Insights into Barley Root Transcriptome under Mild Drought Stress with an Emphasis on Gene Expression Regulatory Mechanisms

 ,
,  ,
,  ,
,
Abstract
1. Introduction
2. Results
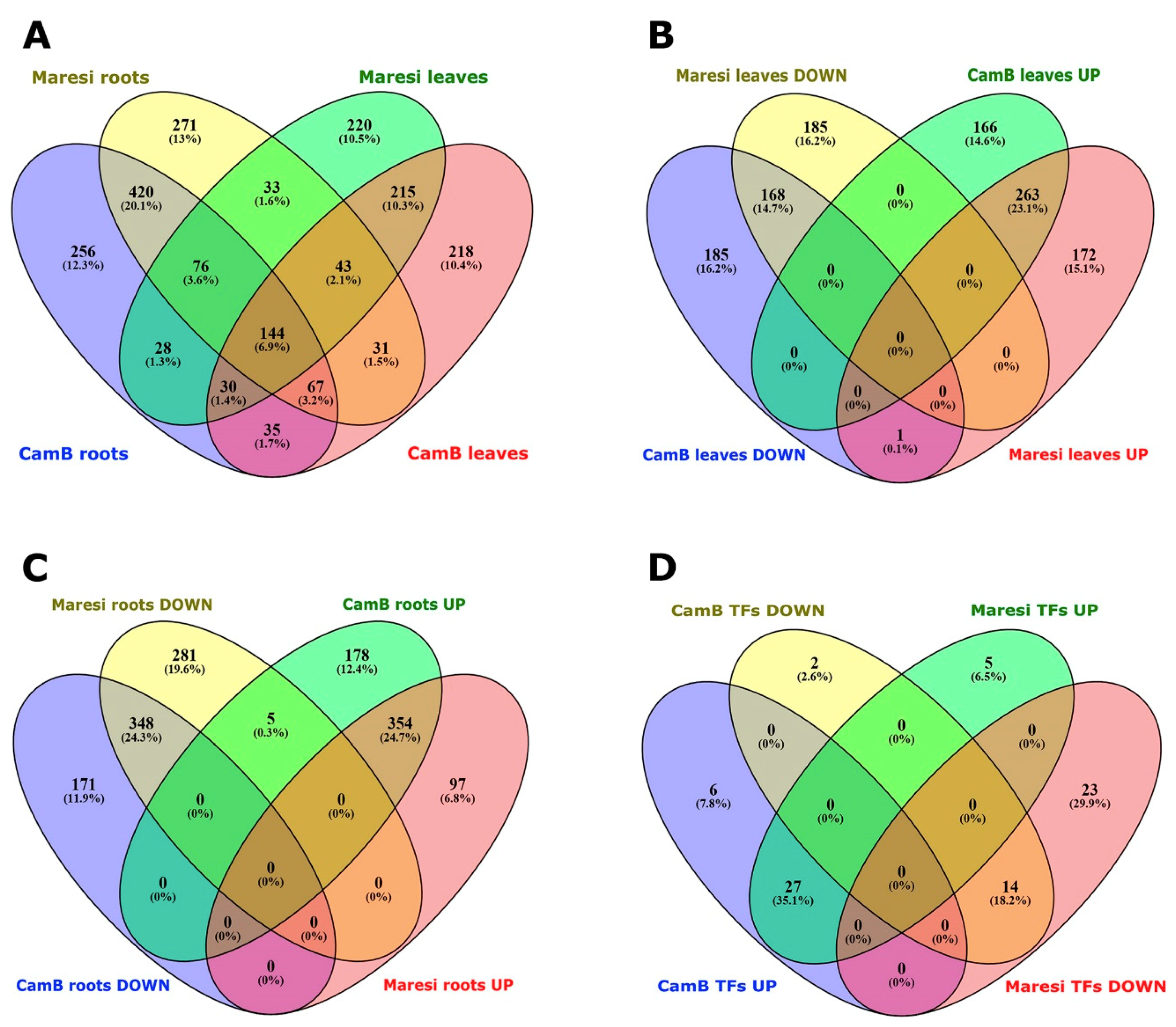
2.1. An Overview of Root and Leaf Transcriptome Changes in Response to Mild Drought Stress
2.2. The Characteristics of Root Transcriptomes of Barley Genotypes Exposed to Mild Drought Stress
2.3. Root DEGs Involved in the Regulation of Gene Expression
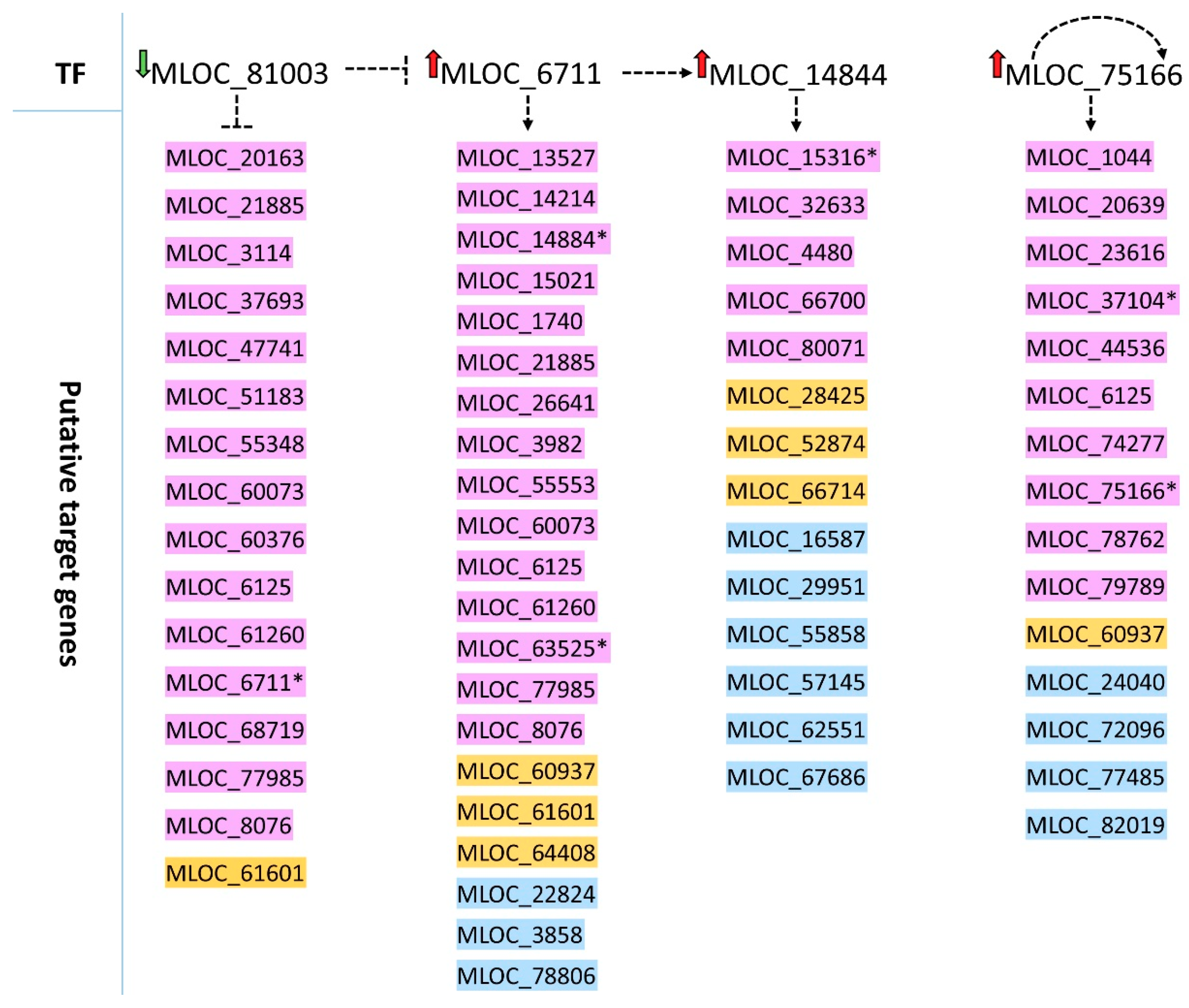
2.4. Target Genes Putatively Regulated by Specific Transcription Factors with Differential Expression in Roots
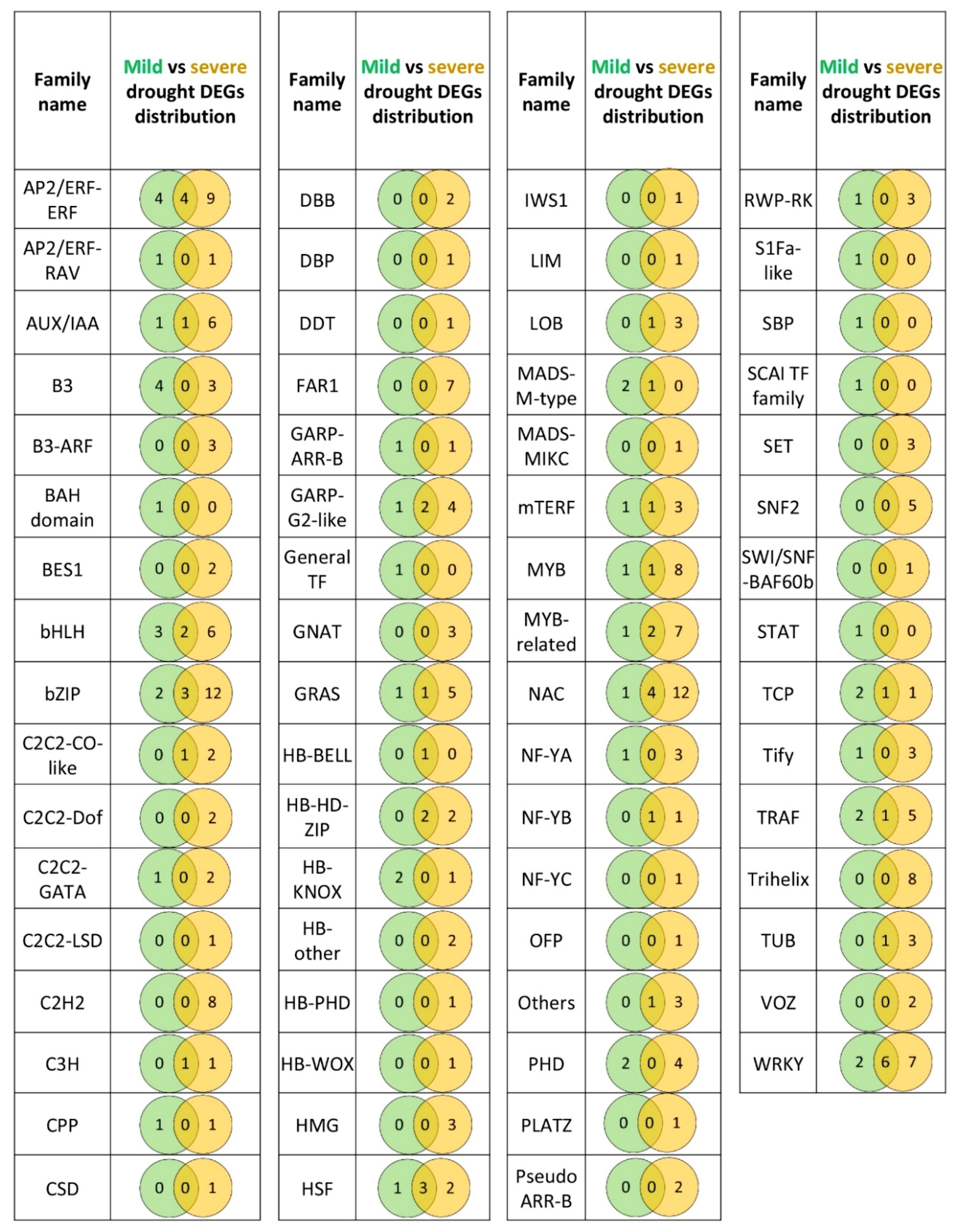
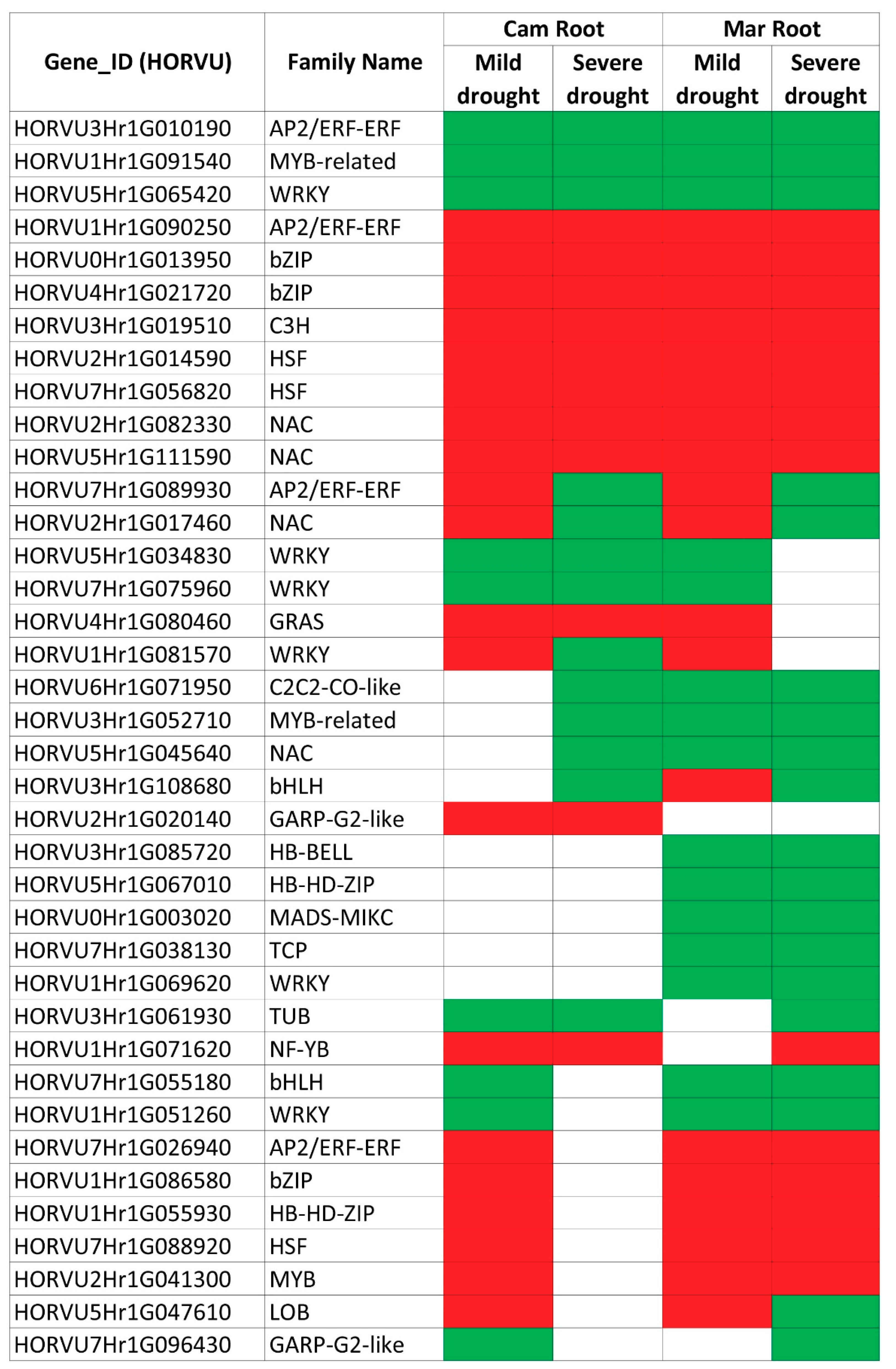
2.5. A Comparison of DEGs Involved in Gene Expression Regulation in Roots during Mild and Severe Drought Stress
3. Discussion
4. Materials and Methods
4.1. Plant Material and STRESS Treatment
4.2. RNA Isolation
4.3. Preparation of Microarrays and Microarray Data Analysis
4.4. Agilent Barley Gene Expression Microarray Annotation, GO Enrichment and Transcription Factor Encoding Genes Analysis
4.5. Quantitative Reverse Transcription (RT)-qPCR
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DEG | Differentially expressed gene |
| GO | Gene ontology |
| PSI | Photosystem I |
| PSII | Photosystem II |
| ROS | Reactive oxygen species |
| TF | Transcription factor |
References
- Schachtman, D.P.; Goodger, J.Q.D. Chemical root to shoot signaling under drought. Trends Plant Sci. 2008, 13, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Comas, L.H.; Becker, S.R.; Cruz, V.M.V.; Byrne, P.F.; Dierig, D.A. Root traits contributing to plant productivity under drought. Front. Plant Sci. 2013, 4, 442. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Xie, Y.; Fan, S.; Wang, Z.; Wang, F.; Zhang, B.; Li, H.; Song, J.; Kong, L. Comparative analysis of root transcriptome profiles between drought-tolerant and susceptible wheat genotypes in response to water stress. Plant Sci. 2018, 272, 276–293. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, L.; Zhang, S.; Zhu, S.; Wu, P.; Chen, Y.; Li, M.; Jiang, H.; Wu, G. Global analysis of gene expression profiles in physic nut (Jatropha curcas L.) seedlings exposed to drought stress. BMC Plant Biol. 2015, 15, 17. [Google Scholar] [CrossRef]
- Sahoo, K.K.; Tripathi, A.K.; Pareek, A.; Singla-Pareek, S.L. Taming drought stress in rice through genetic engineering and transcription factors and protein kinases. Plant Stress 2013, 7, 60–72. [Google Scholar]
- Janiak, A.; Kwaśniewski, M.; Szarejko, I. Gene expression regulation in roots under drought. J. Exp. Bot. 2016, 67, 1003–1014. [Google Scholar] [CrossRef]
- Tripathi, P.; Rabara, R.C.; Rushton, P.J. A systems biology perspective on the role of WRKY transcription factors in drought responses in plants. Planta 2014, 239, 255–266. [Google Scholar] [CrossRef]
- Mun, B.-G.; Lee, S.-U.; Park, E.-J.; Kim, H.-H.; Hussain, A.; Imran, Q.M.; Lee, I.-J.; Yun, B.-W. Analysis of transcription factors among differentially expressed genes induced by drought stress in Populus davidiana. 3 Biotech 2017, 7, 209. [Google Scholar] [CrossRef]
