The Amyloid-Tau-Neuroinflammation Axis in the Context of Cerebral Amyloid Angiopathy


Abstract
1. Introduction
2. The Diversity of Amyloid in CAA
2.1. Aβ Amyloid in CAA
2.2. Non-Aβ Amyloid in CAA
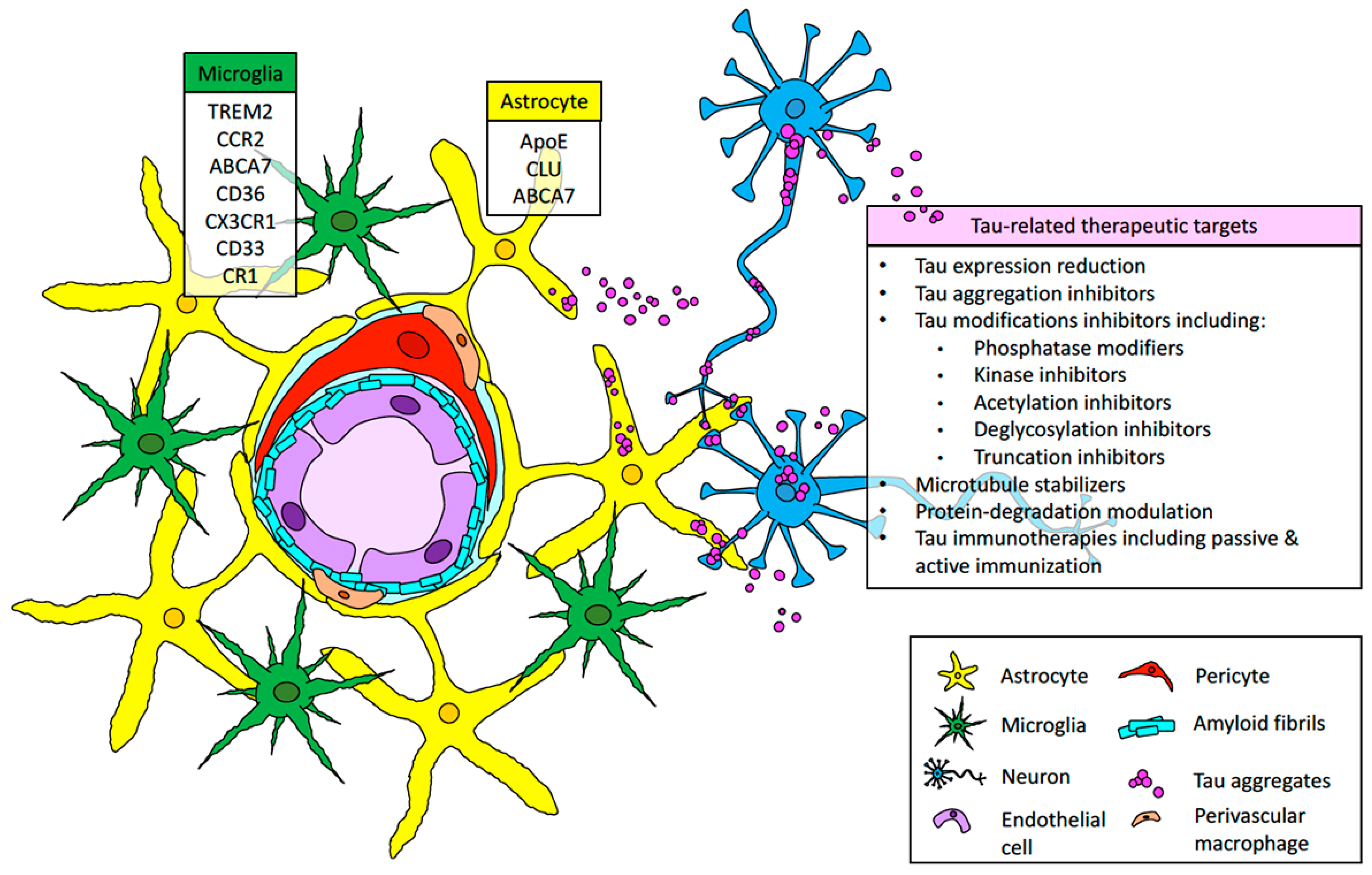
3. Perivascular Tau Aggregation and Its Interplay with Cerebrovascular Damage and CAA
4. The Implication of AD Immune-Risk Factors and Glial Response in CAA
4.1. Microgliosis in CAA
4.2. Astrogliosis in CAA
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Karch, C.M.; Cruchaga, C.; Goate, A.M. Alzheimer’s disease genetics: From the bench to the clinic. Neuron 2014, 83, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Citron, M. Alzheimer’s disease: Strategies for disease modification. Nat. Rev. Drug Discov. 2010, 9, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Attems, J. Sporadic cerebral amyloid angiopathy: Pathology, clinical implications, and possible pathomechanisms. Acta Neuropathol. 2005, 110, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A.; Greenberg, S.M. Cerebral amyloid angiopathy: A systematic review. J. Clin. Neurol. 2011, 7, 1–9. [Google Scholar] [CrossRef]
- Viswanathan, A.; Greenberg, S.M. Cerebral amyloid angiopathy in the elderly. Ann. Neurol. 2011, 70, 871–880. [Google Scholar] [CrossRef]
- Boulouis, G.; Charidimou, A.; Greenberg, S.M. Sporadic Cerebral Amyloid Angiopathy: Pathophysiology, Neuroimaging Features, and Clinical Implications. Semin. Neurol. 2016, 36, 233–243. [Google Scholar] [CrossRef]
- Attems, J.; Jellinger, K.A. Only cerebral capillary amyloid angiopathy correlates with Alzheimer pathology—A pilot study. Acta Neuropathol. 2004, 107, 83–90. [Google Scholar] [CrossRef]
- Tian, J.; Shi, J.; Bailey, K.; Mann, D.M.A. Negative association between amyloid plaques and cerebral amyloid angiopathy in Alzheimer’s disease. Neurosci. Lett. 2003, 352, 137–140. [Google Scholar] [CrossRef]
- Xu, D.; Yang, C.; Wang, L. Cerebral amyloid angiopathy in aged Chinese: A clinico-neuropathological study. Acta Neuropathol. 2003, 106, 89–91. [Google Scholar] [CrossRef]
- Weller, R.O.; Massey, A.; Newman, T.A.; Hutchings, M.; Kuo, Y.M.; Roher, A.E. Cerebral amyloid angiopathy: Amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Am. J. Pathol. 1998, 153, 725–733. [Google Scholar] [CrossRef]
- Weller, R.O.; Subash, M.; Preston, S.; Mazanti, I.; Carare, R. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol. 2008, 18, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.A.; Härtig, W.; Kacza, J.; Schliebs, R.; Weller, R.O.; Nicoll, J.A.; Carare, R.O. Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol. 2011, 121, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Arbel-Ornath, M.; Hudry, E.; Eikermann-Haerter, K.; Hou, S.; Gregory, J.L.; Zhao, L.; Betensky, R.A.; Frosch, M.P.; Greenberg, S.M.; Bacskai, B.J. Interstitial fluid drainage is impaired in ischemic stroke and Alzheimer’s disease mouse models. Acta Neuropathol. 2013, 126, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Chodobski, A.; Zink, B.J.; Szmydynger-Chodobska, J. Blood-brain barrier pathophysiology in traumatic brain injury. Transl. Stroke Res. 2011, 2, 492–516. [Google Scholar] [CrossRef]
- Cadavid, D.; Mena, H.; Koeller, K.; Frommelt, R.A. Cerebral beta amyloid angiopathy is a risk factor for cerebral ischemic infarction. A case control study in human brain biopsies. J. Neuropathol. Exp. Neurol. 2000, 59, 768–773. [Google Scholar] [CrossRef]
- Greenberg, S.M. Cerebral amyloid angiopathy and dementia: Two amyloids are worse than one. Neurology 2002, 58, 1587–1588. [Google Scholar] [CrossRef]
- Natte, R.; Maat-Schieman, M.L.; Haan, J.; Bornebroek, M.; Roos, R.A.; Van Duinen, S.G. Dementia in hereditary cerebral hemorrhage with amyloidosis-Dutch type is associated with cerebral amyloid angiopathy but is independent of plaques and neurofibrillary tangles. Ann. Neurol. 2001, 50, 765–772. [Google Scholar] [CrossRef]
- Garringer, H.J.; Murrell, J.; D’Adamio, L.; Ghetti, B.; Vidal, R. Modeling familial British and Danish dementia. Brain Struct. Funct. 2010, 214, 235–244. [Google Scholar] [CrossRef]
- Vidal, R.; Calero, M.; Piccardo, P.; Farlow, M.R.; Unverzagt, F.W.; Méndez, E.; Jiménez-Huete, A.; Beavis, R.; Gallo, G.; Gomez-Tortosa, E.; et al. Senile dementia associated with amyloid beta protein angiopathy and tau perivascular pathology but not neuritic plaques in patients homozygous for the APOE-epsilon4 allele. Acta Neuropathol. 2000, 100, 1–12. [Google Scholar] [CrossRef]
- Ghetti, B.; Piccardo, P.; Spillantini, M.G.; Ichimiya, Y.; Porro, M.; Perini, F.; Kitamoto, T.; Tateishi, J.; Seiler, C.; Frangione, B.; et al. Vascular variant of prion protein cerebral amyloidosis with tau-positive neurofibrillary tangles: The phenotype of the stop codon 145 mutation in PRNP. Proc. Natl. Acad. Sci. USA 1996, 93, 744–748. [Google Scholar] [CrossRef]
- Revesz, T.; Holton, J.L.; Lashley, T.; Plant, G.; Rostagno, A.; Ghiso, J.; Frangione, B. Sporadic and familial cerebral amyloid angiopathies. Brain Pathol. 2002, 12, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Revesz, T.; Holton, J.L.; Lashley, T.; Plant, G.; Frangione, B.; Rostagno, A.; Ghiso, J. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol. 2009, 118, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Kumar-Singh, S. Hereditary and sporadic forms of abeta-cerebrovascular amyloidosis and relevant transgenic mouse models. Int. J. Mol. Sci. 2009, 10, 1872–1895. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharm. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, C.M.; Ciuchta, J.; Ikonomovic, M.D.; Fish, K.N.; Abrahamson, E.E.; Murray, P.S.; Klunk, W.E.; Sweet, R.A. Dendritic spine density, morphology, and fibrillar actin content surrounding amyloid-beta plaques in a mouse model of amyloid-beta deposition. J. Neuropathol. Exp. Neurol. 2013, 72, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Lemaire, H.G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.H.; Multhaup, G.; Beyreuther, K.; Muller-Hill, B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, T.; Ghiso, J.; Frangione, B. Peptides homologous to the amyloid protein of Alzheimer’s disease containing a glutamine for glutamic acid substitution have accelerated amyloid fibril formation. Biochem. Biophys. Res. Commun. 1991, 180, 1528. [Google Scholar] [CrossRef]
