Integrated Proteomics and Metabolomics Analysis Provides Insights into Ganoderic Acid Biosynthesis in Response to Methyl Jasmonate in Ganoderma Lucidum


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
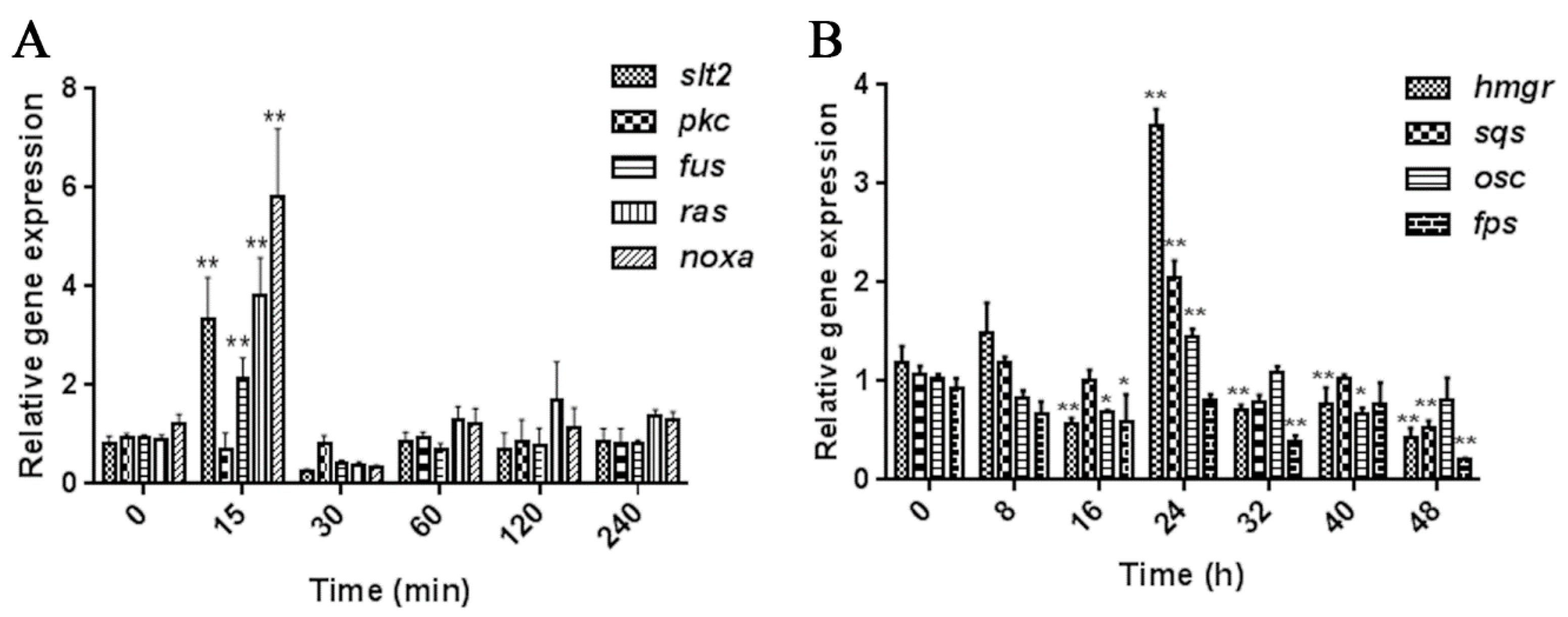
2.1. Screening the Appropriate MeJA Processing Time Points
2.2. Overview of Quantitative Proteomics Analysis
2.3. Characteristics of DAPs in M15
2.4. Characteristics of DAPs in M24
2.5. Characteristics of DAPs in M15 and M24
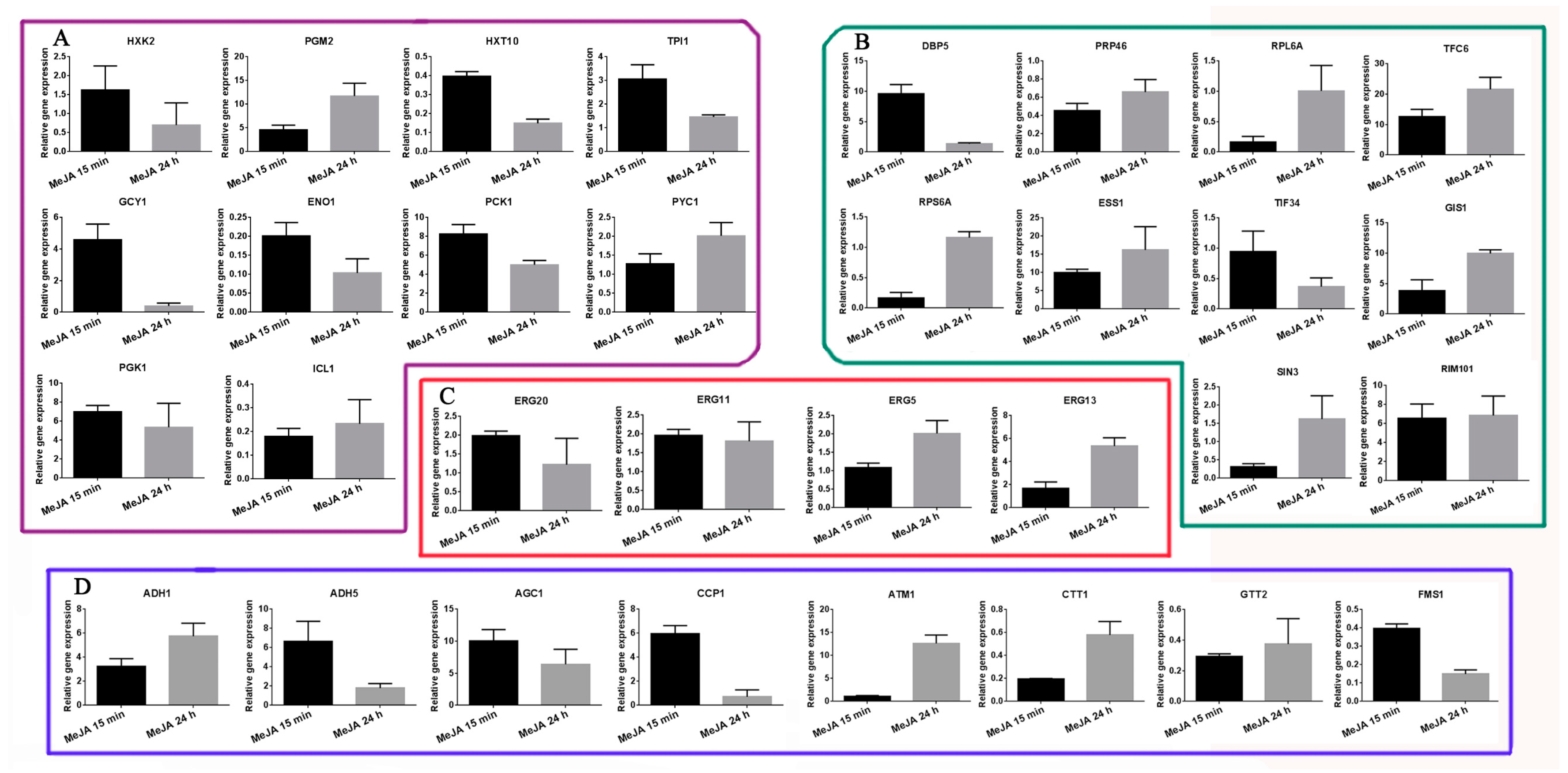
2.6. Transcriptional Expression Analysis of Selected DAPs as Revealed by qRT-PCR
2.7. Metabolic Changes in Response to MeJA
3. Discussion
3.1. Oxidoreduction Process
3.2. Secondary Metabolism
3.3. Energy Metabolism
3.4. Protein Synthesis
4. Materials and Methods
4.1. Fermentation Conditions and Methyl Jasmonate Elicitation of G. lucidum (Fungal Culture and Sample Preparation)
4.2. Protein Extraction
4.3. iTRAQ Labelling and Strong Cation Exchange Fractionation
4.4. Database Search and Quantitative Proteomic Analysis
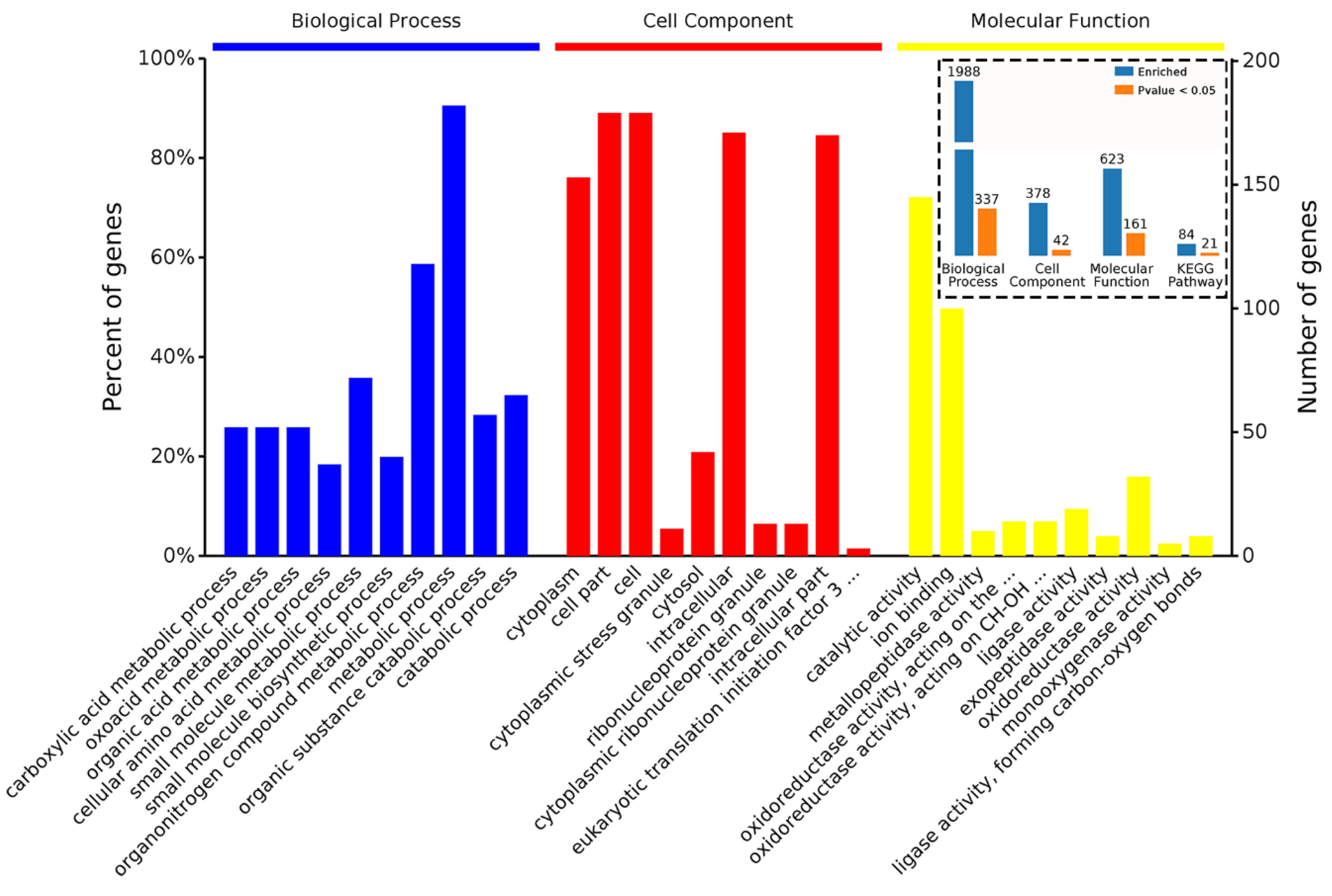
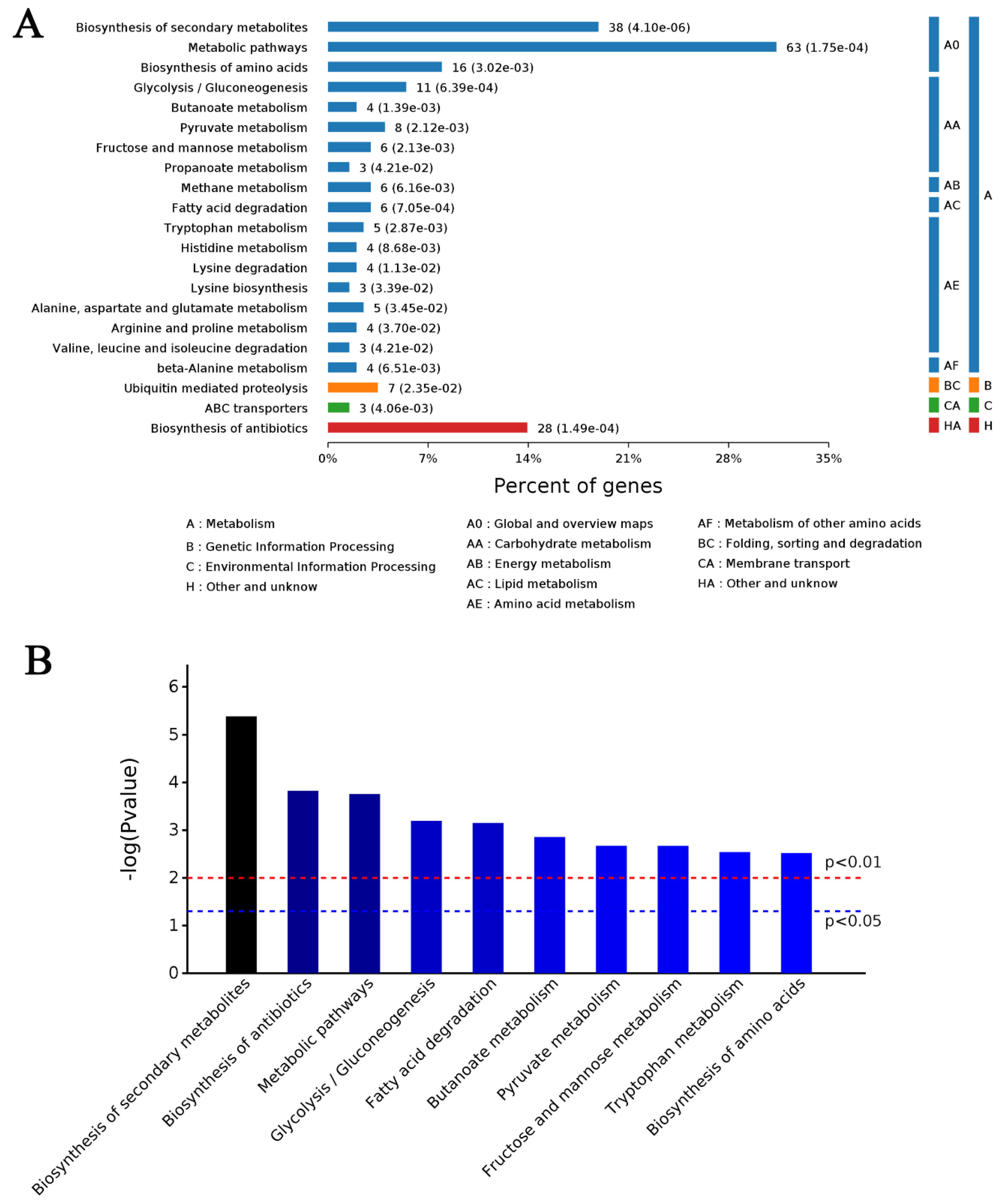
4.5. Bioinformatic Analysis of DAPs
4.6. RNA Extraction Procedure and cDNA Synthesis
4.7. Real-Time RT-PCR Analysis
4.8. GC-MS Data Preprocessing, Statistical Analysis, and Identification of Differential Metabolites
4.9. LC-MS/MS Data Pre-Processing, Statistical Analysis, and Identification of Differential Metabolites
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mizushina, Y.; Hanashima, L.; Yamaguchi, T.; Takemura, M.; Sugawara, F.; Saneyoshi, M.; Matsukage, A.; Yoshida, S.; Sakaguchi, K. A mushroom fruiting body-inducing substance inhibits activities of replicative DNA polymerases. Biochem. Biophys. Res. Commun. 1998, 249, 17–22. [Google Scholar] [CrossRef]