- Wu, J.; Wang, L.; Wang, S. Comprehensive analysis and discovery of drought-related NAC transcription factors in common bean. BMC Plant Biol. 2016, 16, 193. [Google Scholar] [CrossRef]
- Agarwal, P.; Agarwal, P.K.; Joshi, A.J.; Sopory, S.K.; Reddy, M.K. Overexpression of PgDREB2A transcription factor enhances abiotic stress tolerance and activates downstream stress-responsive genes. Mol. Biol. Rep. 2010, 37, 1125–1135. [Google Scholar] [CrossRef]
- Jung, H.; Chung, P.J.; Park, S.-H.; Redillas, M.C.F.R.; Kim, Y.S.; Suh, J.-W.; Kim, J.-K. Overexpression of OsERF48 causes regulation of OsCML16, a calmodulin-like protein gene that enhances root growth and drought tolerance. Plant Biotechnol. J. 2017, 15, 1295–1308. [Google Scholar] [CrossRef]
- Jeong, J.S.; Kim, Y.S.; Redillas, M.C.F.R.; Jang, G.; Jung, H.; Bang, S.W.; Choi, Y.D.; Ha, S.-H.; Reuzeau, C.; Kim, J.-K. OsNAC5 overexpression enlarges root diameter in rice plants leading to enhanced drought tolerance and increased grain yield in the field. Plant Biotechnol. J. 2013, 11, 101–114. [Google Scholar] [CrossRef]
- Lee, D.-K.; Jung, H.; Jang, G.; Jeong, J.S.; Kim, Y.S.; Ha, S.-H.; Do Choi, Y.; Kim, J.-K. Overexpression of the OsERF71 transcription factor alters rice root structure and drought resistance. Plant Physiol. 2016, 172, 575–588. [Google Scholar] [CrossRef]
- Forti, R. (Ed.) Eurostat Report: Agriculture, Forestry and Fishery Statistics; Publications Office of the European Union: Luxembourg, 2017. [Google Scholar]
- Mascher, M.; Gundlach, H.; Himmelbach, A.; Beier, S.; Twardziok, S.O.; Wicker, T.; Radchuk, V.; Dockter, C.; Hedley, P.E.; Russell, J.; et al. A chromosome conformation capture ordered sequence of the barley genome. Nature 2017, 544, 427–433. [Google Scholar] [CrossRef]
- Filek, M.; Łabanowska, M.; Kościelniak, J.; Biesaga-Kościelniak, J.; Kurdziel, M.; Szarejko, I.; Hartikainen, H. Characterization of barley leaf tolerance to drought stress by chlorophyll fluorescence and electron paramagnetic resonance studies. J. Agron. Crop Sci. 2015, 201, 228–240. [Google Scholar] [CrossRef]
- Filek, M.; Łabanowska, M.; Kurdziel, M.; Wesełucha-Birczyńska, A.; Bednarska-Kozakiewicz, E. Structural and biochemical response of chloroplasts in tolerant and sensitive barley genotypes to drought stress. J. Plant Physiol. 2016, 207, 61–72. [Google Scholar] [CrossRef]
- Chmielewska, K.; Rodziewicz, P.; Swarcewicz, B.; Sawikowska, A.; Krajewski, P.; Marczak, Ł.; Ciesiołka, D.; Kuczyńska, A.; Mikołajczak, K.; Ogrodowicz, P.; et al. Analysis of drought-induced proteomic and metabolomic changes in barley (Hordeum vulgare L.) leaves and roots unravels some aspects of biochemical mechanisms involved in drought tolerance. Front. Plant Sci. 2016, 7, 1108. [Google Scholar] [CrossRef]
- Mikołajczak, K.; Kuczyńska, A.; Krajewski, P.; Sawikowska, A.; Surma, M.; Ogrodowicz, P.; Adamski, T.; Krystkowiak, K.; Górny, A.G.; Kempa, M.; et al. Quantitative trait loci for plant height in Maresi × CamB barley population and their associations with yield-related traits under different water regimes. J. Appl. Genet. 2017, 58, 23–35. [Google Scholar] [CrossRef]