- Kumar-Singh, S. Cerebral amyloid angiopathy: Pathogenetic mechanisms and link to dense amyloid plaques. Genes Brain Behav. 2008, 7 (Suppl. 1), 67–82. [Google Scholar] [CrossRef]
- Maat-Schieman, M.L.; van Duinen, S.G.; Bornebroek, M.; Haan, J.; Roos, R.A. Hereditary cerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D): II-A review of histopathological aspects. Brain Pathol. 1996, 6, 115–120. [Google Scholar] [CrossRef]
- Bornebroek, M.; Haan, J.; Maat-Schieman, M.L.; Van Duinen, S.G.; Roos, R.A. Hereditary cerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D): I—A review of clinical, radiologic and genetic aspects. Brain Pathol. 1996, 6, 111–114. [Google Scholar] [CrossRef]
- Levy, E.; Carman, M.D.; Fernandez-Madrid, I.J.; Power, M.D.; Lieberburg, I.; van Duinen, S.G.; Bots, G.T.; Luyendijk, W.; Frangione, B. Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 1990, 248, 1124–1126. [Google Scholar] [CrossRef] [PubMed]
- Miravalle, L.; Tokuda, T.; Chiarle, R.; Giaccone, G.; Bugiani, O.; Tagliavini, F.; Frangione, B.; Ghiso, J. Substitutions at codon 22 of Alzheimer’s abeta peptide induce diverse conformational changes and apoptotic effects in human cerebral endothelial cells. J. Biol. Chem. 2000, 275, 27110–27116. [Google Scholar] [PubMed]
- Bugiani, O.; Giaccone, G.; Rossi, G.; Mangieri, M.; Capobianco, R.; Morbin, M.; Mazzoleni, G.; Cupidi, C.; Marcon, G.; Giovagnoli, A.; et al. Hereditary cerebral hemorrhage with amyloidosis associated with the E693K mutation of APP. Arch. Neurol. 2010, 67, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Nilsberth, C.; Westlind-Danielsson, A.; Eckman, C.B.; Condron, M.M.; Axelman, K.; Forsell, C.; Stenh, C.; Luthman, J.; Teplow, D.B.; Younkin, S.G.; et al. The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Abeta protofibril formation. Nat. Neurosci. 2001, 4, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, T.; Nagata, T.; Shimada, H.; Teraoka, R.; Fukushima, A.; Kanemitsu, H.; Takuma, H.; Kuwano, R.; Imagawa, M.; Ataka, S.; et al. A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. Ann. Neurol. 2008, 63, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Inayathullah, M.; Teplow, D.B. Teplow, Structural dynamics of the DeltaE22 (Osaka) familial Alzheimer’s disease-linked amyloid beta-protein. Amyloid 2011, 18, 98–107. [Google Scholar] [CrossRef]
- De Jonghe, C.; Zehr, C.; Yager, D.; Prada, C.M.; Younkin, S.; Hendriks, L.; Van Broeckhoven, C.; Eckman, C.B. Flemish and Dutch mutations in amyloid beta precursor protein have different effects on amyloid beta secretion. Neurobiol. Dis. 1998, 5, 281–286. [Google Scholar] [CrossRef]
- Grabowski, T.J.; Cho, H.S.; Vonsattel, J.P.; Rebeck, G.W.; Greenberg, S.M. Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann. Neurol. 2001, 49, 697–705. [Google Scholar] [CrossRef]
- Obici, L.; Demarchi, A.; de Rosa, G.; Bellotti, V.; Marciano, S.; Donadei, S.; Arbustini, E.; Palladini, G.; Diegoli, M.; Genovese, E.; et al. A novel AbetaPP mutation exclusively associated with cerebral amyloid angiopathy. Ann. Neurol. 2005, 58, 639–644. [Google Scholar] [CrossRef]
- Rossi, G.; Giaccone, G.; Maletta, R.; Morbin, M.; Capobianco, R.; Mangieri, M.; Giovagnoli, A.R.; Bizzi, A.; Tomaino, C.; Perri, M.; et al. A family with Alzheimer disease and strokes associated with A713T mutation of the APP gene. Neurology 2004, 63, 910–912. [Google Scholar] [CrossRef]
- Conidi, M.E.; Bernardi, L.; Puccio, G.; Smirne, N.; Muraca, M.G.; Curcio, S.A.; Colao, R.; Piscopo, P.; Gallo, M.; Anfossi, M.; et al. Homozygous carriers of APP A713T mutation in an autosomal dominant Alzheimer disease family. Neurology 2015, 84, 2266–2273. [Google Scholar] [CrossRef] [PubMed]
- Kumar-Singh, S.; De Jonghe, C.; Cruts, M.; Kleinert, R.; Wang, R.; Mercken, M.; De Strooper, B.; Vanderstichele, H.; Lofgren, A.; Vanderhoeven, I.; et al. Nonfibrillar diffuse amyloid deposition due to a gamma(42)-secretase site mutation points to an essential role for N-truncated A beta(42) in Alzheimer’s disease. Hum. Mol. Genet. 2000, 9, 2589–2598. [Google Scholar] [CrossRef] [PubMed]
- Pasalar, P.; Najmabadi, H.; Noorian, A.R.; Moghimi, B.; Jannati, A.; Soltanzadeh, A.; Krefft, T.; Crook, R.; Hardy, J. Iranian family with Alzheimer’s disease caused by a novel APP mutation (Thr714Ala). Neurology 2002, 58, 1574–1575. [Google Scholar] [CrossRef] [PubMed]
- Vidal, R.; Frangione, B.; Rostagno, A.; Mead, S.; Révész, T.; Plant, G.; Ghiso, J. A stop-codon mutation in the BRI gene associated with familial British dementia. Nature 1999, 399, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Garringer, H.J.; Sammeta, N.; Oblak, A.; Ghetti, B.; Vidal, R. Amyloid and intracellular accumulation of BRI2. Neurobiol. Aging 2017, 52, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Marcora, M.S. Amyloid peptides ABri and ADan show differential neurotoxicity in transgenic Drosophila models of familial British and Danish dementia. Mol. Neurodegener. 2014, 9, 5. [Google Scholar] [CrossRef]
- Holton, J.L. Regional distribution of amyloid-Bri deposition and its association with neurofibrillary degeneration in familial British dementia. Am. J. Pathol. 2001, 158, 515–526. [Google Scholar] [CrossRef]
- Vidal, R.; Revesz, T.; Rostagno, A.; Kim, E.; Holton, J.L.; Bek, T.; Bojsen-Moller, M.; Braendgaard, H.; Plant, G.; Ghiso, J.; et al. A decamer duplication in the 3′ region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred. Proc. Natl. Acad. Sci. USA 2000, 97, 4920–4925. [Google Scholar] [CrossRef]
