Homologous Recombination under the Single-Molecule Fluorescence Microscope


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
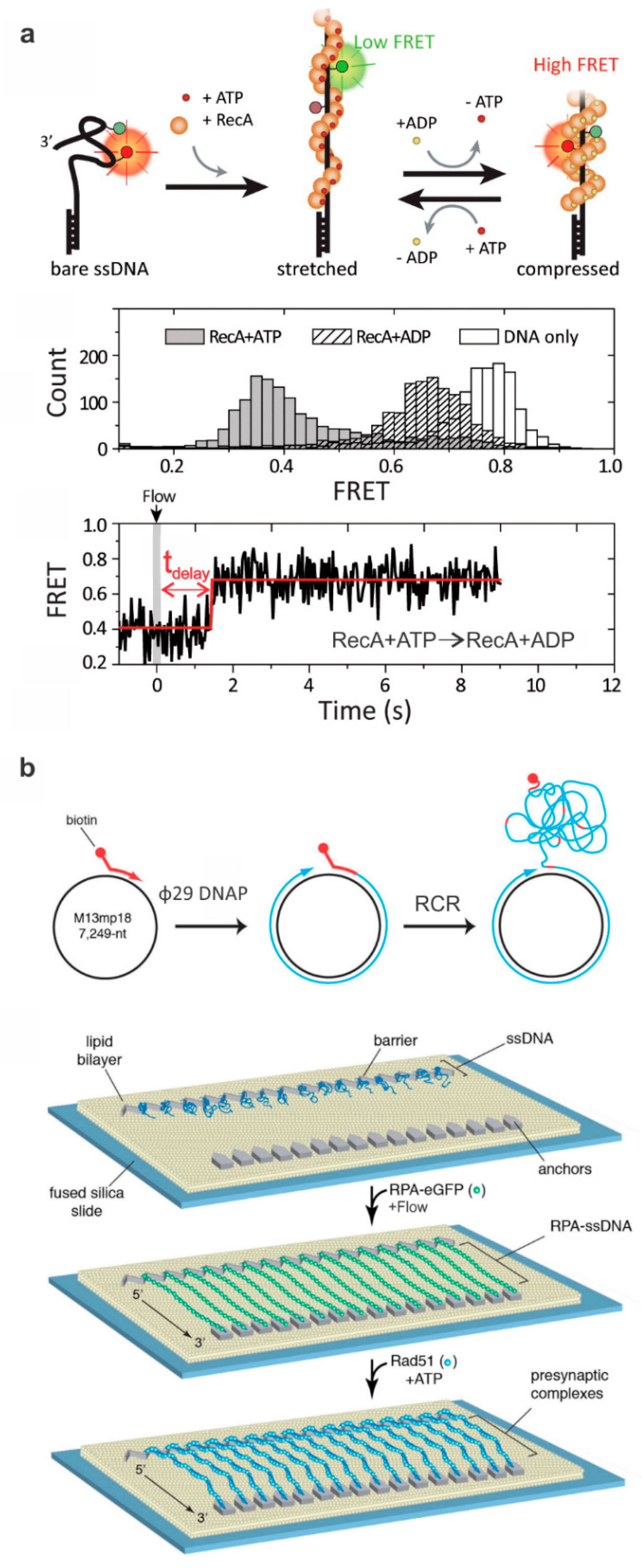
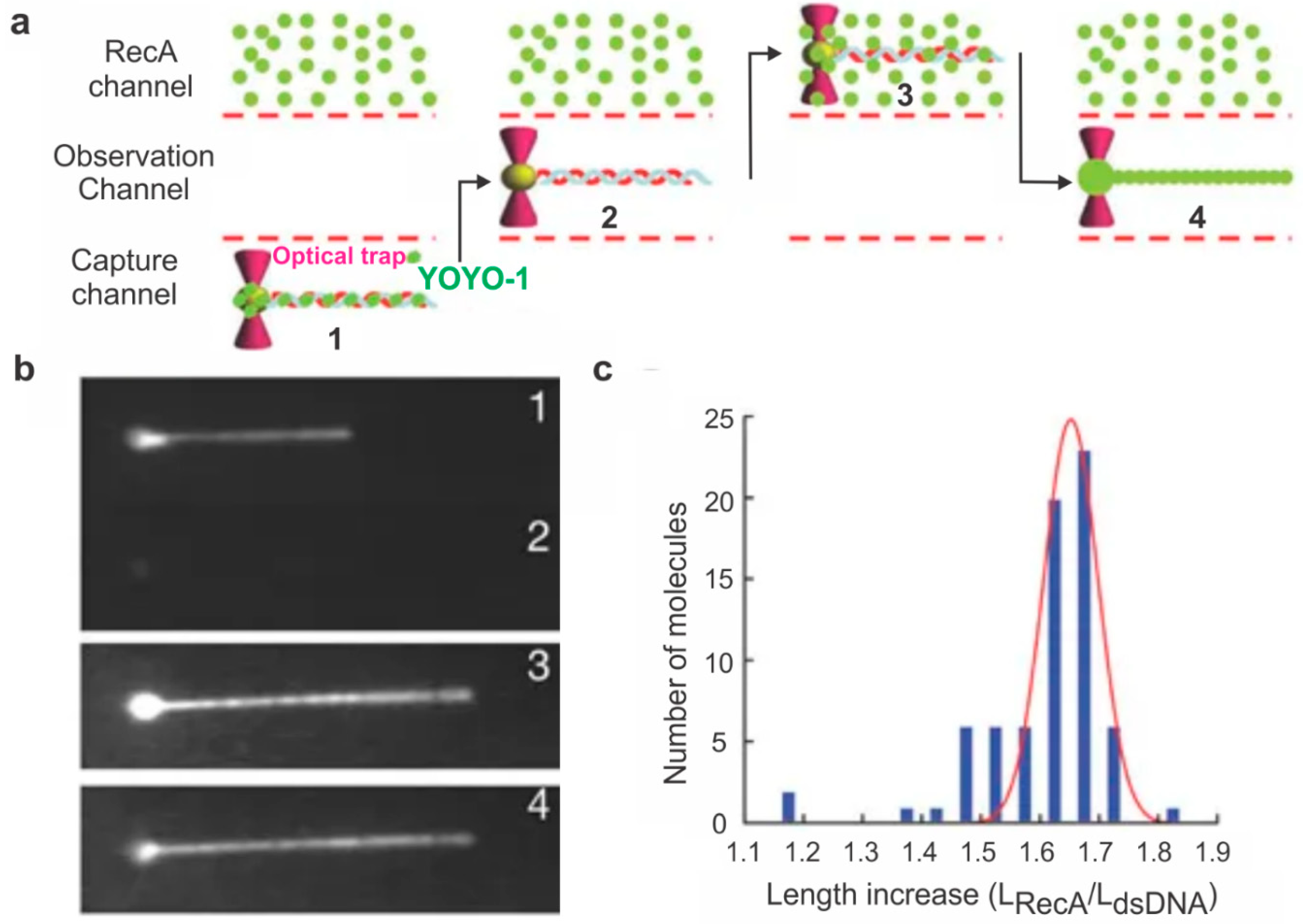
2. Probing HR Proteins Operating on ssDNA
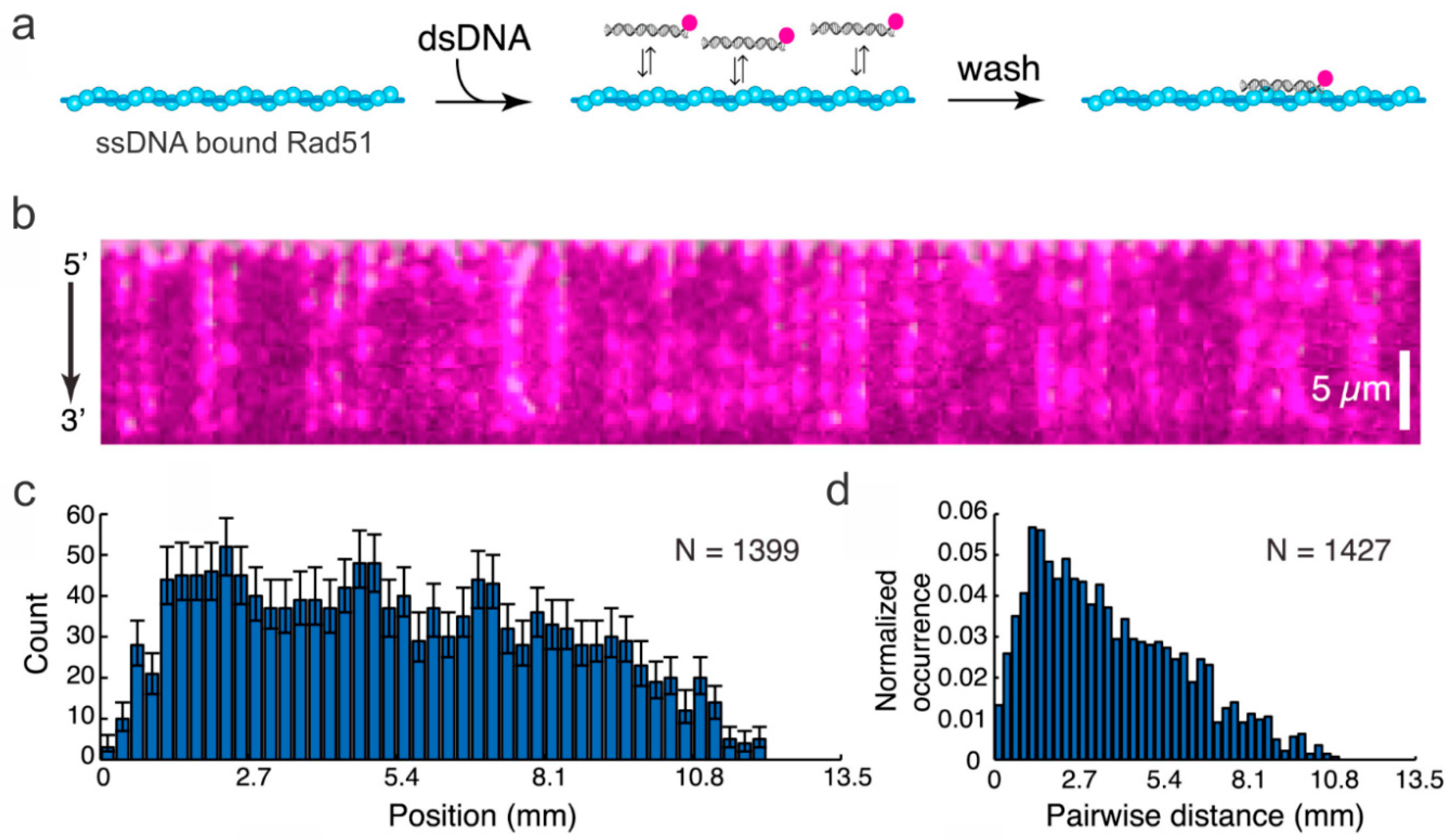
3. Probing HR Proteins Operating on dsDNA
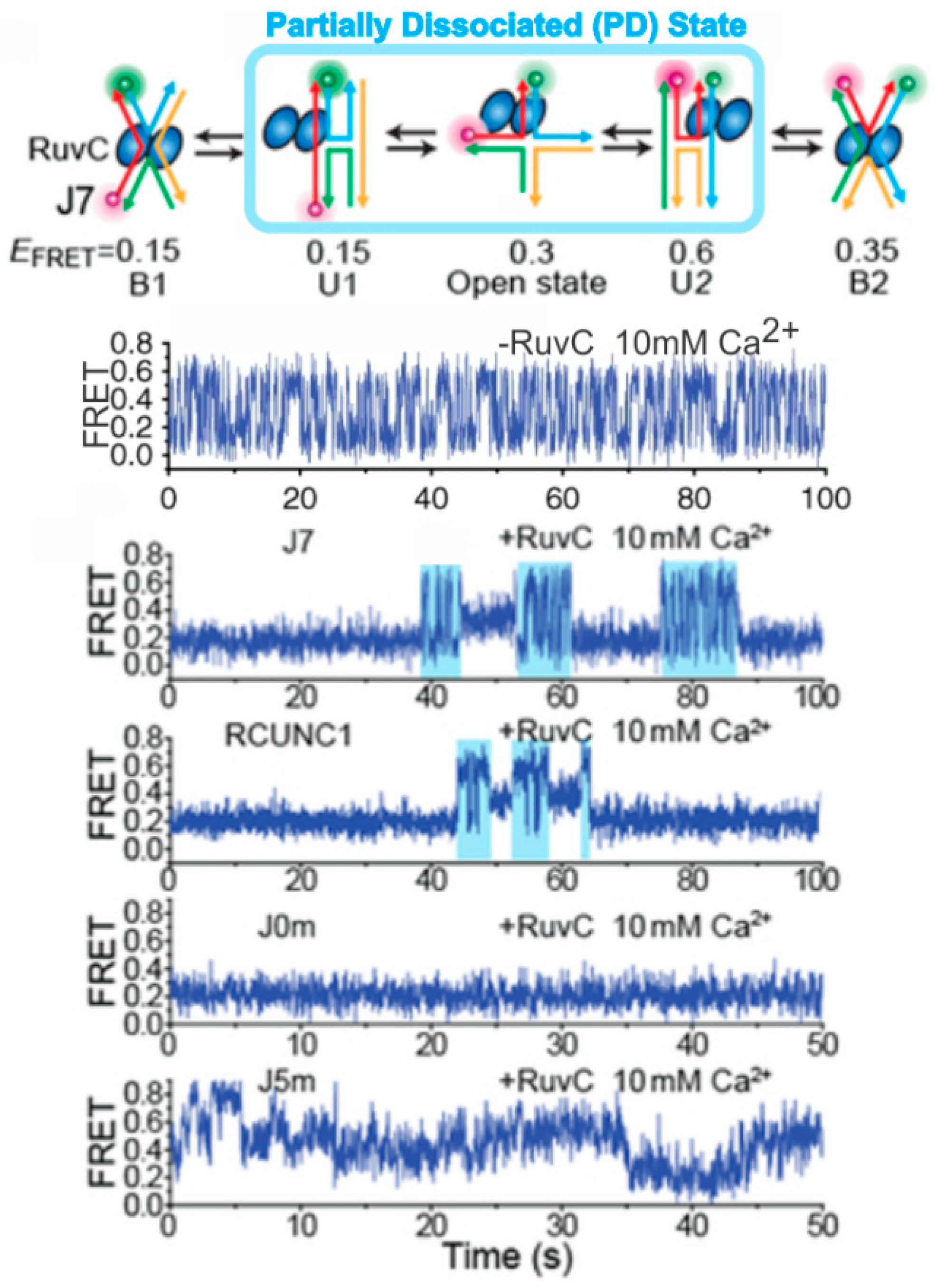
4. Probing HR Proteins Operating on Holliday Junctions
5. Discussion and Future Perspectives
Funding
Conflicts of Interest
References
- Li, X.; Heyer, W.-D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Camerini-Otero, R.D.; Hsieh, P. Homologous Recombination Proteins in Prokaryotes and Eukaryotes. Annu. Rev. Genet. 1995, 29, 509–552. [Google Scholar] [CrossRef] [PubMed]
- Jasin, M.; Rothstein, R. Repair of Strand Breaks by Homologous Recombination. Cold Spring Harb. Perspect. Biol. 2013, 5, a012740. [Google Scholar] [CrossRef] [PubMed]
- Ranjha, L.; Howard, S.M.; Cejka, P. Main steps in DNA double-strand break repair: An introduction to homologous recombination and related processes. Chromosoma 2018, 127, 187–214. [Google Scholar] [CrossRef] [PubMed]
- Yeeles, J.T.P.; Poli, J.; Marians, K.J.; Pasero, P. Rescuing Stalled or Damaged Replication Forks. Cold Spring Harb. Perspect. Biol. 2013, 5, a012815. [Google Scholar] [CrossRef]
- Carvalho, J.F.; Kanaar, R. Targeting homologous recombination-mediated DNA repair in cancer. Expert Opin. Ther. Targets 2014, 18, 427–458. [Google Scholar] [CrossRef]
