelial-dependent vasodilation. Circulation 2003, 107, 803–806. [Google Scholar] [CrossRef] [PubMed]
- Chaisson, N.F.; Hassoun, P.M. Systemic sclerosis-associated pulmonary arterial hypertension. Chest 2013, 144, 1346–1356. [Google Scholar] [CrossRef]
- Hickey, P.M.; Lawrie, A.; Condliffe, R. Circulating Protein Biomarkers in Systemic Sclerosis Related Pulmonary Arterial Hypertension: A Review of Published Data. Front. Med. 2018, 5, 175. [Google Scholar] [CrossRef] [PubMed]
- Manetti, M.; Allanore, Y.; Revillod, L.; Fatini, C.; Guiducci, S.; Cuomo, G.; Bonino, C.; Riccieri, V.; Bazzichi, L.; Liakouli, V.; et al. A genetic variation located in the promoter region of the UPAR (CD87) gene is associated with the vascular complications of systemic sclerosis. Arthritis Rheum. 2011, 63, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Ban, C.; Wang, T.; Zhang, S.; Xin, P.; Liang, L.; Wang, C.; Dai, H. Fibrinolytic system related to pulmonary arterial pressure and lung function of patients with idiopathic pulmonary fibrosis. Clin. Respir. J. 2017, 11, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Moons, L.; Bouché, A.; Shapiro, S.D.; Collen, D.; Carmeliet, P. Deficiency of urokinase-type plasminogen activator-mediated plasmin generation impairs vascular remodeling during hypoxia-induced pulmonary hypertension in mice. Circulation 2001, 103, 2014–2020. [Google Scholar] [CrossRef] [PubMed]
- Jurasz, P.; Ng, D.; Granton, J.T.; Courtman, D.W.; Stewart, D.J. Elevated platelet angiostatin and circulating endothelial microfragments in idiopathic pulmonary arterial hypertension: A preliminary study. Thromb. Res. 2010, 125, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Pascaud, M.A.; Griscelli, F.; Raoul, W.; Marcos, E.; Opolon, P.; Raffestin, B.; Perricaudet, M.; Adnot, S.; Eddahibi, S. Lung overexpression of angiostatin aggravates pulmonary hypertension in chronically hypoxic mice. Am. J. Respir. Cell Mol. Biol. 2003, 29, 449–457. [Google Scholar] [CrossRef]



© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanno, Y. The Role of Fibrinolytic Regulators in Vascular Dysfunction of Systemic Sclerosis. Int. J. Mol. Sci. 2019, 20, 619. https://doi.org/10.3390/ijms20030619
Kanno Y. The Role of Fibrinolytic Regulators in Vascular Dysfunction of Systemic Sclerosis. International Journal of Molecular Sciences. 2019; 20(3):619. https://doi.org/10.3390/ijms20030619
Chicago/Turabian StyleKanno, Yosuke. 2019. "The Role of Fibrinolytic Regulators in Vascular Dysfunction of Systemic Sclerosis" International Journal of Molecular Sciences 20, no. 3: 619. https://doi.org/10.3390/ijms20030619
APA StyleKanno, Y. (2019). The Role of Fibrinolytic Regulators in Vascular Dysfunction of Systemic Sclerosis. International Journal of Molecular Sciences, 20(3), 619. https://doi.org/10.3390/ijms20030619