- Holton, J.L.; Lashley, T.; Ghiso, J.; Braendgaard, H.; Vidal, R.; Guerin, C.J.; Gibb, G.; Hanger, D.P.; Rostagno, A.; Anderton, B.H.; et al. Familial Danish dementia: A novel form of cerebral amyloidosis associated with deposition of both amyloid-Dan and amyloid-beta. J. Neuropathol. Exp. Neurol. 2002, 61, 254–267. [Google Scholar] [CrossRef]
- Vidal, R.; Barbeito, A.G.; Miravalle, L.; Ghetti, B. Cerebral amyloid angiopathy and parenchymal amyloid deposition in transgenic mice expressing the Danish mutant form of human BRI2. Brain Pathol. 2009, 19, 58–68. [Google Scholar] [CrossRef]
- Jurczak, P.; Groves, P.; Szymanska, A.; Rodziewicz-Motowidlo, S. Human cystatin C monomer, dimer, oligomer, and amyloid structures are related to health and disease. FEBS Lett. 2016, 590, 4192–4201. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Z.; Jensson, O.; Thorsteinsson, L.; Vinters, H.V. Microvascular degeneration in hereditary cystatin C amyloid angiopathy of the brain. APMIS 1997, 105, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.; Sastre, M.; Kumar, A.; Gallo, G.; Piccardo, P.; Ghetti, B.; Tagliavini, F. Codeposition of cystatin C with amyloid-beta protein in the brain of Alzheimer disease patients. J. Neuropathol. Exp. Neurol. 2001, 60, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Sato, S.; Takahashi, T.; Nakazaki, H.; Date, Y.; Nakazato, M.; Tominaga, T.; Itoyama, Y.; Ikeda, S. Familial leptomeningeal amyloidosis with a transthyretin variant Asp18Gly representing repeated subarachnoid haemorrhages with superficial siderosis. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1463–1466. [Google Scholar] [CrossRef] [PubMed]
- Garzuly, F.; Vidal, R.; Wisniewski, T.; Brittig, F.; Budka, H. Familial meningocerebrovascular amyloidosis, Hungarian type, with mutant transthyretin (TTR Asp18Gly). Neurology 1996, 47, 1562–1567. [Google Scholar] [CrossRef] [PubMed]
- Garzuly, F. Clinical characteristics of Hungarian-type familial meningo-cerebrovascular amyloidosis. Orv. Hetil. 1996, 137, 2393–2399. [Google Scholar] [PubMed]
- Vidal, R.; Garzuly, F.; Budka, H.; Lalowski, M.; Linke, R.P.; Brittig, F.; Frangione, B.; Wisniewski, T. Meningocerebrovascular amyloidosis associated with a novel transthyretin mis-sense mutation at codon 18 (TTRD 18G). Am. J. Pathol. 1996, 148, 361–366. [Google Scholar]
- Petersen, R.B.; Goren, H.; Cohen, M.; Richardson, S.L.; Tresser, N.; Lynn, A.; Gali, M.; Estes, M.; Gambetti, P. Transthyretin amyloidosis: A new mutation associated with dementia. Ann. Neurol. 1997, 41, 307–313. [Google Scholar] [CrossRef]
- Martin, S.E.; Benson, M.D.; Hattab, E.M. The pathologic spectrum of oculoleptomeningeal amyloidosis with Val30Gly transthyretin gene mutation in a postmortem case. Hum. Pathol. 2014, 45, 1105–1108. [Google Scholar] [CrossRef]
- Roe, R.H.; Fisher, Y.; Eagle, R.C., Jr.; Fine, H.F.; Cunningham, E.T., Jr. Oculoleptomeningeal amyloidosis in a patient with a TTR Val30Gly mutation in the transthyretin gene. Ophthalmology 2007, 114, e33–e37. [Google Scholar] [CrossRef]
- Ziskin, J.L.; Greicius, M.D.; Zhu, W.; Okumu, A.N.; Adams, C.M.; Plowey, E.D. Neuropathologic analysis of Tyr69His TTR variant meningovascular amyloidosis with dementia. Acta Neuropathol. Commun. 2015, 3, 43. [Google Scholar] [CrossRef] [PubMed]
- Blevins, G.; Macaulay, R.; Harder, S.; Fladeland, D.; Yamashita, T.; Yazaki, M.; Hamidi Asl, K.; Benson, M.D.; Donat, J.R. Oculoleptomeningeal amyloidosis in a large kindred with a new transthyretin variant Tyr69His. Neurology 2003, 60, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, K.; Ehmann, D.; Garcia, R.; Alport, E. Oculoleptomeningeal amyloidosis in 3 individuals with the transthyretin variant Tyr69His. Can. J. Ophthalmol. 2009, 44, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Suhr, O.B.; Andersen, O.; Aronsson, T.; Jonasson, J.; Kalimo, H.; Lundahl, C.; Lundgren, H.E.; Melberg, A.; Nyberg, J.; Olsson, M.; et al. Report of five rare or previously unknown amyloidogenic transthyretin mutations disclosed in Sweden. Amyloid 2009, 16, 208–214. [Google Scholar] [CrossRef]
- Hagiwara, K.; Ochi, H.; Suzuki, S.; Shimizu, Y.; Tokuda, T.; Murai, H.; Shigeto, H.; Ohyagi, Y.; Iwata, M.; Iwaki, T.; et al. Highly selective leptomeningeal amyloidosis with transthyretin variant Ala25Thr. Neurology 2009, 72, 1358–1360. [Google Scholar] [CrossRef]
- Shimizu, Y.; Takeuchi, M.; Matsumura, M.; Tokuda, T.; Iwata, M. A case of biopsy-proven leptomeningeal amyloidosis and intravenous Ig-responsive polyneuropathy associated with the Ala25Thr transthyretin gene mutation. Amyloid 2006, 13, 37–41. [Google Scholar] [CrossRef]
- Herrick, M.K.; DeBruyne, K.; Horoupian, D.S.; Skare, J.; Vanefsky, M.A.; Ong, T. Massive leptomeningeal amyloidosis associated with a Val30Met transthyretin gene. Neurology 1996, 47, 988–992. [Google Scholar] [CrossRef]
- Maia, L.F.; Magalhaes, R.; Freitas, J.; Taipa, R.; Pires, M.M.; Osorio, H.; Dias, D.; Pessegueiro, H.; Correia, M.; Coelho, T. CNS involvement in V30M transthyretin amyloidosis: Clinical, neuropathological and biochemical findings. J. Neurol. Neurosurg. Psychiatry 2015, 86, 159–167. [Google Scholar] [CrossRef]
- Nakagawa, K.; Sheikh, S.I.; Snuderl, M.; Frosch, M.P.; Greenberg, S.M. A new Thr49Pro transthyretin gene mutation associated with leptomeningeal amyloidosis. J. Neurol. Sci. 2008, 272, 186–190. [Google Scholar] [CrossRef]
- Motozaki, Y.; Sugiyama, Y.; Ishida, C.; Komai, K.; Matsubara, S.; Yamada, M. Phenotypic heterogeneity in a family with FAP due to a TTR Leu58Arg mutation: A clinicopathologic study. J. Neurol. Sci. 2007, 260, 236–239. [Google Scholar] [CrossRef][Green Version]