- Janiak, A.; Kwasniewski, M.; Sowa, M.; Gajek, K.; Żmuda, K.; Kościelniak, J.; Szarejko, I. No time to waste: Transcriptome study reveals that drought tolerance in barley may be attributed to stressed-like expression patterns that exist before the occurrence of stress. Front. Plant Sci. 2017, 8, 2212. [Google Scholar] [CrossRef]
- Olesen, C.; Møller, M.; Byskov, A.G. Tesmin transcription is regulated differently during male and female meiosis. Mol. Reprod. Dev. 2004, 67, 116–126. [Google Scholar] [CrossRef]
- Andersen, S.U.; Algreen-Petersen, R.G.; Hoedl, M.; Jurkiewicz, A.; Cvitanich, C.; Braunschweig, U.; Schauser, L.; Oh, S.-A.; Twell, D.; Jensen, E.Ø. The conserved cysteine-rich domain of a tesmin/TSO1-like protein binds zinc in vitro and TSO1 is required for both male and female fertility in Arabidopsis thaliana. J. Exp. Bot. 2007, 58, 3657–3670. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Sijacic, P.; Xu, P.; Lian, H.; Liu, Z. Arabidopsis TSO1 and MYB3R1 form a regulatory module to coordinate cell proliferation with differentiation in shoot and root. Proc. Natl. Acad. Sci. USA 2018, 115, E3045–E3054. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Konishi, M.; Kiba, T.; Sakuraba, Y.; Sawaki, N.; Kurai, T.; Ueda, Y.; Sakakibara, H.; Yanagisawa, S. A NIGT1-centred transcriptional cascade regulates nitrate signalling and incorporates phosphorus starvation signals in Arabidopsis. Nat. Commun. 2018, 9, 1376. [Google Scholar] [CrossRef] [PubMed]
- Kiba, T.; Inaba, J.; Kudo, T.; Ueda, N.; Konishi, M.; Mitsuda, N.; Takiguchi, Y.; Kondou, Y.; Yoshizumi, T.; Ohme-Takagi, M.; et al. Repression of nitrogen starvation responses by members of the arabidopsis GARP-Type transcription factor NIGT1/HRS1 subfamily. Plant Cell 2018, 30, 925–945. [Google Scholar] [CrossRef] [PubMed]
- Ruan, W.; Guo, M.; Wu, P.; Yi, K. Phosphate starvation induced OsPHR4 mediates Pi-signaling and homeostasis in rice. Plant Mol. Biol. 2017, 93, 327–340. [Google Scholar] [CrossRef] [PubMed]
- López-Bucio, J.; Cruz-Ramírez, A.; Herrera-Estrella, L. The role of nutrient availability in regulating root architecture. Curr. Opin. Plant Biol. 2003, 6, 280–287. [Google Scholar] [CrossRef]
- Deak, K.I.; Malamy, J. Osmotic regulation of root system architecture. Plant J. 2005, 43, 17–28. [Google Scholar] [CrossRef]
- Brandt, D.T.; Baarlink, C.; Kitzing, T.M.; Kremmer, E.; Ivaska, J.; Nollau, P.; Grosse, R. SCAI acts as a suppressor of cancer cell invasion through the transcriptional control of beta1-integrin. Nat. Cell Biol. 2009, 11, 557–568. [Google Scholar] [CrossRef]
- Brauchle, M.; Yao, Z.; Arora, R.; Thigale, S.; Clay, I.; Inverardi, B.; Fletcher, J.; Taslimi, P.; Acker, M.G.; Gerrits, B.; et al. Protein complex interactor analysis and differential activity of KDM3 subfamily members towards H3K9 methylation. PLoS ONE 2013, 8, e60549. [Google Scholar] [CrossRef]
- Kreßner, C.; Nollau, P.; Grosse, R.; Brandt, D.T. Functional interaction of SCAI with the SWI/SNF complex for transcription and tumor cell invasion. PLoS ONE 2013, 8, e69947. [Google Scholar] [CrossRef]
