The Mechanism of Bisphenol A Atherogenicity Involves Apolipoprotein A-I Downregulation through NF-κB Activation

 ,
, 
Abstract
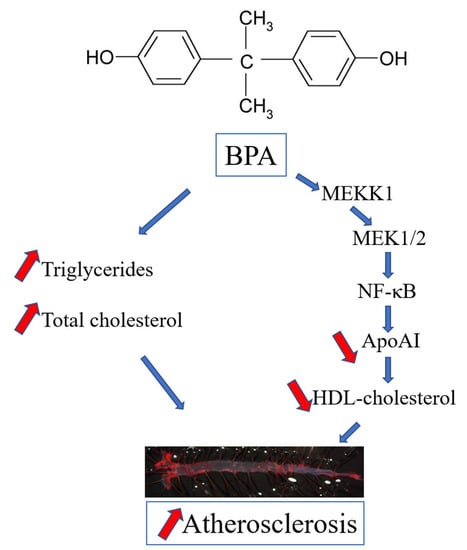
1. Introduction
2. Results
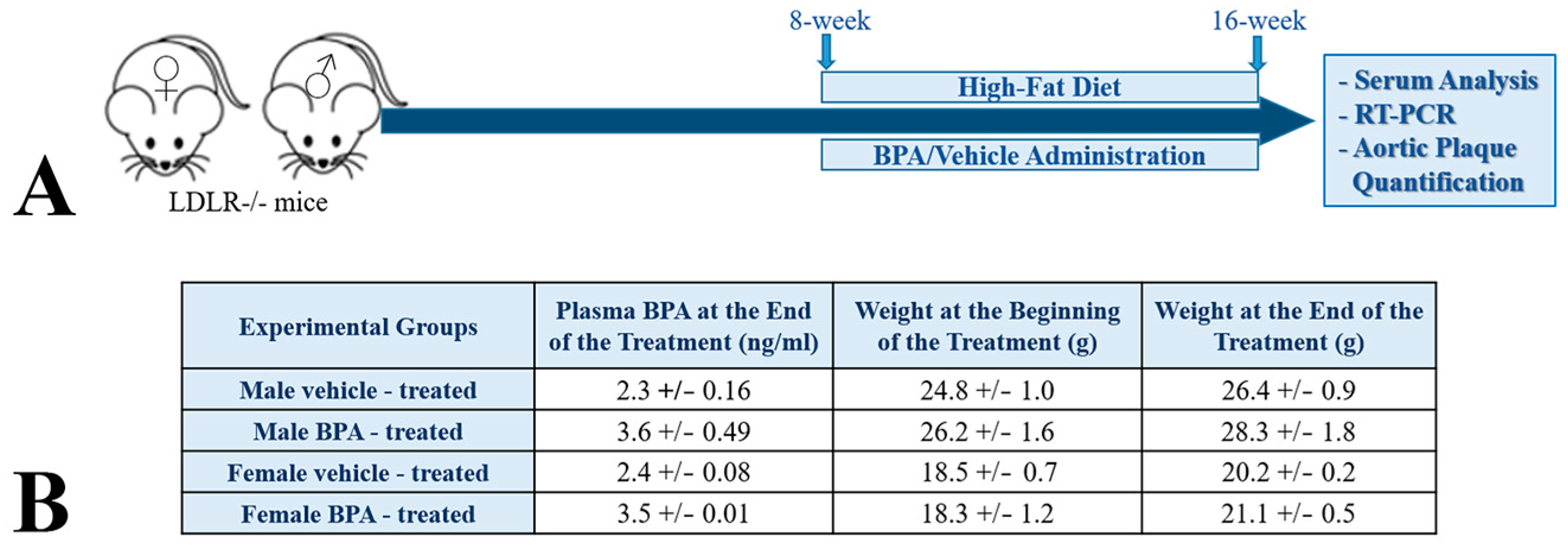
2.1. BPA Increases Plasma Lipid Levels and Atherosclerotic Lesions in LDLR−/− Mice
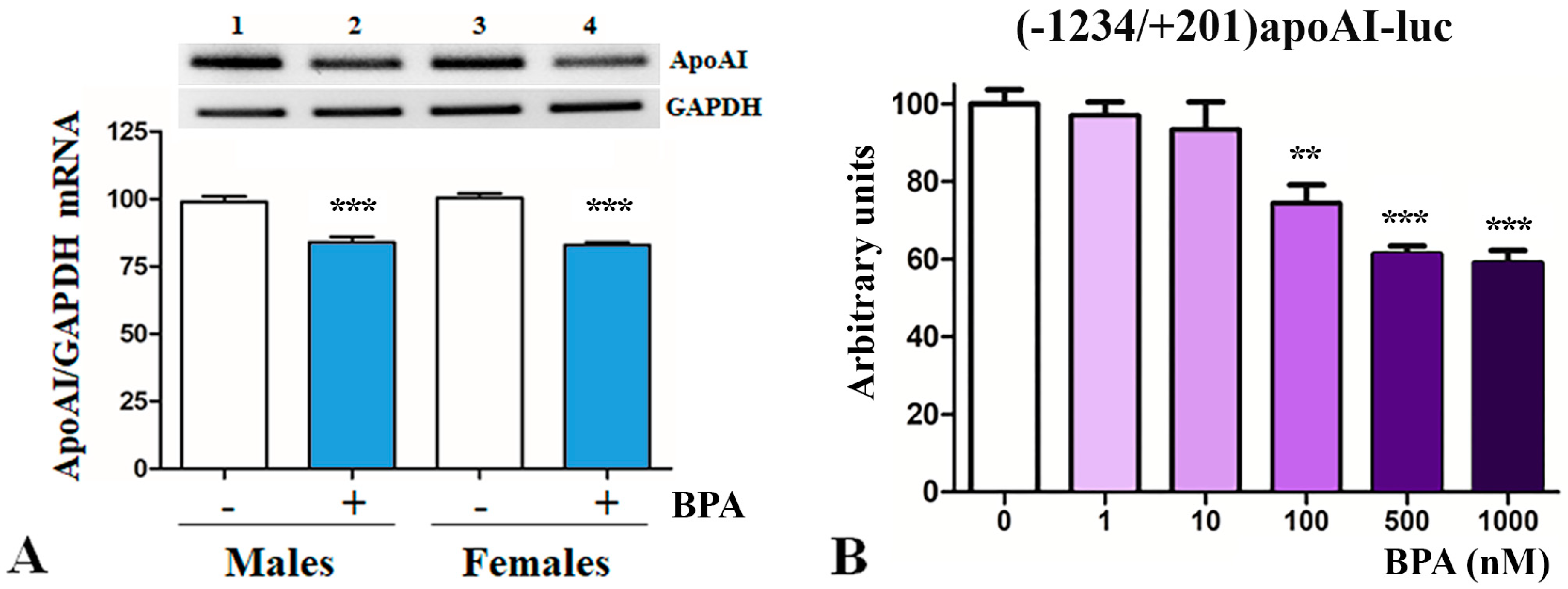
2.2. BPA Downregulates apoA-I in Hepatocytes
2.3. The Mechanisms of apoA-I Downregulation by BPA
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| apoA-I | apolipoprotein A-I |
| APOAI | apolipoprotein A-I gene |
| BPA | Bisphenol A |
| NF-κB | Nuclear Factor Kappa B |
| JNK | c-Jun N-terminal kinase |
| MEKK1 | Mitogen-activated protein kinase kinase kinase 1 |
| LPS | Lipopolysaccharide |
| HDL | High Density Lipoprotein |
| LDL | Low Density Lipoprotein |
| VLDL | Very Low-Density Lipoprotein |
| LCAT | Lecithin Cholesterol Acyltransferase |
| ChIP | Chromatin immunoprecipitation |
| DNAP | DNA pull-down assays |
| Luc | Luciferase |
| HNF4 | Hepatocyte nuclear factor 4 |
| RT-PCR | Reverse Transcription-Polymerase Chain Reaction |
| IKKB | IκB kinase |
| IKb DN | IKb dominant-negative |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| PFA | Paraformaldehyde |
References
- Frank, P.G.; Marcel, Y.L. Apolipoprotein A-I: Structure-function relationships. J. Lipid. Res. 2000, 41, 853–872. [Google Scholar] [PubMed]
- Kontush, A.; Lindahl, M.; Lhomme, M.; Calabresi, L.; Chapman, M.J.; Davidson, W.S. Structure of HDL: Particle subclasses and molecular components. In High Density Lipoproteins: From Biological Understanding to Clinical Exploitation; von Eckardstein, A., Kardassis, D., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 3–51. [Google Scholar] [CrossRef]
- Cooke, A.L.; Morris, J.; Melchior, J.T.; Street, S.E.; Jerome, W.G.; Huang, R.; Herr, A.B.; Smith, L.E.; Segrest, J.P.; Remaley, A.T.; et al. A thumbwheel mechanism for APOA1 activation of LCAT activity in HDL. J. Lipid Res. 2018, 59, 1244–1255. [Google Scholar] [CrossRef] [PubMed]
- Curtiss, L.K.; Valenta, D.T.; Hime, N.J.; Rye, K.A. What is so special about apolipoprotein AI in reverse cholesterol transport? Arter. Thromb. Vasc. Biol. 2006, 26, 12–19. [Google Scholar] [CrossRef]
- Voyiaziakis, E.; Goldberg, I.J.; Plump, A.S.; Rubin, E.M.; Breslow, J.L.; Huang, L.S. ApoA-I deficiency causes both hypertriglyceridemia and increased atherosclerosis in human apoB transgenic mice. J. Lipid Res. 1998, 39, 313–321. [Google Scholar]