- Calvino, E.; Manjon, J.L.; Sancho, P.; Tejedor, M.C.; Herraez, A.; Diez, J.C. Ganoderma lucidum induced apoptosis in NB4 human leukemia cells: Involvement of Akt and Erk. J. Ethnopharmacol. 2010, 128, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.L.; Yen, G.C. Ganoderic Acid and Lucidenic Acid (Triterpenoid). Enzymes 2014, 36, 33–56. [Google Scholar] [PubMed]
- Shiao, M.S. Natural products of the medicinal fungus Ganoderma lucidum: Occurrence, biological activities, and pharmacological functions. Chem. Rec. 2003, 3, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Ren, A.; Mu, D.; Zhao, M. Current progress in the study on biosynthesis and regulation of ganoderic acids. Appl. Microbiol. Biotechnol. 2010, 88, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Zapata, P.; Rojas, D.; Atehortua, L. Production of biomass, polysaccharides, and ganoderic acid using non-conventional carbon sources under submerged culture of the Lingzhi or Reishi medicinal mushroom, Ganoderma lucidum (W.Curt.:Fr.)P. Karst. (higher Basidiomycetes). Int. J. Med. Mushrooms 2012, 14, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Papinutti, L. Effects of nutrients, pH and water potential on exopolysaccharides production by a fungal strain belonging to Ganoderma lucidum complex. Bioresour. Technol. 2010, 101, 1941–1946. [Google Scholar] [CrossRef]
- Liang, C.X.; Li, Y.B.; Xu, J.W.; Wang, J.L.; Miao, X.L.; Tang, Y.J.; Gu, T.; Zhong, J.J. Enhanced biosynthetic gene expressions and production of ganoderic acids in static liquid culture of Ganoderma lucidum under phenobarbital induction. Appl. Microbiol. Biotechnol. 2010, 86, 1367–1374. [Google Scholar] [CrossRef]
- Ren, A.; Li, X.B.; Miao, Z.G.; Shi, L.; Jaing, A.L.; Zhao, M.W. Transcript and metabolite alterations increase ganoderic acid content in Ganoderma lucidum using acetic acid as an inducer. Biotechnol. Lett. 2014, 36, 2529–2536. [Google Scholar] [CrossRef]