- Yang, N.; Xu, R.-M. Structure and function of the BAH domain in chromatin biology. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; Grafi, G. Methyl-CpG-binding domain proteins in plants: Interpreters of DNA methylation. Trends Plant Sci. 2007, 12, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Licausi, F.; Ohme-Takagi, M.; Perata, P. APETALA2/Ethylene Responsive Factor (AP2/ERF) transcription factors: Mediators of stress responses and developmental programs. New Phytol. 2013, 199, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Lata, C.; Mishra, A.K.; Muthamilarasan, M.; Bonthala, V.S.; Khan, Y.; Prasad, M. Genome-wide investigation and expression profiling of AP2/ERF transcription factor superfamily in foxtail millet (Setaria italica L.). PLoS ONE 2014, 9, e113092. [Google Scholar] [CrossRef]
- Fernández-Calvo, P.; Chini, A.; Fernández-Barbero, G.; Chico, J.-M.; Gimenez-Ibanez, S.; Geerinck, J.; Eeckhout, D.; Schweizer, F.; Godoy, M.; Franco-Zorrilla, J.M.; et al. The Arabidopsis bHLH transcription factors MYC3 and MYC4 are targets of JAZ repressors and act additively with MYC2 in the activation of jasmonate responses. Plant Cell 2011, 23, 701–715. [Google Scholar] [CrossRef]
- Ye, H.; Du, H.; Tang, N.; Li, X.; Xiong, L. Identification and expression profiling analysis of TIFY family genes involved in stress and phytohormone responses in rice. Plant Mol. Biol. 2009, 71, 291–305. [Google Scholar] [CrossRef]
- Gao, Q.-M.; Venugopal, S.; Navarre, D.; Kachroo, A. Low oleic acid-derived repression of jasmonic acid-inducible defense responses requires the WRKY50 and WRKY51 proteins. Plant Physiol. 2011, 155, 464–476. [Google Scholar] [CrossRef]
- Pan, L.; Zhang, X.; Wang, J.; Ma, X.; Zhou, M.; Huang, L.; Nie, G.; Wang, P.; Yang, Z.; Li, J. Transcriptional profiles of drought-related genes in modulating metabolic processes and antioxidant defenses in Lolium multiflorum. Front. Plant Sci. 2016, 7, 519. [Google Scholar] [CrossRef]
- Srinivasan, T.; Kumar, K.R.R.; Meur, G.; Kirti, P.B. Heterologous expression of Arabidopsis NPR1 (AtNPR1) enhances oxidative stress tolerance in transgenic tobacco plants. Biotechnol. Lett. 2009, 31, 1343–1351. [Google Scholar] [CrossRef]
- Bolduc, N.; Hake, S. The maize transcription factor KNOTTED1 directly regulates the gibberellin catabolism gene ga2ox1. Plant Cell 2009, 21, 1647–1658. [Google Scholar] [CrossRef]
- Ubeda-Tomás, S.; Federici, F.; Casimiro, I.; Beemster, G.T.S.; Bhalerao, R.; Swarup, R.; Doerner, P.; Haseloff, J.; Bennett, M.J. Gibberellin signaling in the endodermis controls Arabidopsis root meristem size. Curr. Biol. 2009, 19, 1194–1199. [Google Scholar] [CrossRef]
- Huang, H.; Liu, B.; Liu, L.; Song, S. Jasmonate action in plant growth and development. J. Exp. Bot. 2017, 68, 1349–1359. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, J.; Li, X.; Wang, S.; Wu, J. Salt and drought stress and ABA responses related to bZIP genes from V. radiata and V. angularis. Gene 2018, 651, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Nijhawan, A.; Jain, M.; Tyagi, A.K.; Khurana, J.P. Genomic survey and gene expression analysis of the basic leucine zipper transcription factor family in rice. Plant Physiol. 2008, 146, 333–350. [Google Scholar] [CrossRef] [PubMed]