- Moore, R.E.; Kawashiri, M.A.; Kitajima, K.; Secreto, A.; Millar, J.S.; Pratico, D.; Rader, D.J. Apolipoprotein A-I deficiency results in markedly increased atherosclerosis in mice lacking the LDL receptor. Arter. Thromb. Vasc. Biol. 2003, 23, 1914–1920. [Google Scholar] [CrossRef]
- Paszty, C.; Maeda, N.; Verstuyft, J.; Rubin, E.M. Apolipoprotein AI transgene corrects apolipoprotein E deficiency-induced atherosclerosis in mice. J. Clin. Investig. 1994, 94, 899–903. [Google Scholar] [CrossRef]
- Li, H.; Gu, S.; Cao, X.; Wang, Z.; She, M. Suppression of induced atherosclerosis in h-apo AI transgenic mice by overexpression of human apo AI in the aortic wall. Chin. Med. J. (Engl.) 2000, 113, 657–661. [Google Scholar]
- Plump, A.S.; Scott, C.J.; Breslow, J.L. Human apolipoprotein A-I gene expression increases high density lipoprotein and suppresses atherosclerosis in the apolipoprotein E-deficient mouse. Proc. Natl. Acad. Sci. USA 1994, 91, 9607–9611. [Google Scholar] [CrossRef]
- Pastore, L.; Belalcazar, L.M.; Oka, K.; Cela, R.; Lee, B.; Chan, L.; Beaudet, A.L. Helper-dependent adenoviral vector-mediated long-term expression of human apolipoprotein A-I reduces atherosclerosis in apo E-deficient mice. Gene 2004, 327, 153–160. [Google Scholar] [CrossRef]
- Benoit, P.; Emmanuel, F.; Caillaud, J.M.; Bassinet, L.; Castro, G.; Gallix, P.; Fruchart, J.C.; Branellec, D.; Denefle, P.; Duverger, N. Somatic gene transfer of human ApoA-I inhibits atherosclerosis progression in mouse models. Circulation 1999, 99, 105–110. [Google Scholar] [CrossRef][Green Version]
- Rubin, E.M.; Krauss, R.M.; Spangler, E.A.; Verstuyft, J.G.; Clift, S.M. Inhibition of early atherogenesis in transgenic mice by human apolipoprotein AI. Nature 1991, 353, 265–267. [Google Scholar] [CrossRef]
- Belalcazar, L.M.; Merched, A.; Carr, B.; Oka, K.; Chen, K.H.; Pastore, L.; Beaudet, A.; Chan, L. Long-term stable expression of human apolipoprotein A-I mediated by helper-dependent adenovirus gene transfer inhibits atherosclerosis progression and remodels atherosclerotic plaques in a mouse model of familial hypercholesterolemia. Circulation 2003, 107, 2726–2732. [Google Scholar] [CrossRef]
- Valenta, D.T.; Bulgrien, J.J.; Banka, C.L.; Curtiss, L.K. Overexpression of human ApoAI transgene provides long-term atheroprotection in LDL receptor-deficient mice. Atherosclerosis 2006, 189, 255–263. [Google Scholar] [CrossRef]
- Rhee, E.J.; Byrne, C.D.; Sung, K.C. The HDL cholesterol/apolipoprotein A-I ratio: An indicator of cardiovascular disease. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 148–153. [Google Scholar] [CrossRef]