- Kaniecki, K.; Tullio, L.D.; Greene, E.C. A change of view: Homologous recombination at single-molecule resolution. Nat. Rev. Genet. 2018, 19, 191–207. [Google Scholar] [CrossRef]
- Erie, D.A.; Weninger, K.R. Single molecule studies of DNA mismatch repair. DNA Repair (Amst.) 2014, 20, 71–81. [Google Scholar] [CrossRef]
- Gauer, J.W.; LeBlanc, S.; Hao, P.; Qiu, R.; Case, B.C.; Sakato, M.; Hingorani, M.M.; Erie, D.A.; Weninger, K.R. Single-Molecule FRET to Measure Conformational Dynamics of DNA Mismatch Repair Proteins. Methods Enzymol. 2016, 581, 285–315. [Google Scholar]
- Dupaigne, P.; Tavares, E.M.; Piétrement, O.; Le Cam, E. Recombinases and Related Proteins in the Context of Homologous Recombination Analyzed by Molecular Microscopy. Methods Mol. Biol. 2018, 1805, 251–270. [Google Scholar]
- Bell, J.C.; Kowalczykowski, S.C. Mechanics and Single-Molecule Interrogation of DNA Recombination. Annu. Rev. Biochem. 2016, 85, 193–226. [Google Scholar] [CrossRef] [PubMed]
- Roe, S.M.; Barlow, T.; Brown, T.; Oram, M.; Keeley, A.; Tsaneva, I.R.; Pearl, L.H. Crystal Structure of an Octameric RuvA–Holliday Junction Complex. Mol. Cell 1998, 2, 361–372. [Google Scholar] [CrossRef]
- Ariyoshi, M.; Vassylyev, D.G.; Iwasaki, H.; Nakamura, H.; Shinagawa, H.; Morikawa, K. Atomic structure of the RuvC resolvase: A holliday junction-specific endonuclease from E. coli. Cell 1994, 78, 1063–1072. [Google Scholar] [CrossRef]