- Shaikhali, J.; Norén, L.; de Dios Barajas-López, J.; Srivastava, V.; König, J.; Sauer, U.H.; Wingsle, G.; Dietz, K.-J.; Strand, Å. Redox-mediated mechanisms regulate DNA binding activity of the G-group of basic region leucine zipper (bZIP) transcription factors in Arabidopsis. J. Biol. Chem. 2012, 287, 27510–27525. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Mitsuda, N.; Ohme-Takagi, M. Arabidopsis HsfB1 and HsfB2b act as repressors of the expression of heat-inducible Hsfs but positively regulate the acquired thermotolerance. Plant Physiol. 2011, 157, 1243–1254. [Google Scholar] [CrossRef]
- Chiu, R.S.; Nahal, H.; Provart, N.J.; Gazzarrini, S. The role of the Arabidopsis FUSCA3 transcription factor during inhibition of seed germination at high temperature. BMC Plant Biol. 2012, 12, 15. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, X.; Zhang, D.; Tang, H.; Sun, B.; Li, C.; Hao, L.; Liu, C.; Li, Y.; Shi, Y.; et al. Genome-wide identification of gene expression in contrasting maize inbred lines under field drought conditions reveals the significance of transcription factors in drought tolerance. PLoS ONE 2017, 12, e0179477. [Google Scholar] [CrossRef]
- Seo, P.J.; Park, C.-M. Auxin homeostasis during lateral root development under drought condition. Plant Signal. Behav. 2009, 4, 1002–1004. [Google Scholar] [CrossRef]
- Hirsch, S.; Oldroyd, G.E.D. GRAS-domain transcription factors that regulate plant development. Plant Signal. Behav. 2009, 4, 698–700. [Google Scholar] [CrossRef]
- Chung, H.S.; Howe, G.A. A critical role for the TIFY motif in repression of jasmonate signaling by a stabilized splice variant of the JASMONATE ZIM-domain protein JAZ10 in Arabidopsis. Plant Cell 2009, 21, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, A.; Dixit, S.; Jetter, R.; Thoenes, E.; van Arkel, G.; Pereira, A. The SHINE clade of AP2 domain transcription factors activates wax biosynthesis, alters cuticle properties, and confers drought tolerance when overexpressed in Arabidopsis. Plant Cell 2004, 16, 2463–2480. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.X.; Malitsky, S.; De Oliveira, S.; Branigan, C.; Franke, R.B.; Schreiber, L.; Aharoni, A. SHINE transcription factors act redundantly to pattern the archetypal surface of Arabidopsis flower organs. PLoS Genet. 2011, 7, e1001388. [Google Scholar] [CrossRef] [PubMed]
- Kreszies, T.; Schreiber, L.; Ranathunge, K. Suberized transport barriers in Arabidopsis, barley and rice roots: From the model plant to crop species. J. Plant Physiol. 2018, 227, 75–83. [Google Scholar] [CrossRef]
- Baxter, I.; Hosmani, P.S.; Rus, A.; Lahner, B.; Borevitz, J.O.; Muthukumar, B.; Mickelbart, M.V.; Schreiber, L.; Franke, R.B.; Salt, D.E. Root suberin forms an extracellular barrier that affects water relations and mineral nutrition in Arabidopsis. PLoS Genet. 2009, 5, e1000492. [Google Scholar] [CrossRef]