- Uemichi, T.; Uitti, R.J.; Koeppen, A.H.; Donat, J.R.; Benson, M.D. Oculoleptomeningeal amyloidosis associated with a new transthyretin variant Ser64. Arch. Neurol. 1999, 56, 1152–1155. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Yamashita, T.; Ueda, M.; Obayashi, K.; Sato, T.; Ikeda, T.; Washimi, Y.; Hirai, T.; Kuwahara, Y.; Yamamoto, M.T.; et al. Neuroradiologic and clinicopathologic features of oculoleptomeningeal type amyloidosis. Neurology 2005, 65, 1051–1056. [Google Scholar] [CrossRef] [PubMed]
- Brett, M.; Persey, M.R.; Reilly, M.M.; Revesz, T.; Booth, D.R.; Booth, S.E.; Hawkins, P.N.; Pepys, M.B.; Morgan-Hughes, J.A. Transthyretin Leu12Pro is associated with systemic, neuropathic and leptomeningeal amyloidosis. Brain 1999, 122 Pt 2, 183–190. [Google Scholar] [CrossRef]
- Liepnieks, J.J.; Dickson, D.W.; Benson, M.D. A new transthyretin mutation associated with leptomeningeal amyloidosis. Amyloid 2011, 18 (Suppl. 1), 160–162. [Google Scholar] [CrossRef] [PubMed]
- Solomon, J.P.; Page, L.J.; Balch, W.E.; Kelly, J.W. Gelsolin amyloidosis: Genetics, biochemistry, pathology and possible strategies for therapeutic intervention. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 282–296. [Google Scholar] [CrossRef] [PubMed]
- Kiuru, S. Gelsolin-related familial amyloidosis, Finnish type (FAF), and its variants found worldwide. Amyloid 1998, 5, 55–66. [Google Scholar] [CrossRef]
- Ghiso, J.; Haltia, M.; Prelli, F.; Novello, J.; Frangione, B. Gelsolin variant (Asn-187) in familial amyloidosis, Finnish type. Biochem. J. 1990, 272, 827–830. [Google Scholar] [CrossRef]
- Mead, S.; Gandhi, S.; Beck, J.; Caine, D.; Gallujipali, D.; Carswell, C.; Hyare, H.; Joiner, S.; Ayling, H.; Lashley, T.; et al. A novel prion disease associated with diarrhea and autonomic neuropathy. N. Engl. J. Med. 2013, 369, 1904–1914. [Google Scholar] [CrossRef]
- Weller, R.O.; Preston, S.D.; Subash, M.; Carare, R.O. Cerebral amyloid angiopathy in the aetiology and immunotherapy of Alzheimer disease. Alzheimers Res. Ther. 2009, 1, 6. [Google Scholar] [CrossRef]
- Revesz, T.; Ghiso, J.; Lashley, T.; Plant, G.; Rostagno, A.; Frangione, B.; Holton, J.L. Cerebral amyloid angiopathies: A pathologic, biochemical, and genetic view. J. Neuropathol. Exp. Neurol. 2003, 62, 885–898. [Google Scholar] [CrossRef]
- Frangione, B.; Revesz, T.; Vidal, R.; Holton, J.; Lashley, T.; Houlden, H.; Wood, N.; Rostagno, A.; Plant, G.; Ghiso, J. Familial cerebral amyloid angiopathy related to stroke and dementia. Amyloid 2001, 8 (Suppl. 1), 36–42. [Google Scholar] [PubMed]
- Garringer, H.J.; Murrell, J.; Sammeta, N.; Gnezda, A.; Ghetti, B.; Vidal, R. Increased tau phosphorylation and tau truncation, and decreased synaptophysin levels in mutant BRI2/tau transgenic mice. PLoS ONE 2013, 8, e56426. [Google Scholar] [CrossRef] [PubMed]
- Kolodziejczyk, R.; Michalska, K.; Hernandez-Santoyo, A.; Wahlbom, M.; Grubb, A.; Jaskolski, M. Crystal structure of human cystatin C stabilized against amyloid formation. Febs J. 2010, 277, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.I.; Kuwata, K. Formation and properties of amyloid fibrils of prion protein. Biophys. Rev. 2018, 10, 517–525. [Google Scholar] [CrossRef]
- Ghetti, B.; Piccardo, P.; Frangione, B.; Bugiani, O.; Giaccone, G.; Young, K.; Prelli, F.; Farlow, M.R.; Dlouhy, S.R.; Tagliavini, F. Prion protein amyloidosis. Brain Pathol. 1996, 6, 127–145. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The many faces of tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef] [PubMed]
- Nation, D.A.; Edmonds, E.C.; Bangen, K.J.; Delano-Wood, L.; Scanlon, B.K.; Han, S.D.; Edland, S.D.; Salmon, D.P.; Galasko, D.R.; Bondi, M.W.; et al. Pulse pressure in relation to tau-mediated neurodegeneration, cerebral amyloidosis, and progression to dementia in very old adults. JAMA Neurol. 2015, 72, 546–553. [Google Scholar] [CrossRef]
- Kim, H.J.; Park, S.; Cho, H.; Jang, Y.K.; San Lee, J.; Jang, H.; Kim, Y.; Kim, K.W.; Ryu, Y.H.; Choi, J.Y.; et al. Assessment of Extent and Role of Tau in Subcortical Vascular Cognitive Impairment Using 18F-AV1451 Positron Emission Tomography Imaging. JAMA Neurol. 2018. [Google Scholar] [CrossRef]
- Bennett, R.E.; Robbins, A.B.; Hu, M.; Cao, X.; Betensky, R.A.; Clark, T.; Das, S.; Hyman, B.T. Tau induces blood vessel abnormalities and angiogenesis-related gene expression in P301L transgenic mice and human Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E1289–E1298. [Google Scholar] [CrossRef]
- Merlini, M.; Wanner, D.; Nitsch, R.M. Tau pathology-dependent remodelling of cerebral arteries precedes Alzheimer’s disease-related microvascular cerebral amyloid angiopathy. Acta Neuropathol. 2016, 131, 737–752. [Google Scholar] [CrossRef]
- Bartels, A.L.; Willemsen, A.T.; Kortekaas, R.; de Jong, B.M.; de Vries, R.; de Klerk, O.; van Oostrom, J.C.; Portman, A.; Leenders, K.L. Decreased blood-brain barrier P-glycoprotein function in the progression of Parkinson’s disease, PSP and MSA. J. Neural Transm. 2008, 115, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Carranza, D.L.; Nilson, A.N.; Van Skike, C.E.; Jahrling, J.B.; Patel, K.; Garach, P.; Gerson, J.E.; Sengupta, U.; Abisambra, J.; Nelson, P.; et al. Cerebral Microvascular Accumulation of Tau Oligomers in Alzheimer’s Disease and Related Tauopathies. Aging Dis. 2017, 8, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Blair, L.J.; Frauen, H.D.; Zhang, B.; Nordhues, B.A.; Bijan, S.; Lin, Y.C.; Zamudio, F.; Hernandez, L.D.; Sabbagh, J.J.; Selenica, M.L.; et al. Tau depletion prevents progressive blood-brain barrier damage in a mouse model of tauopathy. Acta Neuropathol. Commun. 2015, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Oshima, K.; Uchikado, H.; Dickson, D.W. Perivascular neuritic dystrophy associated with cerebral amyloid angiopathy in Alzheimer’s disease. Int. J. Clin. Exp. Pathol. 2008, 1, 403–408. [Google Scholar]
- You, Y.; Perkins, A.; Cisternas, P.; Munoz, B.; Taylor, X.; You, Y.; Garringer, H.J.; Oblak, A.L.; Atwood, B.K.; Vidal, R.; et al. Tau as a mediator of neurotoxicity associated to cerebral amyloid angiopathy. Acta Neuropathol. Commun. 2019, 7, 26. [Google Scholar] [CrossRef]