- Liu, R.; Zhang, X.; Ren, A.; Shi, D.K.; Shi, L.; Zhu, J.; Yu, H.S.; Zhao, M.W. Heat stress-induced reactive oxygen species participate in the regulation of HSP expression, hyphal branching and ganoderic acid biosynthesis in Ganoderma lucidum. Microbiol. Res. 2018, 209, 43–54. [Google Scholar] [CrossRef]
- Liu, Y.N.; Lu, X.X.; Ren, A.; Shi, L.; Zhu, J.; Jiang, A.L.; Yu, H.S.; Zhao, M.W. Conversion of phosphatidylinositol (PI) to PI4-phosphate (PI4P) and then to PI(4,5)P-2 is essential for the cytosolic Ca2+ concentration under heat stress in Ganoderma lucidum. Environ. Microbiol. 2018, 20, 2456–2468. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ren, A.; Li, M.J.; Cao, P.F.; Chen, T.X.; Zhang, G.; Shi, L.; Jiang, A.L.; Zhao, M.W. Heat Stress Modulates Mycelium Growth, Heat Shock Protein Expression, Ganoderic Acid Biosynthesis, and Hyphal Branching of Ganoderma lucidum via Cytosolic Ca2+. Appl. Environ. Microb. 2016, 82, 4112–4125. [Google Scholar] [CrossRef]
- Liu, Y.N.; Tong, T.; Zhang, R.R.; Liu, L.M.; Shi, M.L.; Ma, Y.C.; Liu, G.Q. Interdependent nitric oxide and hydrogen peroxide independently regulate the coix seed oil-induced triterpene acid accumulation in Ganoderma lingzhi. Mycologia 2019, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.L.; Ren, A.; Wang, T.; Zhu, J.; Hu, Y.R.; Shi, L.; Yu, H.S.; Zhao, M.W. Hydrogen sulfide, a novel small molecule signalling agent, participates in the regulation of ganoderic acids biosynthesis induced by heat stress in Ganoderma lucidum. Fungal Genet. Biol. 2019, 130, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Shi, L.; Zhu, T.; Yang, T.; Ren, A.; Zhu, J.; Zhao, M.W. Cross-talk between nitric oxide and calcium-calmodulin regulate ganoderic acid biosynthesis in Ganoderma lucidum under heat stress. Appl. Environ. Microbiol. 