- Shuai, B.; Reynaga-Pena, C.G.; Springer, P.S. The LATERAL ORGAN BOUNDARIES gene defines a novel, plant-specific gene family. Plant Physiol. 2002, 129, 747–761. [Google Scholar] [CrossRef]
- Ariel, F.D.; Diet, A.; Crespi, M.; Chan, R.L. The LOB-like transcription factor MtLBD1 controls Medicago truncatula root architecture under salt stress. Plant Signal. Behav. 2010, 5, 1666–1668. [Google Scholar] [CrossRef]
- Xu, C.; Tai, H.; Saleem, M.; Ludwig, Y.; Majer, C.; Berendzen, K.W.; Nagel, K.A.; Wojciechowski, T.; Meeley, R.B.; Taramino, G.; et al. Cooperative action of the paralogous maize lateral organ boundaries (LOB) domain proteins RTCS and RTCL in shoot-borne root formation. New Phytol. 2015, 207, 1123–1133. [Google Scholar] [CrossRef]
- Taramino, G.; Sauer, M.; Stauffer, J.L.; Multani, D.; Niu, X.; Sakai, H.; Hochholdinger, F. The maize (Zea mays L.) RTCS gene encodes a LOB domain protein that is a key regulator of embryonic seminal and post-embryonic shoot-borne root initiation. Plant J. 2007, 50, 649–659. [Google Scholar] [CrossRef]
- Liu, H.; Wang, S.; Yu, X.; Yu, J.; He, X.; Zhang, S.; Shou, H.; Wu, P. ARL1, a LOB-domain protein required for adventitious root formation in rice. Plant J. 2005, 43, 47–56. [Google Scholar] [CrossRef]
- Ranjan, A.; Sawant, S. Genome-wide transcriptomic comparison of cotton (Gossypium herbaceum) leaf and root under drought stress. 3 Biotech 2015, 5, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Molina, C.; Rotter, B.; Horres, R.; Udupa, S.M.; Besser, B.; Bellarmino, L.; Baum, M.; Matsumura, H.; Terauchi, R.; Kahl, G.; et al. SuperSAGE: The drought stress-responsive transcriptome of chickpea roots. BMC Genom. 2008, 9, 553. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.; Bogeat-Triboulot, M.-B.; Tisserant, E.; Balzergue, S.; Martin-Magniette, M.-L.; Lelandais, G.; Ningre, N.; Renou, J.-P.; Tamby, J.-P.; Le Thiec, D.; et al. Comparative transcriptomics of drought responses in Populus: A meta-analysis of genome-wide expression profiling in mature leaves and root apices across two genotypes. BMC Genom. 2010, 11, 630. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, W.W.; Alba, R.; Yu, Y.-S.; Bordeaux, J.M.; Simões, M.; Dean, J.F.D. Microarray analysis and scale-free gene networks identify candidate regulators in drought-stressed roots of loblolly pine (P. taeda L.). BMC Genom. 2011, 12, 264. [Google Scholar] [CrossRef]
- Kwasniewski, M.; Daszkowska-Golec, A.; Janiak, A.; Chwialkowska, K.; Nowakowska, U.; Sablok, G.; Szarejko, I. Transcriptome analysis reveals the role of the root hairs as environmental sensors to maintain plant functions under water-deficiency conditions. J. Exp. Bot. 2016, 67, 1079–1094. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B Met. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. ITAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef]
- Oliveros, J.C. Venny. An interactive tool for comparing lists with Venn’s diagrams. Available online: http://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 4 December 2019).