- Stoekenbroek, R.M.; Stroes, E.S.; Hovingh, G.K. ApoA-I mimetics. Handb. Exp. Pharm. 2015, 224, 631–648. [Google Scholar] [CrossRef]
- Valanti, E.K.; Dalakoura-Karagkouni, K.; Sanoudou, D. Current and emerging reconstituted HDL-apoA-I and HDL-apoE approaches to treat atherosclerosis. J. Pers. Med. 2018, 8, 34. [Google Scholar] [CrossRef]
- Karathanasis, S.K.; Ferris, E.; Haddad, I.A. DNA inversion within the apolipoproteins AI/CIII/AIV-encoding gene cluster of certain patients with premature atherosclerosis. Proc. Natl. Acad. Sci. USA 1987, 84, 7198–7202. [Google Scholar] [CrossRef]
- Kardassis, D.; Gafencu, A.; Zannis, V.I.; Davalos, A. Regulation of HDL genes: Transcriptional, posttranscriptional, and posttranslational. Handb. Exp. Pharm. 2015, 224, 113–179. [Google Scholar] [CrossRef]
- Haas, M.J.; Reinacher, D.; Li, J.P.; Wong, N.C.; Mooradian, A.D. Regulation of apoA1 gene expression with acidosis: Requirement for a transcriptional repressor. J. Mol. Endocrinol. 2001, 27, 43–57. [Google Scholar] [CrossRef]
- Hargrove, G.M.; Junco, A.; Wong, N.C. Hormonal regulation of apolipoprotein AI. J. Mol. Endocrinol. 1999, 22, 103–111. [Google Scholar] [CrossRef]
- Murao, K.; Wada, Y.; Nakamura, T.; Taylor, A.H.; Mooradian, A.D.; Wong, N.C. Effects of glucose and insulin on rat apolipoprotein A-I gene expression. J. Biol. Chem. 1998, 273, 18959–18965. [Google Scholar] [CrossRef]
- Staels, B.; Auwerx, J. Regulation of apo A-I gene expression by fibrates. Atherosclerosis 1998, 137, S19–S23. [Google Scholar] [CrossRef]
- Kagawa, A.; Azuma, H.; Akaike, M.; Kanagawa, Y.; Matsumoto, T. Aspirin reduces apolipoprotein(a) (apo(a)) production in human hepatocytes by suppression of apo(a) gene transcription. J. Biol. Chem. 1999, 274, 34111–34115. [Google Scholar] [CrossRef][Green Version]
- Vandenberg, L.N.; Maffini, M.V.; Sonnenschein, C.; Rubin, B.S.; Soto, A.M. Bisphenol-A and the great divide: A review of controversies in the field of endocrine disruption. Endocrinol. Rev. 2009, 30, 75–95. [Google Scholar] [CrossRef]
- Ribeiro, E.; Ladeira, C.; Viegas, S. Occupational exposure to bisphenol A (BPA): A reality that still needs to be unveiled. Toxics 2017, 5, 22. [Google Scholar] [CrossRef]
- Dekant, W.; Volkel, W. Human exposure to bisphenol A by biomonitoring: Methods, results and assessment of environmental exposures. Toxicol. Appl. Pharm. 2008, 228, 114–134. [Google Scholar] [CrossRef]
- Vandenberg, L.N.; Hauser, R.; Marcus, M.; Olea, N.; Welshons, W.V. Human exposure to bisphenol A (BPA). Reprod. Toxicol. 2007, 24, 139–177. [Google Scholar] [CrossRef]