2018, 84, e00043-18. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.J.; An, C.H.; Park, S.C.; Pyun, J.W.; Lee, J.; Kim, S.W.; Kim, H.S.; Kim, H.; Jeong, J.C.; Kim, C.Y. Methyl Jasmonate Increases Isoflavone Production in Soybean Cell Cultures by Activating Structural Genes Involved in Isoflavonoid Biosynthesis. J. Agric. Food Chem. 2018, 66, 4099–4105. [Google Scholar] [CrossRef]
- Thines, B.; Mandaokar, A.; Browse, J. Characterizing jasmonate regulation of male fertility in Arabidopsis. Methods Mol. Biol. 2013, 1011, 13–23. [Google Scholar]
- Yuan, Z.; Zhang, D. Roles of jasmonate signalling in plant inflorescence and flower development. Curr. Opin. Plant Biol. 2015, 27, 44–51. [Google Scholar] [CrossRef]
- Widemann, E.; Smirnova, E.; Aubert, Y.; Miesch, L.; Heitz, T. Dynamics of Jasmonate Metabolism upon Flowering and across Leaf Stress Responses in Arabidopsis thaliana. Plants 2016, 5, 4. [Google Scholar] [CrossRef]
- Fingrut, O.; Flescher, E. Plant stress hormones suppress the proliferation and induce apoptosis in human cancer cells. Leukemia 2002, 16, 608–616. [Google Scholar] [CrossRef]
- Kniazhanski, T.; Jackman, A.; Heyfets, A.; Gonen, P.; Flescher, E.; Sherman, L. Methyl jasmonate induces cell death with mixed characteristics of apoptosis and necrosis in cervical cancer cells. Cancer Lett. 2008, 271, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Milrot, E.; Jackman, A.; Kniazhanski, T.; Gonen, P.; Flescher, E.; Sherman, L. Methyl jasmonate reduces the survival of cervical cancer cells and downregulates HPV E6 and E7, and survivin. Cancer Lett. 2012, 319, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.J.; Tian, S.P. Effects of a biocontrol agent and methyl jasmonate on postharvest diseases of peach fruit and the possible mechanisms involved. J. Appl. Microbiol. 