- Rapacz, M.; Stępień, A.; Skorupa, A. Internal standards for quantitative RT-PCR studies of gene expression under drought treatment in barley (Hordeum vulgare L.): The effects of developmental stage and leaf age. Acta Phys. Plant. 2012, 34, 1723–1733. [Google Scholar] [CrossRef]
- Ramakers, C.; Ruijter, J.M.; Deprez, R.H.L.; Moorman, A.F.M. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci. Lett. 2003, 339, 62–66. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST©) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Organ | No. of Probes | No. of Genes with Known Annotations * | ||
|---|---|---|---|---|---|
| Down-Regulation | Up-Regulation | Down-Regulation | Up-Regulation | ||
| CamB | Roots | 792 | 1019 | 519 | 536 |
| CamB | Leaves | 597 | 705 | 354 | 429 |
| Maresi | Roots | 999 | 884 | 634 | 452 |
| Maresi | Leaves | 602 | 717 | 353 | 436 |
| Horvu ID | Expression Change | Family Name | Gene Description | |||
|---|---|---|---|---|---|---|
| CamB Roots | CamB Leaves | Mar Roots | Mar Leaves | |||
| Transcription factors: | ||||||
| HORVU4HR1G087580 | up | n/c | n/c | n/c | bHLH | Uncharacterized |
| HORVU3Hr1G073470 | up | n/c | n/c | n/c | CPP | Protein tesmin/TSO1-like CXC 5 |
| HORVU2Hr1G020140 | up | n/c | n/c | up | GARP-G2-like | Two-component response regulator ARR18 |
| HORVU7Hr1G096430 | down | n/c | n/c | n/c | GARP-G2-like | Myb-like transcription factor family protein |
| HORVU5Hr1G110960 | up | n/c | n/c | n/c | General TF | Transcription initiation factor IIE subunit beta |
| HORVU1Hr1G071620 | up | n/c | n/c | n/c | NF-YB | Nuclear transcription factor Y subunit B |
| HORVU5Hr1G062040 | up | n/c | n/c | n/c | SCAI TF family | Protein SCAI |
| HORVU3Hr1G061930 | down | down | n/c | down | TUB | Tubby-like F-box protein 1 |
| Transcriptional Regulators: | ||||||
| HORVU5Hr1G061120 | down | n/c | n/c | n/c | BAH domain | Bromo-adjacent homology (BAH) domain-containing protein |
| HORVU6Hr1G034680 | up | n/c | n/c | n/c | PHD | methyl-CPG-binding domain 9 |
| Horvu ID | Expression change | Family Name | Gene Description | |||
|---|---|---|---|---|---|---|
| CamB Roots | CamB Leaves | Mar Roots | Mar Leaves | |||
| Transcription factors: | ||||||
| HORVU2Hr1G050260 | n/c | n/c | up | n/c | AP2/ERF-ERF | Ethylene-responsive transcription factor |
| HORVU4Hr1G077310 | n/c | n/c | down | n/c | AP2/ERF-ERF | Ethylene-responsive transcription factor |
| HORVU5Hr1G080300 | n/c | n/c | down | down | AP2/ERF-ERF | Dehydration-responsive element-binding protein 1B |
| HORVU6Hr1G080340 | n/c | up | up | up | AP2/ERF-ERF | Ethylene-responsive transcription factor 5 |
| HORVU3Hr1G105720 | n/c | n/c | down | n/c | B3 | B3 domain-containing protein |
| HORVU5Hr1G017450 | n/c | n/c | up | n/c | B3 | B3 domain-containing protein |
| HORVU3Hr1G108680 | n/c | n/c | up | n/c | bHLH | Transcription factor ORG2 |
| HORVU5Hr1G002090 | n/c | n/c | down | n/c | bHLH | Basic helix-loop-helix (bHLH) DNA-binding superfamily protein |
| HORVU6Hr1G071950 | n/c | n/c | down | n/c | C2C2-CO-like | Zinc finger protein CONSTANS-LIKE 10 |
| HORVU4Hr1G088280 | n/c | up | up | up | C2C2-GATA | GATA transcription factor 2 |
| HORVU3Hr1G085720 | n/c | n/c | down | n/c | HB-BELL | BEL1-like homeodomain 8 |
| HORVU5Hr1G067010 | n/c | n/c | down | n/c | HB-HD-ZIP | Homeobox-leucine zipper protein family |
| HORVU2Hr1G061320 | n/c | n/c | down | n/c | HB-KNOX | Homeobox protein knotted-1-like 12 |
| HORVU5Hr1G098570 | n/c | n/c | down | n/c | HB-KNOX | Homeobox protein knotted-1-like 12 |
| HORVU0Hr1G003020 | n/c | n/c | down | n/c | MADS-M-type | MADS-box transcription factor 18 |
| HORVU3Hr1G095090 | n/c | n/c | down | n/c | MADS-M-type | MADS-box transcription factor family protein |
| HORVU3Hr1G052710 | n/c | n/c | down | n/c | MYB-related | Myb-like transcription factor family protein |
| HORVU6Hr1G066000 | n/c | down | down | n/c | MYB-related | Myb-like transcription factor family protein |