- Ransom, B.; Behar, T.; Nedergaard, M. New roles for astrocytes (stars at last). Trends Neurosci. 2003, 26, 520–522. [Google Scholar] [CrossRef]
- Komori, T. Tau-positive glial inclusions in progressive supranuclear palsy, corticobasal degeneration and Pick’s disease. Brain Pathol. 1999, 9, 663–679. [Google Scholar] [CrossRef]
- Ikeda, K.; Akiyama, H.; Kondo, H.; Haga, C.; Tanno, E.; Tokuda, T.; Ikeda, S. Thorn-shaped astrocytes: Possibly secondarily induced tau-positive glial fibrillary tangles. Acta Neuropathol. 1995, 90, 620–625. [Google Scholar] [CrossRef]
- Kanaan, N.M.; Cox, K.; Alvarez, V.E.; Stein, T.D.; Poncil, S.; McKee, A.C. Characterization of Early Pathological Tau Conformations and Phosphorylation in Chronic Traumatic Encephalopathy. J. Neuropathol. Exp. Neurol. 2016, 75, 19–34. [Google Scholar] [CrossRef]
- Mez, J.; Daneshvar, D.H.; Kiernan, P.T.; Abdolmohammadi, B.; Alvarez, V.E.; Huber, B.R.; Alosco, M.L.; Solomon, T.M.; Nowinski, C.J.; McHale, L.; et al. Clinicopathological Evaluation of Chronic Traumatic Encephalopathy in Players of American Football. JAMA 2017, 318, 360–370. [Google Scholar] [CrossRef]
- Andrews-Zwilling, Y.; Bien-Ly, N.; Xu, Q.; Li, G.; Bernardo, A.; Yoon, S.Y.; Zwilling, D.; Yan, T.X.; Chen, L.; Huang, Y. Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 13707–13717. [Google Scholar] [CrossRef]
- Roberson, E.D.; Halabisky, B.; Yoo, J.W.; Yao, J.; Chin, J.; Yan, F.; Wu, T.; Hamto, P.; Devidze, N.; Yu, G.Q.; et al. Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci. 2011, 31, 700–711. [Google Scholar] [CrossRef]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef]
- Singh, B.; Covelo, A.; Martell-Martinez, H.; Nanclares, C.; Sherman, M.A.; Okematti, E.; Meints, J.; Teravskis, P.J.; Gallardo, C.; Savonenko, A.V.; et al. Tau is required for progressive synaptic and memory deficits in a transgenic mouse model of alpha-synucleinopathy. Acta Neuropathol. 2019, 138, 551–574. [Google Scholar] [CrossRef]
- Fernandez-Nogales, M.; Cabrera, J.R.; Santos-Galindo, M.; Hoozemans, J.J.; Ferrer, I.; Rozemuller, A.J.; Hernandez, F.; Avila, J.; Lucas, J.J. Huntington’s disease is a four-repeat tauopathy with tau nuclear rods. Nat. Med. 2014, 20, 881–885. [Google Scholar] [CrossRef]
- Cheng, J.S.; Craft, R.; Yu, G.Q.; Ho, K.; Wang, X.; Mohan, G.; Mangnitsky, S.; Ponnusamy, R.; Mucke, L. Tau reduction diminishes spatial learning and memory deficits after mild repetitive traumatic brain injury in mice. PLoS ONE 2014, 9, e115765. [Google Scholar] [CrossRef]
- DeVos, S.L.; Goncharoff, D.K.; Chen, G.; Kebodeaux, C.S.; Yamada, K.; Stewart, F.R.; Schuler, D.R.; Maloney, S.E.; Wozniak, D.F.; Rigo, F.; et al. Antisense reduction of tau in adult mice protects against seizures. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 12887–12897. [Google Scholar] [CrossRef]
- Holth, J.K.; Bomben, V.C.; Reed, J.G.; Inoue, T.; Younkin, L.; Younkin, S.G.; Pautler, R.G.; Botas, J.; Noebels, J.L. Tau loss attenuates neuronal network hyperexcitability in mouse and Drosophila genetic models of epilepsy. J. Neurosci. 2013, 33, 1651–1659. [Google Scholar] [CrossRef]
- Gheyara, A.L.; Ponnusamy, R.; Djukic, B.; Craft, R.J.; Ho, K.; Guo, W.; Finucane, M.M.; Sanchez, P.E.; Mucke, L. Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann. Neurol. 2014, 76, 443–456. [Google Scholar] [CrossRef]
- Faraco, G.; Hochrainer, K.; Segarra, S.G.; Schaeffer, S.; Santisteban, M.M.; Menon, A.; Jiang, H.; Holtzman, D.M.; Anrather, J.; Iadecola, C. Dietary salt promotes cognitive impairment through tau phosphorylation. Nature 2019, 574, 686–690. [Google Scholar] [CrossRef]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef]
- Bertram, L.; Lange, C.; Mullin, K.; Parkinson, M.; Hsiao, M.; Hogan, M.F.; Schjeide, B.M.; Hooli, B.; Divito, J.; Ionita, I.; et al. Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am. J. Hum. Genet. 2008, 83, 623–632. [Google Scholar] [CrossRef]
- Ramirez, L.M.; Goukasian, N.; Porat, S.; Hwang, K.S.; Eastman, J.A.; Hurtz, S.; Wang, B.; Vang, N.; Sears, R.; Klein, E.; et al. Common variants in ABCA7 and MS4A6A are associated with cortical and hippocampal atrophy. Neurobiol. Aging 2016, 39, 82–89. [Google Scholar] [CrossRef]
- Kimbrough, I.F.; Robel, S.; Roberson, E.D.; Sontheimer, H. Vascular amyloidosis impairs the gliovascular unit in a mouse model of Alzheimer’s disease. Brain 2015, 138, 3716–3733. [Google Scholar] [CrossRef]
- Thomsen, M.S.; Routhe, L.J.; Moos, T. The vascular basement membrane in the healthy and pathological brain. J. Cereb. Blood Flow Metab. 2017, 37, 3300–3317. [Google Scholar] [CrossRef]
- Magaki, S.; Tang, Z.; Tung, S.; Williams, C.K.; Lo, D.; Yong, W.H.; Khanlou, N.; Vinters, H.V. The effects of cerebral amyloid angiopathy on integrity of the blood-brain barrier. Neurobiol. Aging 2018, 70, 70–77. [Google Scholar] [CrossRef]
- Yamada, M.; Itoh, Y.; Shintaku, M.; Kawamura, J.; Jensson, O.; Thorsteinsson, L.; Suematsu, N.; Matsushita, M.; Otomo, E. Immune reactions associated with cerebral amyloid angiopathy. Stroke 1996, 27, 1155–1162. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- Misra, A.; Chakrabarti, S.S.; Gambhir, I.S. New genetic players in late-onset Alzheimer’s disease: Findings of genome-wide association studies. Indian J. Med. Res. 2018, 148, 135–144. [Google Scholar] [CrossRef]
- Brown, L.S.; Foster, C.G.; Courtney, J.M.; King, N.E.; Howells, D.W.; Sutherland, B.A. Pericytes and Neurovascular Function in the Healthy and Diseased Brain. Front. Cell. Neurosci. 2019, 13, 282. [Google Scholar] [CrossRef]
- Winkler, E.A.; Sagare, A.P.; Zlokovic, B.V. The pericyte: A forgotten cell type with important implications for Alzheimer’s disease? Brain Pathol. 2014, 24, 371–386. [Google Scholar] [CrossRef]
- Cheng, J.; Korte, N.; Nortley, R.; Sethi, H.; Tang, Y.; Attwell, D. Targeting pericytes for therapeutic approaches to neurological disorders. Acta Neuropathol. 2018, 136, 507–523. [Google Scholar] [CrossRef]