- Hargreaves, D.; Rice, D.W.; Sedelnikova, S.E.; Artymiuk, P.J.; Lloyd, R.G.; Rafferty, J.B. Crystal structure of E.coli RuvA with bound DNA Holliday junction at 6 A resolution. Nat. Struct. Mol. Biol. 1998, 5, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, D.A.; Zittel, M.C.; Keck, J.L. High-resolution structure of the E. coli RecQ helicase catalytic core. Embo J. 2003, 22, 4910–4921. [Google Scholar] [CrossRef] [PubMed]
- Xing, X.; Bell, C.E. Crystal structures of Escherichia coli RecA in a compressed helical filament. J. Mol. Biol. 2004, 342, 1471–1485. [Google Scholar] [CrossRef]
- Brouwer, I.; Moschetti, T.; Candelli, A.; Garcin, E.B.; Modesti, M.; Pellegrini, L.; Wuite, G.J.; Peterman, E.J. Two distinct conformational states define the interaction of human RAD51-ATP with single-stranded DNA. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Osipiuk, J.; Li, H.; Bargassa, M.; Sahi, C.; Craig, E.A.; Joachimiak, A. Crystal structure of ATPase domain of Ssb1 chaperone, member of the HSP70 family from Saccharomyces cerevisiae. To be published, PDB 3GL1.
- Churchill, M.E.; Tullius, T.D.; Kallenbach, N.R.; Seeman, N.C. A Holliday recombination intermediate is twofold symmetric. Proc. Natl. Acad. Sci. USA 1988, 85, 4653–4656. [Google Scholar] [CrossRef]