2005, 98, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Vergopoulou, S.; Galanopoulou, D.; Markaki, P. Methyl jasmonate stimulates aflatoxin B1 biosynthesis by Aspergillus parasiticus. J. Agric. Food Chem. 2001, 49, 3494–3498. [Google Scholar] [CrossRef]
- Ren, A.; Qin, L.; Shi, L.; Dong, X.; Mu, D.S.; Li, Y.X.; Zhao, M.W. Methyl jasmonate induces ganoderic acid biosynthesis in the basidiomycetous fungus Ganoderma lucidum. Bioresour. Technol. 2010, 101, 6785–6790. [Google Scholar] [CrossRef]
- Ren, A.; Li, M.J.; Shi, L.; Mu, D.S.; Jiang, A.L.; Han, Q.; Zhao, M.W. Profiling and quantifying differential gene transcription provide insights into ganoderic acid biosynthesis in Ganoderma lucidum in response to methyl jasmonate. PLoS ONE 2013, 8, e65027. [Google Scholar] [CrossRef]
- Shi, L.; Gong, L.; Zhang, X.; Ren, A.; Gao, T.; Zhao, M. The regulation of methyl jasmonate on hyphal branching and GA biosynthesis in Ganoderma lucidum partly via ROS generated by NADPH oxidase. Fungal Genet. Biol. 2015, 81, 201–211. [Google Scholar] [CrossRef]
- Al-Obaidi, J.R.; Saidi, N.B.; Usuldin, S.R.; Hussin, S.N.; Yusoff, N.M.; Idris, A.S. Comparison of Different Protein Extraction Methods for Gel-Based Proteomic Analysis of Ganoderma spp. Protein J. 2016, 35, 100–106. [Google Scholar] [CrossRef]
- Manavalan, T.; Manavalan, A.; Thangavelu, K.P.; Heese, K. Secretome analysis of Ganoderma lucidum cultivated in sugarcane bagasse. J. Proteom. 2012, 77, 298–309. [Google Scholar] [CrossRef]
- Yao, L.; Zhu, L.P.; Xu, X.Y.; Tan, L.L.; Sadilek, M.; Fan, H.; Hu, B.; Shen, X.T.; Yang, J.; Qiao, B.; et al. Discovery of novel xylosides in co-culture of basidiomycetes Trametes versicolor and Ganoderma applanatum by integrated metabolomics and bioinformatics. Sci. Rep. 2016, 6, 33237. [Google Scholar] [CrossRef]