- Sagare, A.P.; Bell, R.D.; Zhao, Z.; Ma, Q.; Winkler, E.A.; Ramanathan, A.; Zlokovic, B.V. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 2013, 4, 2932. [Google Scholar] [CrossRef]
- Zehendner, C.M.; Sebastiani, A.; Hugonnet, A.; Bischoff, F.; Luhmann, H.J.; Thal, S.C. Traumatic brain injury results in rapid pericyte loss followed by reactive pericytosis in the cerebral cortex. Sci. Rep. 2015, 5, 13497. [Google Scholar] [CrossRef]
- Lindahl, P.; Johansson, B.R.; Leveen, P.; Betsholtz, C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 1997, 277, 242–245. [Google Scholar] [CrossRef]
- Schultz, N.; Brännström, K.; Byman, E.; Moussaud, S.; Nielsen, H.M.; Olofsson, A.; Wennström, M. Amyloid-beta 1-40 is associated with alterations in NG2+ pericyte population ex vivo and in vitro. Aging Cell 2018, 17, e12728. [Google Scholar] [CrossRef]
- Thomas, W.E. Brain macrophages: On the role of pericytes and perivascular cells. Brain Res. Brain Res. Rev. 1999, 31, 42–57. [Google Scholar] [CrossRef]
- Faraco, G.; Park, L.; Anrather, J.; Iadecola, C. Brain perivascular macrophages: Characterization and functional roles in health and disease. J. Mol. Med. 2017, 95, 1143–1152. [Google Scholar] [CrossRef]
- Williams, K.; Alvarez, X.; Lackner, A.A. Central nervous system perivascular cells are immunoregulatory cells that connect the CNS with the peripheral immune system. Glia 2001, 36, 156–164. [Google Scholar] [CrossRef]
- Bechmann, I.; Priller, J.; Kovac, A.; Bontert, M.; Wehner, T.; Klett, F.F.; Bohsung, J.; Stuschke, M.; Dirnagl, U.; Nitsch, R. Immune surveillance of mouse brain perivascular spaces by blood-borne macrophages. Eur. J. Neurosci. 2001, 14, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Polfliet, M.M.; Goede, P.H.; van Kesteren-Hendrikx, E.M.; van Rooijen, N.; Dijkstra, C.D.; van den Berg, T.K. A method for the selective depletion of perivascular and meningeal macrophages in the central nervous system. J. Neuroimmunol. 2001, 116, 188–195. [Google Scholar] [CrossRef]
- Hawkes, C.A.; McLaurin, J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc. Natl. Acad. Sci. USA 2009, 106, 1261–1266. [Google Scholar] [CrossRef]
- Hawkes, C.; Brown, M.; Williams, K.; McLaurin, J. Perivascular macrophages and cerebral amyloid angiopathy in CRND8 mice. J. Neurol. Sci. 2009, 283, 289–290. [Google Scholar] [CrossRef]
- Mildner, A.; Schlevogt, B.; Kierdorf, K.; Bottcher, C.; Erny, D.; Kummer, M.P.; Quinn, M.; Bruck, W.; Bechmann, I.; Heneka, M.T.; et al. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 11159–11171. [Google Scholar] [CrossRef]
- Meyer-Luehmann, M.; Spires-Jones, T.L.; Prada, C.; Garcia-Alloza, M.; de Calignon, A.; Rozkalne, A.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Bacskai, B.J.; Hyman, B.T. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature 2008, 451, 720–724. [Google Scholar] [CrossRef]
- Bolmont, T.; Haiss, F.; Eicke, D.; Radde, R.; Mathis, C.A.; Klunk, W.E.; Kohsaka, S.; Jucker, M.; Calhoun, M.E. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 4283–4292. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Spangenberg, E.E.; Lee, R.J.; Najafi, A.R.; Rice, R.A.; Elmore, M.R.; Blurton-Jones, M.; West, B.L.; Green, K.N. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology. Brain 2016, 139, 1265–1281. [Google Scholar] [CrossRef]
- Spangenberg, E.; Severson, P.L.; Hohsfield, L.A.; Crapser, J.; Zhang, J.; Burton, E.A.; Zhang, Y.; Spevak, W.; Lin, J.; Phan, N.Y.; et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 3758. [Google Scholar] [CrossRef]
- Koenigsknecht-Talboo, J.; Meyer-Luehmann, M.; Parsadanian, M.; Garcia-Alloza, M.; Finn, M.B.; Hyman, B.T.; Bacskai, B.J.; Holtzman, D.M. Rapid microglial response around amyloid pathology after systemic anti-Abeta antibody administration in PDAPP mice. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 14156–14164. [Google Scholar] [CrossRef] [PubMed]
- Bard, F.; Cannon, C.; Barbour, R.; Burke, R.L.; Games, D.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 2000, 6, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, D.M.; Rojiani, A.; Rosenthal, A.; Levkowitz, G.; Subbarao, S.; Alamed, J.; Wilson, D.; Wilson, N.; Freeman, M.J.; Gordon, M.N.; et al. Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 6144–6151. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, D.M.; Alamed, J.; Gottschall, P.E.; Grimm, J.; Rosenthal, A.; Pons, J.; Ronan, V.; Symmonds, K.; Gordon, M.N.; Morgan, D. Deglycosylated anti-amyloid-beta antibodies eliminate cognitive deficits and reduce parenchymal amyloid with minimal vascular consequences in aged amyloid precursor protein transgenic mice. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 5340–5346. [Google Scholar] [CrossRef]
- Vasilevko, V.; Xu, F.; Previti, M.L.; Van Nostrand, W.E.; Cribbs, D.H. Experimental investigation of antibody-mediated clearance mechanisms of amyloid-beta in CNS of Tg-SwDI transgenic mice. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 13376–13383. [Google Scholar] [CrossRef]
- Boche, D.; Zotova, E.; Weller, R.O.; Love, S.; Neal, J.W.; Pickering, R.M.; Wilkinson, D.; Holmes, C.; Nicoll, J.A. Consequence of Abeta immunization on the vasculature of human Alzheimer’s disease brain. Brain 2008, 131, 3299–3310. [Google Scholar] [CrossRef]
- Wilcock, D.M.; Rojiani, A.; Rosenthal, A.; Subbarao, S.; Freeman, M.J.; Gordon, M.N.; Morgan, D. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J. Neuroinflam. 2004, 1, 24. [Google Scholar] [CrossRef]
- Patton, R.L.; Kalback, W.M.; Esh, C.L.; Kokjohn, T.A.; Van Vickle, G.D.; Luehrs, D.C.; Kuo, Y.M.; Lopez, J.; Brune, D.; Ferrer, I.; et al. Amyloid-beta peptide remnants in AN-1792-immunized Alzheimer’s disease patients: A biochemical analysis. Am. J. Pathol. 2006, 169, 1048–1063. [Google Scholar] [CrossRef]