| HORVU5Hr1G045640 | n/c | n/c | down | down | NAC | NAC domain protein, |
| HORVU2Hr1G072420 | n/c | n/c | down | n/c | S1Fa-like | DNA-binding protein S1FA2 |
| HORVU3Hr1G094730 | n/c | n/c | down | n/c | SBP | Squamosa promoter-binding-like protein 2 |
| HORVU1Hr1G054620 | n/c | n/c | down | n/c | STAT | SH2 domain protein B |
| HORVU3Hr1G095400 | n/c | n/c | down | down | TCP | TCP family transcription factor |
| HORVU6Hr1G093960 | n/c | n/c | down | n/c | TCP | TCP family transcription factor |
| HORVU7Hr1G038130 | n/c | n/c | down | n/c | TCP | TCP family transcription factor |
| HORVU7Hr1G041230 | n/c | n/c | down | up | Tify | Protein TIFY 3A |
| HORVU1Hr1G069620 | n/c | n/c | down | n/c | WRKY | WRKY DNA-binding protein 54 |
| HORVU3Hr1G050590 | n/c | n/c | down | n/c | WRKY | WRKY DNA-binding protein 50 |
| Transcriptional regulators: | ||||||
| HORVU6Hr1G091700 | n/c | n/c | down | n/c | Others | Ethylene receptor 3 |
| HORVU2Hr1G017680 | n/c | up | down | up | PHD | E3 ubiquitin-protein ligase SHPRH |
| HORVU4Hr1G003040 | n/c | n/c | down | n/c | TRAF | Regulatory protein (NPR1) |
| Horvu ID | MLOC ID | Background-All 1 | Background-Bind 2 | Query-All 3 | Query-Bind 4 | p-Value | q-Value | Description (IPK) |
|---|---|---|---|---|---|---|---|---|
| HORVU1Hr1G090030 | MLOC_6711 | 24306 | 470 | 546 | 21 | 1.059 × 10−3 | 3.352 × 10−2 | G-box binding factor 2 |
| HORVU5Hr1G014170 | MLOC_81003 | 24306 | 388 | 546 | 16 | 7.159 × 10−3 | 9.715 × 10−2 | Basic-leucine zipper (bZIP) TF family protein |
| HORVU7Hr1G056820 | MLOC_75166 | 24306 | 368 | 546 | 15 | 9.549 × 10−3 | 1.210 × 10−1 | Heat stress transcription factor B-2b |
| HORVU6Hr1G068100 | MLOC_14844 | 24306 | 386 | 546 | 14 | 2.906 × 10−2 | 2.301 × 10−1 | Myb-like TF family protein |
| Horvu ID | MLOC ID | Family Name | Mild Drought | Severe Drought | Description | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Roots | Leaves | Roots | Leaves | ||||||||
| CamB | Mar | CamB | Mar | CamB | Mar | CamB | Mar | ||||
| HORVU7Hr1G089930 | MLOC_59305 | AP2/ERF-ERF | up | up | n/c | n/c | down | down | n/c | n/c | Ethylene-responsive transcription factor 1 |
| HORVU5Hr1G047610 | MLOC_5148 | LOB | up | up | n/c | n/c | n/c | down | n/c | n/c | LOB domain-containing protein 4 |
| HORVU2Hr1G017460 | MLOC_65101 | NAC | up | up | n/c | up | down | down | up | NAC domain protein | |
| HORVU1Hr1G081570 | MLOC_32433 | WRKY | up | up | n/c | n/c | down | n/c | n/c | n/c | WRKY DNA-binding protein 24 |
| HORVU3Hr1G108680 | MLOC_36351 | bHLH | n/c | up | n/c | n/c | down | down | n/c | n/c | Transcription factor ORG2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janiak, A.; Kwasniewski, M.; Sowa, M.; Kuczyńska, A.; Mikołajczak, K.; Ogrodowicz, P.; Szarejko, I. Insights into Barley Root Transcriptome under Mild Drought Stress with an Emphasis on Gene Expression Regulatory Mechanisms. Int. J. Mol. Sci. 2019, 20, 6139. https://doi.org/10.3390/ijms20246139
Janiak A, Kwasniewski M, Sowa M, Kuczyńska A, Mikołajczak K, Ogrodowicz P, Szarejko I. Insights into Barley Root Transcriptome under Mild Drought Stress with an Emphasis on Gene Expression Regulatory Mechanisms. International Journal of Molecular Sciences. 2019; 20(24):6139. https://doi.org/10.3390/ijms20246139
Chicago/Turabian StyleJaniak, Agnieszka, Miroslaw Kwasniewski, Marta Sowa, Anetta Kuczyńska, Krzysztof Mikołajczak, Piotr Ogrodowicz, and Iwona Szarejko. 2019. "Insights into Barley Root Transcriptome under Mild Drought Stress with an Emphasis on Gene Expression Regulatory Mechanisms" International Journal of Molecular Sciences 20, no. 24: 6139. https://doi.org/10.3390/ijms20246139
APA StyleJaniak, A., Kwasniewski, M., Sowa, M., Kuczyńska, A., Mikołajczak, K., Ogrodowicz, P., & Szarejko, I. (2019). Insights into Barley Root Transcriptome under Mild Drought Stress with an Emphasis on Gene Expression Regulatory Mechanisms. International Journal of Molecular Sciences, 20(24), 6139. https://doi.org/10.3390/ijms20246139