- Duckett, D.R.; Murchie, A.I.H.; Diekmann, S.; von Kitzing, E.; Kemper, B.; Lilley, D.M.J. The structure of the Holliday junction, and its resolution. Cell 1988, 55, 79–89. [Google Scholar] [CrossRef]
- Petrillo, M.L.; Newton, C.J.; Cunningham, R.P.; Ma, R.-I.; Kallenbach, N.R.; Seeman, N.C. The ligation and flexibility of four-arm DNA junctions. Biopolymers 1988, 27, 1337–1352. [Google Scholar] [CrossRef]
- Mueller, J.E.; Newton, C.J.; Jensch, F.; Kemper, B.; Cunningham, R.P.; Kallenbach, N.R.; Seeman, N.C. Resolution of Holliday junction analogs by T4 endonuclease VII can be directed by substrate structure. J. Biol. Chem. 1990, 265, 13918–13924. [Google Scholar] [PubMed]
- Stasiak, A.; Tsaneva, I.R.; West, S.C.; Benson, C.J.; Yu, X.; Egelman, E.H. The Escherichia coli RuvB branch migration protein forms double hexameric rings around DNA. Proc. Natl. Acad. Sci. USA 1994, 91, 7618–7622. [Google Scholar] [CrossRef] [PubMed]
- Van Gool, A.J.; Hajibagheri, N.M.A.; Stasiak, A.; West, S.C. Assembly of the Escherichia coli RuvABC resolvasome directs the orientation of Holliday junction resolution. Genes Dev. 1999, 13, 1861–1870. [Google Scholar] [CrossRef] [PubMed]
- Jachymczyk, W.J.; von Borstel, R.C.; Mowat, M.R.; Hastings, P.J. Repair of interstrand cross-links in DNA of Saccharomyces cerevisiae requires two systems for DNA repair: The RAD3 system and the RAD51 system. Mol. Gen. Genet. 1981, 182, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Masson, J.