- Nicoll, J.A.; Barton, E.; Boche, D.; Neal, J.W.; Ferrer, I.; Thompson, P.; Vlachouli, C.; Wilkinson, D.; Bayer, A.; Games, D.; et al. Abeta species removal after abeta42 immunization. J. Neuropathol. Exp. Neurol. 2006, 65, 1040–1048. [Google Scholar] [CrossRef]
- Park, L.; Zhou, J.; Zhou, P.; Pistick, R.; El Jamal, S.; Younkin, L.; Pierce, J.; Arreguin, A.; Anrather, J.; Younkin, S.G.; et al. Innate immunity receptor CD36 promotes cerebral amyloid angiopathy. Proc. Natl. Acad. Sci. USA 2013, 110, 3089–3094. [Google Scholar] [CrossRef]
- Chun, H.; Lee, C.J. Reactive astrocytes in Alzheimer’s disease: A double-edged sword. Neurosci. Res. 2018, 126, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Farina, C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef] [PubMed]
- al-Ali, S.Y.; al-Hussain, S.M. An ultrastructural study of the phagocytic activity of astrocytes in adult rat brain. J. Anat. 1996, 188 Pt 2, 257–262. [Google Scholar]
- Montgomery, D.L. Astrocytes: Form, functions, and roles in disease. Vet. Pathol. 1994, 31, 145–167. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.M.; Rebeck, G.W.; Vonsattel, J.P.; Gomez-Isla, T.; Hyman, B.T. Apolipoprotein E epsilon 4 and cerebral hemorrhage associated with amyloid angiopathy. Ann. Neurol. 1995, 38, 254–259. [Google Scholar] [CrossRef]
- Mahley, R.W. Central Nervous System Lipoproteins: ApoE and Regulation of Cholesterol Metabolism. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1305–1315. [Google Scholar] [CrossRef]
- Rannikmae, K.; Kalaria, R.N.; Greenberg, S.M.; Chui, H.C.; Schmitt, F.A.; Samarasekera, N.; Al-Shahi Salman, R.; Sudlow, C.L. APOE associations with severe CAA-associated vasculopathic changes: Collaborative meta-analysis. J. Neurol. Neurosurg. Psychiatry 2014, 85, 300–305. [Google Scholar] [CrossRef]
- Koistinaho, M.; Lin, S.; Wu, X.; Esterman, M.; Koger, D.; Hanson, J.; Higgs, R.; Liu, F.; Malkani, S.; Bales, K.R.; et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat. Med. 2004, 10, 719–726. [Google Scholar] [CrossRef]
- Matsunaga, W.; Shirokawa, T.; Isobe, K. Specific uptake of Abeta1-40 in rat brain occurs in astrocyte, but not in microglia. Neurosci. Lett. 2003, 342, 129–131. [Google Scholar] [CrossRef]
- Funato, H.; Yoshimura, M.; Yamazaki, T.; Saido, T.C.; Ito, Y.; Yokofujita, J.; Okeda, R.; Ihara, Y. Astrocytes containing amyloid beta-protein (Abeta)-positive granules are associated with Abeta40-positive diffuse plaques in the aged human brain. Am. J. Pathol. 1998, 152, 983–992. [Google Scholar] [PubMed]
- Fryer, J.D.; Simmons, K.; Parsadanian, M.; Bales, K.R.; Paul, S.M.; Sullivan, P.M.; Holtzman, D.M. Human apolipoprotein E4 alters the amyloid-beta 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 2803–2810. [Google Scholar] [CrossRef] [PubMed]
- Harr, S.D.; Uint, L.; Hollister, R.; Hyman, B.T.; Mendez, A.J. Brain expression of apolipoproteins E, J, and A-I in Alzheimer’s disease. J. Neurochem. 1996, 66, 2429–2435. [Google Scholar] [CrossRef]
- Nelson, A.R.; Sagare, A.P.; Zlokovic, B.V. Role of clusterin in the brain vascular clearance of amyloid-beta. Proc. Natl. Acad. Sci. USA 2017, 114, 8681–8682. [Google Scholar] [CrossRef]
- Wojtas, A.M.; Kang, S.S.; Olley, B.M.; Gatherer, M.; Shinohara, M.; Lozano, P.A.; Liu, C.C.; Kurti, A.; Baker, K.E.; Dickson, D.W.; et al. Loss of clusterin shifts amyloid deposition to the cerebrovasculature via disruption of perivascular drainage pathways. Proc. Natl. Acad. Sci. USA 2017, 114, e6962–e6971. [Google Scholar] [CrossRef]
- Koudinov, A.R.; Berezov, T.T.; Kumar, A.; Koudinova, N.V. Alzheimer’s amyloid beta interaction with normal human plasma high density lipoprotein: Association with apolipoprotein and lipids. Clin. Chim. Acta Int. J. Clin. Chem. 1998, 270, 75–84. [Google Scholar] [CrossRef]
- Button, E.B.; Boyce, G.K.; Wilkinson, A.; Stukas, S.; Hayat, A.; Fan, J.; Wadsworth, B.J.; Robert, J.; Martens, K.M.; Wellington, C.L. ApoA-I deficiency increases cortical amyloid deposition, cerebral amyloid angiopathy, cortical and hippocampal astrogliosis, and amyloid-associated astrocyte reactivity in APP/PS1 mice. Alzheimers Res. 2019, 11, 44. [Google Scholar] [CrossRef]
- Handattu, S.P.; Garber, D.W.; Monroe, C.E.; van Groen, T.; Kadish, I.; Nayyar, G.; Cao, D.; Palgunachari, M.N.; Li, L.; Anantharamaiah, G.M. Oral apolipoprotein A-I mimetic peptide improves cognitive function and reduces amyloid burden in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2009, 34, 525–534. [Google Scholar] [CrossRef]
- Fernandez-de Retana, S.; Montanola, A.; Marazuela, P.; De La Cuesta, M.; Batlle, A.; Fatar, M.; Grudzenski, S.; Montaner, J.; Hernandez-Guillamon, M. Intravenous treatment with human recombinant ApoA-I Milano reduces beta amyloid cerebral deposition in the APP23-transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 2017, 60, 116–128. [Google Scholar] [CrossRef]
- Robert, J.; Stukas, S.; Button, E.; Cheng, W.H.; Lee, M.; Fan, J.; Wilkinson, A.; Kulic, I.; Wright, S.D.; Wellington, C.L. Reconstituted high-density lipoproteins acutely reduce soluble brain Abeta levels in symptomatic APP/PS1 mice. Biochim. Biophys. Acta 2016, 1862, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Karch, C.M.; Goate, A.M. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 2015, 77, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Giri, M.; Zhang, M.; Lu, Y. Genes associated with Alzheimer’s disease: An overview and current status. Clin. Interv. Aging 2016, 11, 665–681. [Google Scholar] [CrossRef] [PubMed]
- Beecham, G.W.; Hamilton, K.; Naj, A.C.; Martin, E.R.; Huentelman, M.; Myers, A.J.; Corneveaux, J.J.; Hardy, J.; Vonsattel, J.P.; Younkin, S.G.; et al. Genome-wide association meta-analysis of neuropathologic features of Alzheimer’s disease and related dementias. PLoS Genet. 2014, 10, e1004606. [Google Scholar] [CrossRef]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 2016, 90, 724–739. [Google Scholar] [CrossRef]