- Calafat, A.M.; Ye, X.; Wong, L.Y.; Reidy, J.A.; Needham, L.L. Exposure of the U.S. population to bisphenol A and 4-tertiary-octylphenol: 2003–2004. Environ. Health Perspect. 2008, 116, 39–44. [Google Scholar] [CrossRef]
- Jalal, N.; Surendranath, A.R.; Pathak, J.L.; Yu, S.; Chung, C.Y. Bisphenol A (BPA) the mighty and the mutagenic. Toxicol. Rep. 2018, 5, 76–84. [Google Scholar] [CrossRef]
- Han, C.; Hong, Y.C. Bisphenol A, Hypertension, and cardiovascular diseases: Epidemiological, laboratory, and clinical trial evidence. Curr. Hypertens. Rep. 2016, 18, 11. [Google Scholar] [CrossRef]
- Bae, S.; Hong, Y.C. Exposure to bisphenol A from drinking canned beverages increases blood pressure: Randomized crossover trial. Hypertension 2015, 65, 313–319. [Google Scholar] [CrossRef]
- Lind, P.M.; Lind, L. Circulating levels of bisphenol A and phthalates are related to carotid atherosclerosis in the elderly. Atherosclerosis 2011, 218, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Melzer, D.; Rice, N.E.; Lewis, C.; Henley, W.E.; Galloway, T.S. Association of urinary bisphenol a concentration with heart disease: Evidence from NHANES 2003/06. PLoS ONE 2010, 5, e8673. [Google Scholar] [CrossRef]
- Melzer, D.; Osborne, N.J.; Henley, W.E.; Cipelli, R.; Young, A.; Money, C.; McCormack, P.; Luben, R.; Khaw, K.T.; Wareham, N.J.; et al. Urinary bisphenol A concentration and risk of future coronary artery disease in apparently healthy men and women. Circulation 2012, 125, 1482–1490. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.; Park, S.H.; Wang, F.; Zhou, C. Perinatal bisphenol A exposure increases atherosclerosis in adult male PXR-humanized mice. Endocrinology 2018, 159, 1595–1608. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.; Park, S.H.; Helsley, R.N.; Sunkara, M.; Gonzalez, F.J.; Morris, A.J.; Zhou, C. Bisphenol A increases atherosclerosis in pregnane X receptor-humanized ApoE deficient mice. J. Am. Heart Assoc. 2014, 3, e000492. [Google Scholar] [CrossRef]
- Kim, M.J.; Moon, M.K.; Kang, G.H.; Lee, K.J.; Choi, S.H.; Lim, S.; Oh, B.C.; Park, D.J.; Park, K.S.; Jang, H.C.; et al. Chronic exposure to bisphenol A can accelerate atherosclerosis in high-fat-fed apolipoprotein E knockout mice. Cardiovasc. Toxicol. 2014, 14, 120–128. [Google Scholar] [CrossRef]
- Fang, C.; Ning, B.; Waqar, A.B.; Niimi, M.; Li, S.; Satoh, K.; Shiomi, M.; Ye, T.; Dong, S.; Fan, J. Bisphenol A exposure enhances atherosclerosis in WHHL rabbits. PLoS ONE 2014, 9, e110977. [Google Scholar] [CrossRef]