- Zhang, G.; Sun, Z.; Ren, A.; Shi, L.; Shi, D.; Li, X.; Zhao, M. The mitogen-activated protein kinase GlSlt2 regulates fungal growth, fruiting body development, cell wall integrity, oxidative stress and ganoderic acid biosynthesis in Ganoderma lucidum. Fungal Genet. Biol. 2017, 104, 6–15. [Google Scholar] [CrossRef]
- Maeder, C.I.; Hink, M.A.; Kinkhabwala, A.; Mayr, R.; Bastiaens, P.I.; Knop, M. Spatial regulation of Fus3 MAP kinase activity through a reaction-diffusion mechanism in yeast pheromone signalling. Nat. Cell Biol. 2007, 9, 1319–1326. [Google Scholar] [CrossRef]
- Heinisch, J.J.; Rodicio, R. Protein kinase C in fungi-more than just cell wall integrity. FEMS Microbiol. Rev. 2018, 42. [Google Scholar] [CrossRef]
- Guan, Y.; Wang, D.Y.; Ying, S.H.; Feng, M.G. A novel Ras GTPase (Ras3) regulates conidiation, multi-stress tolerance and virulence by acting upstream of Hog1 signaling pathway in Beauveria bassiana. Fungal Genet. Biol. 2015, 82, 85–94. [Google Scholar] [CrossRef]
- Park, J.C.; Kim, Y.; Lee, H.S. Involvement of the NADH oxidase-encoding noxA gene in oxidative stress responses in Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 2015, 99, 1363–1374. [Google Scholar] [CrossRef]
- Huang, A.C.; Jiang, T.; Liu, Y.-X.; Bai, Y.-C.; Reed, J.; Qu, B.; Goossens, A.; Nützmann, H.-W.; Bai, Y.; Osbourn, A. A specialized metabolic network selectively modulates Arabidopsis root microbiota. Science 2019, 364, eaau6389. [Google Scholar] [CrossRef]
- Werck-Reichhart, D.; Feyereisen, R. Cytochromes P450: A success story. Genome Biol. 2000, 1, REVIEWS3003. [Google Scholar] [CrossRef]
- Hirotani, M.; Asaka, I.; Furuya, T. Investigation of the biosynthesis of 3?-hydroxy triterpenoids, ganoderic acids T and S, by application of a feeding experiment using [1,2-13C2]acetate. J. Chem. Soc. Perkin Trans. 1990, 10, 2751–2754. [Google Scholar] [CrossRef]