- Ulland, T.K.; Song, W.M.; Huang, S.C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649–663.e613. [Google Scholar] [CrossRef]
- Jay, T.R.; Miller, C.M.; Cheng, P.J.; Graham, L.C.; Bemiller, S.; Broihier, M.L.; Xu, G.; Margevicius, D.; Karlo, J.C.; Sousa, G.L.; et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med. 2015, 212, 287–295. [Google Scholar] [CrossRef]
- Efthymiou, A.G.; Goate, A.M. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 2017, 12, 43. [Google Scholar] [CrossRef]
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Mäkelä, M.; Kaivola, K.; Valori, M.; Paetau, A.; Polvikoski, T.; Singleton, A.B.; Traynor, B.J.; Stone, D.J.; Peuralinna, T.; Tienari, P.J.; et al. Alzheimer risk loci and associated neuropathology in a population-based study (Vantaa 85+). Neurol. Genet. 2018, 4, e211. [Google Scholar] [CrossRef] [PubMed]
- Wes, P.D.; Sayed, F.A.; Bard, F.; Gan, L. Targeting microglia for the treatment of Alzheimer’s Disease. Glia 2016, 64, 1710–1732. [Google Scholar] [CrossRef] [PubMed]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Castillo-Carranza, D.L.; Guerrero-Munoz, M.J.; Sengupta, U.; Hernandez, C.; Barrett, A.D.; Dineley, K.; Kayed, R. Tau immunotherapy modulates both pathological tau and upstream amyloid pathology in an Alzheimer’s disease mouse model. J. Neurosci. 2015, 35, 4857–4868. [Google Scholar] [CrossRef]
- Vidal, R.; Ghetti, B. Hereditary Cerebral Amyloid Angiopathies. In Neuropathology of Neurodegenerative Diseases: A Practical Guide; Kovacs, G., Ed.; Cambridge University Press: Cambridge, UK, 2015; Volume 13, pp. 249–258. [Google Scholar]
- Herzig, M.C.; Winkler, D.T.; Burgermeister, P.; Pfeifer, M.; Kohler, E.; Schmidt, S.D.; Danner, S.; Abramowski, D.; Sturchler-Pierrat, C.; Burki, K.; et al. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat. Neurosci. 2004, 7, 954–960. [Google Scholar] [CrossRef]


{kind=link}
| Amyloid | Gene | Precursor Protein | Mutation | Disease |
|---|---|---|---|---|
| Aβ | APP | Amyloid precursor protein | none | Sporadic CAA [21,22] |
| Aβ | APP | Amyloid precursor protein | E693Q | Hereditary Cerebral Hemorrhage with Amyloidosis Dutch type [29,30,31,32] |
| Aβ | APP | Amyloid precursor protein | E693K | Hereditary Cerebral Hemorrhage with Amyloidosis Italian type [33] |
| Aβ | APP | Amyloid precursor protein | E693G | Hereditary Cerebral Hemorrhage with Amyloidosis Arctic type [34] |
| Aβ | APP | Amyloid precursor protein | E693Δ | Hereditary Cerebral Hemorrhage with Amyloidosis Osaka type [35,36] |
| Aβ | APP | Amyloid precursor protein | A692G | Hereditary Cerebral Hemorrhage with Amyloidosis Flemish type [37] |
| Aβ | APP | Amyloid precursor protein | D694N | Hereditary Cerebral Hemorrhage with Amyloidosis Iowa type [38] |
| Aβ | APP | Amyloid precursor protein | L705V | Hereditary Cerebral Hemorrhage with Amyloidosis Piedmont type [39] |
| Aβ | APP | Amyloid precursor protein | A713T | Hereditary Cerebral Hemorrhage with Amyloidosis Italian type [40,41] |
| Aβ | APP | Amyloid precursor protein | T714I | Hereditary Cerebral Hemorrhage with Amyloidosis Austrian type [42] |
| Aβ | APP | Amyloid precursor protein | T714A | Hereditary Cerebral Hemorrhage with Amyloidosis Iranian type [43] |
| ABri | BRI2 | British Amyloid protein | 799A>T | Familial British Dementia [18,44,45,46,47] |
| ADan | BRI2 | Danish Amyloid protein | 787_796dupTTTAATTTGT | Familial Danish Dementia [18,45,46,48,49,50] |
| ACys | CST3 | Cystatin C | L68Q | Hereditary Cerebral Hemorrhage with Amyloidosis Islandic type [51,52,53] |
| ATTR | TTR | Transthyretin | D18G | Meningovascular amyloidosis Hungarian variant [54,55,56,57] |
| ATTR | TTR | Transthyretin | V30G | Meningovascular amyloidosis Ohio variant [58,59,60] |
| ATTR | TTR | Transthyretin | Y69H | Meningovascular amyloidosis rare variant [61,62,63,64] |
| ATTR | TTR | Transthyretin | A25T | Meningovascular amyloidosis rare variant [65,66] |
| ATTR | TTR | Transthyretin | V30M | Meningovascular amyloidosis rare variant [67,68] |
| ATTR | TTR | Transthyretin | T49P | Meningovascular amyloidosis rare variant [69] |
| ATTR | TTR | Transthyretin | L58R | Meningovascular amyloidosis rare variant [70] |
| ATTR | TTR | Transthyretin | F64S | Meningovascular amyloidosis rare variant [71] |
| ATTR | TTR | Transthyretin | Y114C | Meningovascular amyloidosis rare variant [72] |
| ATTR | TTR | Transthyretin | L12P | Meningovascular amyloidosis rare variant [73] |
| ATTR | TTR | Transthyretin | G53R | Meningovascular amyloidosis rare variant [74] |
| AGel | GSN | Gelsolin | D187N or D187Y | Hereditary gelsolin amyloidosis or familial amyloidosis Finnish type [75,76,77] |
| PrPSc | PRNP | Prion protein | Y145stop | Gerstmann–Sträussler–Scheinker syndrome variant [20] |
| PrPSc | PRNP | Prion protein | Y163stop | Gerstmann–Sträussler–Scheinker syndrome variant [78] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cisternas, P.; Taylor, X.; A. Lasagna-Reeves, C. The Amyloid-Tau-Neuroinflammation Axis in the Context of Cerebral Amyloid Angiopathy. Int. J. Mol. Sci. 2019, 20, 6319. https://doi.org/10.3390/ijms20246319
Cisternas P, Taylor X, A. Lasagna-Reeves C. The Amyloid-Tau-Neuroinflammation Axis in the Context of Cerebral Amyloid Angiopathy. International Journal of Molecular Sciences. 2019; 20(24):6319. https://doi.org/10.3390/ijms20246319
Chicago/Turabian StyleCisternas, Pablo, Xavier Taylor, and Cristian A. Lasagna-Reeves. 2019. "The Amyloid-Tau-Neuroinflammation Axis in the Context of Cerebral Amyloid Angiopathy" International Journal of Molecular Sciences 20, no. 24: 6319. https://doi.org/10.3390/ijms20246319
APA StyleCisternas, P., Taylor, X., & A. Lasagna-Reeves, C. (2019). The Amyloid-Tau-Neuroinflammation Axis in the Context of Cerebral Amyloid Angiopathy. International Journal of Molecular Sciences, 20(24), 6319. https://doi.org/10.3390/ijms20246319