- Fang, C.; Ning, B.; Waqar, A.B.; Niimi, M.; Li, S.; Satoh, K.; Shiomi, M.; Ye, T.; Dong, S.; Fan, J. Bisphenol A exposure induces metabolic disorders and enhances atherosclerosis in hyperlipidemic rabbits. J. Appl. Toxicol. 2015, 35, 1058–1070. [Google Scholar] [CrossRef]
- Hassan, Z.K.; Elobeid, M.A.; Virk, P.; Omer, S.A.; ElAmin, M.; Daghestani, M.H.; AlOlayan, E.M. Bisphenol A induces hepatotoxicity through oxidative stress in rat model. Oxid. Med. Cell Longev. 2012, 2012, 194829. [Google Scholar] [CrossRef]
- Amraoui, W.; Adjabi, N.; Bououza, F.; Boumendjel, M.; Taibi, F.; Boumendjel, A.; Abdennour, C.; Messarah, M. Modulatory role of selenium and vitamin E, natural antioxidants, against bisphenol A-induced oxidative stress in Wistar albinos rats. Toxicol. Res. 2018, 34, 231–239. [Google Scholar] [CrossRef]
- Bindhumol, V.; Chitra, K.C.; Mathur, P.P. Bisphenol A induces reactive oxygen species generation in the liver of male rats. Toxicology 2003, 188, 117–124. [Google Scholar] [CrossRef]
- Jiang, Y.; Xia, W.; Zhu, Y.; Li, X.; Wang, D.; Liu, J.; Chang, H.; Li, G.; Xu, B.; Chen, X.; et al. Mitochondrial dysfunction in early life resulted from perinatal bisphenol A exposure contributes to hepatic steatosis in rat offspring. Toxicol. Lett. 2014, 228, 85–92. [Google Scholar] [CrossRef]
- Martella, A.; Silvestri, C.; Maradonna, F.; Gioacchini, G.; Allara, M.; Radaelli, G.; Overby, D.R.; Di Marzo, V.; Carnevali, O. Bisphenol A induces fatty liver by an endocannabinoid-mediated positive feedback loop. Endocrinology 2016, 157, 1751–1763. [Google Scholar] [CrossRef]
- Moon, M.K.; Kim, M.J.; Jung, I.K.; Koo, Y.D.; Ann, H.Y.; Lee, K.J.; Kim, S.H.; Yoon, Y.C.; Cho, B.J.; Park, K.S.; et al. Bisphenol A impairs mitochondrial function in the liver at doses below the no observed adverse effect level. J. Korean Med. Sci. 2012, 27, 644–652. [Google Scholar] [CrossRef]
- Rezg, R.; El-Fazaa, S.; Gharbi, N.; Mornagui, B. Bisphenol A and human chronic diseases: Current evidences, possible mechanisms, and future perspectives. Environ. Int. 2014, 64, 83–90. [Google Scholar] [CrossRef]
- Tsunoda, T.; Takagi, T. Estimating transcription factor bindability on DNA. Bioinformatics 1999, 15, 622–630. [Google Scholar] [CrossRef]
- Davidson, W.S.; Thompson, T.B. The structure of apolipoprotein A-I in high density lipoproteins. J. Biol. Chem. 2007, 282, 22249–22253. [Google Scholar] [CrossRef]
- Lund-Katz, S.; Phillips, M.C. High density lipoprotein structure-function and role in reverse cholesterol transport. Subcell Biochem. 2010, 51, 183–227. [Google Scholar] [CrossRef]