- Xu, J.W.; Zhao, W.; Zhong, J.J. Biotechnological production and application of ganoderic acids. Appl. Microbiol. Biotechnol. 2010, 87, 457–466. [Google Scholar] [CrossRef]
- Chen, S.L.; Xu, J.; Liu, C.; Zhu, Y.J.; Nelson, D.R.; Zhou, S.G.; Li, C.F.; Wang, L.Z.; Guo, X.; Sun, Y.Z.; et al. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat. Commun. 2012, 3, 913. [Google Scholar] [CrossRef]
- Wang, W.F.; Xiao, H.; Zhong, J.J. Biosynthesis of a ganoderic acid in Saccharomyces cerevisiae by expressing a cytochrome P450 gene from Ganoderma lucidum. Biotechnol. Bioeng. 2018, 115, 1842–1854. [Google Scholar] [CrossRef] [PubMed]
- Holthuis, J.C.M.; Menon, A.K. Lipid landscapes and pipelines in membrane homeostasis. Nature 2014, 510, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.N.; Zhang, T.J.; Lu, X.X.; Ma, B.L.; Ren, A.; Liang, S.; Jiang, A.L.; Yu, H.S.; Zhao, M.W. Membrane fluidity is involved in the regulation of heat stress induced secondary metabolism in Ganoderma lucidum: Membrane fluidity regulates secondary metabolism. Environ. Microbiol. 2017, 19, 1653–1668. [Google Scholar] [CrossRef] [PubMed]
- Aghdam, M.S.; Bodbodak, S. Physiological and biochemical mechanisms regulating chilling tolerance in fruits and vegetables under postharvest salicylates and jasmonates treatments. Sci. Hortic. 2013, 156, 73–85. [Google Scholar] [CrossRef]
- Lu, M.; Zhang, Y.Y.; Tang, S.K.; Pan, J.B.; Yu, Y.K.; Han, J.; Li, Y.Y.; Du, X.H.; Nan, Z.J.; Sun, Q.P. AtCNGC2 is involved in jasmonic acid-induced calcium mobilization. J. Exp. Bot. 2016, 67, 809–819. [Google Scholar] [CrossRef]
- Cesari, I.M.; Carvalho, E.; Figueiredo Rodrigues, M.; Mendonca Bdos, S.; Amoedo, N.D.; Rumjanek, F.D. Methyl jasmonate: Putative mechanisms of action on cancer cells cycle, metabolism, and apoptosis. Int. J. Cell Biol. 2014, 2014, 572097. [Google Scholar] [CrossRef]