-Y.; Shah, R.; O’Regan, P.; West, S.C. RAD51C Is Required for Holliday Junction Processing in Mammalian Cells. Science 2004, 303, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, H.D.M.; West, S.C. Holliday Junction Resolvases. Cold Spring Harb. Perspect. Biol. 2014, 6, a023192. [Google Scholar] [CrossRef]
- Ogawa, T.; Shinohara, A.; Nabetani, A.; Ikeya, T.; Yu, X.; Egelman, E.H.; Ogawa, H. RecA-like Recombination Proteins in Eukaryotes: Functions and Structures of RAD51 Genes. Cold Spring Harb. Symp. Quant. Biol. 1993, 58, 567–576. [Google Scholar] [CrossRef]
- Shinohara, A.; Ogawa, H.; Ogawa, T. Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell 1992, 69, 457–470. [Google Scholar] [CrossRef]
- Broussard, J.A.; Green, K.J. Research Techniques Made Simple: Methodology and Applications of Förster Resonance Energy Transfer (FRET) Microscopy. J. Investig. Dermatol. 2017, 137, e185–e191. [Google Scholar] [CrossRef]
- Roy, R.; Hohng, S.; Ha, T. A practical guide to single-molecule FRET. Nat. Methods 2008, 5, 507–516. [Google Scholar] [CrossRef]
- Marceau, A.H. Functions of Single-Strand DNA-Binding Proteins in DNA Replication, Recombination, and Repair. In Single-Stranded DNA Binding Proteins: Methods and Protocols; Keck, J.L., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, 2012; pp. 1–21. ISBN 978–1-62703–032-8. [Google Scholar]