- Mooradian, A.D.; Haas, M.J.; Wong, N.C. Transcriptional control of apolipoprotein A-I gene expression in diabetes. Diabetes 2004, 53, 513–520. [Google Scholar] [CrossRef]
- Song, H.; Saito, K.; Fujigaki, S.; Noma, A.; Ishiguro, H.; Nagatsu, T.; Seishima, M. IL-1 beta and TNF-alpha suppress apolipoprotein (apo) E secretion and apo A-I expression in HepG2 cells. Cytokine 1998, 10, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Orlov, S.V.; Mogilenko, D.A.; Shavva, V.S.; Dizhe, E.B.; Ignatovich, I.A.; Perevozchikov, A.P. Effect of TNFalpha on activities of different promoters of human apolipoprotein A-I gene. Biochem. Biophys. Res. Commun. 2010, 398, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Pritchett, J.J.; Kuester, R.K.; Sipes, I.G. Metabolism of bisphenol a in primary cultured hepatocytes from mice, rats, and humans. Drug Metab. Dispos. 2002, 30, 1180–1185. [Google Scholar] [CrossRef] [PubMed]
- Rochester, J.R. Bisphenol A and human health: A review of the literature. Reprod. Toxicol. 2013, 42, 132–155. [Google Scholar] [CrossRef]
- Ke, Z.H.; Pan, J.X.; Jin, L.Y.; Xu, H.Y.; Yu, T.T.; Ullah, K.; Rahman, T.U.; Ren, J.; Cheng, Y.; Dong, X.Y.; et al. Bisphenol A exposure may induce hepatic lipid accumulation via reprogramming the DNA methylation patterns of genes involved in lipid metabolism. Sci. Rep. 2016, 6, 31331. [Google Scholar] [CrossRef]
- Marmugi, A.; Lasserre, F.; Beuzelin, D.; Ducheix, S.; Huc, L.; Polizzi, A.; Chetivaux, M.; Pineau, T.; Martin, P.; Guillou, H.; et al. Adverse effects of long-term exposure to bisphenol A during adulthood leading to hyperglycaemia and hypercholesterolemia in mice. Toxicology 2014, 325, 133–143. [Google Scholar] [CrossRef]
- Ronn, M.; Kullberg, J.; Karlsson, H.; Berglund, J.; Malmberg, F.; Orberg, J.; Lind, L.; Ahlstrom, H.; Lind, P.M. Bisphenol A exposure increases liver fat in juvenile fructose-fed Fischer 344 rats. Toxicology 2013, 303, 125–132. [Google Scholar] [CrossRef]
- Yang, M.; Lee, H.S.; Pyo, M.Y. Proteomic biomarkers for prenatal bisphenol A-exposure in mouse immune organs. Environ. Mol. Mutagen. 2008, 49, 368–373. [Google Scholar] [CrossRef]
- Huang, F.M.; Chang, Y.C.; Lee, S.S.; Yang, M.L.; Kuan, Y.H. Expression of pro-inflammatory cytokines and mediators induced by Bisphenol A via ERK-NFκB and JAK1/2-STAT3 pathways in macrophages. Environ. Toxicol. 2019, 34, 486–494. [Google Scholar] [CrossRef]