- Lu, X.; Liu, Y.; Zhao, L.; Liu, Y.; Zhao, M. Selection of reliable reference genes for RT-qPCR during methyl jasmonate, salicylic acid and hydrogen peroxide treatments in Ganoderma lucidum. World J. Microbiol. Biotechnol. 2018, 34, 92. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, A.-L.; Liu, Y.-N.; Liu, R.; Ren, A.; Ma, H.-Y.; Shu, L.-B.; Shi, L.; Zhu, J.; Zhao, M.-W. Integrated Proteomics and Metabolomics Analysis Provides Insights into Ganoderic Acid Biosynthesis in Response to Methyl Jasmonate in Ganoderma Lucidum. Int. J. Mol. Sci. 2019, 20, 6116. https://doi.org/10.3390/ijms20246116
Jiang A-L, Liu Y-N, Liu R, Ren A, Ma H-Y, Shu L-B, Shi L, Zhu J, Zhao M-W. Integrated Proteomics and Metabolomics Analysis Provides Insights into Ganoderic Acid Biosynthesis in Response to Methyl Jasmonate in Ganoderma Lucidum. International Journal of Molecular Sciences. 2019; 20(24):6116. https://doi.org/10.3390/ijms20246116
Chicago/Turabian StyleJiang, Ai-Liang, Yong-Nan Liu, Rui Liu, Ang Ren, Hong-Yu Ma, Lie-Bo Shu, Liang Shi, Jing Zhu, and Ming-Wen Zhao. 2019. "Integrated Proteomics and Metabolomics Analysis Provides Insights into Ganoderic Acid Biosynthesis in Response to Methyl Jasmonate in Ganoderma Lucidum" International Journal of Molecular Sciences 20, no. 24: 6116. https://doi.org/10.3390/ijms20246116
APA StyleJiang, A.-L., Liu, Y.-N., Liu, R., Ren, A., Ma, H.-Y., Shu, L.-B., Shi, L., Zhu, J., & Zhao, M.-W. (2019). Integrated Proteomics and Metabolomics Analysis Provides Insights into Ganoderic Acid Biosynthesis in Response to Methyl Jasmonate in Ganoderma Lucidum. International Journal of Molecular Sciences, 20(24), 6116. https://doi.org/10.3390/ijms20246116