- Valentino, R.; D’Esposito, V.; Passaretti, F.; Liotti, A.; Cabaro, S.; Longo, M.; Perruolo, G.; Oriente, F.; Beguinot, F.; Formisano, P. Bisphenol-A impairs insulin action and up-regulates inflammatory pathways in human subcutaneous adipocytes and 3T3-L1 cells. PLoS ONE 2013, 8, e82099. [Google Scholar] [CrossRef]
- Morishima, A.; Ohkubo, N.; Maeda, N.; Miki, T.; Mitsuda, N. NFkappaBregulates plasma apolipoprotein A-I and high density lipoprotein cholesterol through inhibition of peroxisome proliferator-activated receptor alpha. J. Biol. Chem. 2003, 278, 38188–38193. [Google Scholar] [CrossRef]
- Haas, M.J.; Jurado-Flores, M.; Hammoud, R.; Plazarte, G.; Onstead-Haas, L.; Wong, N.C.W.; Mooradian, A.D. Regulation of apolipoprotein A-I gene expression by the histamine H1 receptor: Requirement for NF-κB. Life Sci. 2018, 208, 102–110. [Google Scholar] [CrossRef]
- Zhu, J.; Jiang, L.; Liu, Y.; Qian, W.; Liu, J.; Zhou, J.; Gao, R.; Xiao, H.; Wang, J. MAPK and NF-κB pathways are involved in bisphenol A-induced TNF-α and IL-6 production in BV2 microglial cells. Inflammation 2015, 38, 637–648. [Google Scholar] [CrossRef]
- Trusca, V.G.; Fuior, E.V.; Kardassis, D.; Simionescu, M.; Gafencu, A.V. The opposite effect of c-Jun transcription factor on apolipoprotein E gene regulation in hepatocytes and macrophages. Int. J. Mol. Sci. 2019, 20, 1471. [Google Scholar] [CrossRef]
- Trusca, V.G.; Fuior, E.V.; Fenyo, I.M.; Kardassis, D.; Simionescu, M.; Gafencu, A.V. Differential action of glucocorticoids on apolipoprotein E gene expression in macrophages and hepatocytes. PLoS ONE 2017, 12, e0174078. [Google Scholar] [CrossRef]
- Roman, C.; Fuior, E.V.; Trusca, V.G.; Kardassis, D.; Simionescu, M.; Gafencu, A.V. Thyroid hormones upregulate apolipoprotein E gene expression in astrocytes. Biochem. Biophys. Res. Commun. 2015, 468, 190–195. [Google Scholar] [CrossRef]
- Stavri, S.; Simionescu, M.; Kardassis, D.; Gafencu, A.V. Kruppel-like factor 4 synergizes with CREB to increase the activity of apolipoprotein E gene promoter in macrophages. Biochem. Biophys. Res. Commun. 2015, 468, 66–72. [Google Scholar] [CrossRef]
- Gafencu, A.V.; Robciuc, M.R.; Fuior, E.; Zannis, V.I.; Kardassis, D.; Simionescu, M. Inflammatory signaling pathways regulating ApoE gene expression in macrophages. J. Biol. Chem. 2007, 282, 21776–21785. [Google Scholar] [CrossRef]
- Trusca, V.G.; Fuior, E.V.; Florea, I.C.; Kardassis, D.; Simionescu, M.; Gafencu, A.V. Macrophage-specific up-regulation of apolipoprotein E gene expression by STAT1 is achieved via long range genomic interactions. J. Biol. Chem. 2011, 286, 13891–13904. [Google Scholar] [CrossRef]
- Stavri, S.; Trusca, V.G.; Simionescu, M.; Gafencu, A.V. Metformin reduces the endotoxin-induced down-regulation of apolipoprotein E gene expression in macrophages. Biochem. Biophys. Res. Commun. 2015, 461, 435–440. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers for Human apoA-I Promoter Cloning: | |
| Forward apoA-I promoter−1234 | 5′-AAAGTCCCTCTGTGCTCTTCAGTC |
| Forward apoA-I promoter−406 | 5′-GGAATGCTGGTGGTGGGGGAG |
| Reverse apoA-I promoter+201 | 5′-AAAACTCGAGGAAGGGCCTGGCTGAGTGGGGTGC |
| Primers for Real-Time PCR | |
| Mouse apoA-I Forward | 5′-CAAGACAGCGGCAGAGAC |
| Mouse apoA-I Reverse | 5′-CACCTTCTGTTTCACTTCC |
| GAPDH Forward | 5′-ACCACAGTCCATGCCATCAC |
| GAPDH Reverse | 5′-TCCACCACCCTGTTGCTGTA |
| Primers for DNA Pull-Down Assays | |
| RV3 Forward-Biotin | 5′-Biotin-CTAGCAAAATAGGCTGTCCC |
| ApoA-I Reverse-873 | 5′-GGGCAAAGGAAGTGGCAGGAG |
| ApoA-I Reverse-424 | 5′-GACTACTTCACTCCCCTCCCCC |
| ApoA-I Reverse-156 | 5′-GGGCAAATAGAGTGGGCAAACAGC |
| Primers for Chromatin Immunoprecipitation | |
| Forward apoA-I promoter | 5′-AAAGTCCCTCTGTGCTCTTCAGTC |
| Reverse apoA-I promoter | 5′-GGGCAAAGGAAGTGGCAGGAG |
| Primers for Sequencing | |
| RV3 Forward | 5′-CTAGCAAAATAGGCTGTCCC |
| RVprimer4 Reverse | 5′-GACGATAGTCATGCCCCGCG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trusca, V.G.; Dumitrescu, M.; Fenyo, I.M.; Tudorache, I.F.; Simionescu, M.; Gafencu, A.V. The Mechanism of Bisphenol A Atherogenicity Involves Apolipoprotein A-I Downregulation through NF-κB Activation. Int. J. Mol. Sci. 2019, 20, 6281. https://doi.org/10.3390/ijms20246281
Trusca VG, Dumitrescu M, Fenyo IM, Tudorache IF, Simionescu M, Gafencu AV. The Mechanism of Bisphenol A Atherogenicity Involves Apolipoprotein A-I Downregulation through NF-κB Activation. International Journal of Molecular Sciences. 2019; 20(24):6281. https://doi.org/10.3390/ijms20246281
Chicago/Turabian StyleTrusca, Violeta G., Madalina Dumitrescu, Ioana M. Fenyo, Irina F. Tudorache, Maya Simionescu, and Anca V. Gafencu. 2019. "The Mechanism of Bisphenol A Atherogenicity Involves Apolipoprotein A-I Downregulation through NF-κB Activation" International Journal of Molecular Sciences 20, no. 24: 6281. https://doi.org/10.3390/ijms20246281
APA StyleTrusca, V. G., Dumitrescu, M., Fenyo, I. M., Tudorache, I. F., Simionescu, M., & Gafencu, A. V. (2019). The Mechanism of Bisphenol A Atherogenicity Involves Apolipoprotein A-I Downregulation through NF-κB Activation. International Journal of Molecular Sciences, 20(24), 6281. https://doi.org/10.3390/ijms20246281