- Smith, G.R. How RecBCD enzyme and Chi promote DNA break repair and recombination: A molecular biologist’s view. Microbiol. Mol. Biol. Rev. 2012, 76, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Croteau, D.L.; Popuri, V.; Opresko, P.L.; Bohr, V.A. Human RecQ Helicases in DNA Repair, Recombination, and Replication. Annu. Rev. Biochem. 2014, 83, 519–552. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.R. Homologous Recombination in Procaryotes. Microbiol. Rev. 1988, 52, 28. [Google Scholar]
- Henrikus, S.S.; Henry, C.; Ghodke, H.; Wood, E.A.; Mbele, N.; Saxena, R.; Basu, U.; van Oijen, A.M.; Cox, M.M.; Robinson, A. RecFOR epistasis group: RecF and RecO have distinct localizations and functions in Escherichia coli. Nucleic Acids Res. 2019, 47, 2946–2965. [Google Scholar] [CrossRef]
- Bell, J.C.; Plank, J.L.; Dombrowski, C.C.; Kowalczykowski, S.C. Direct imaging of RecA nucleation and growth on single molecules of SSB-coated ssDNA. Nature 2012, 491, 274–278. [Google Scholar] [CrossRef]
- Lilley, D.M.J. Holliday junction-resolving enzymes—Structures and mechanisms. FEBS Lett. 2017, 591, 1073–1082. [Google Scholar] [CrossRef]
- Joo, C.; McKinney, S.A.; Nakamura, M.; Rasnik, I.; Myong, S.; Ha, T. Real-Time Observation of RecA Filament Dynamics with Single Monomer Resolution. Cell 2006, 126, 515–527. [Google Scholar] [CrossRef]
- Kim, S.H.; Ragunathan, K.; Park, J.; Joo, C.; Kim, D.; Ha, T. Cooperative Conformational Transitions Keep RecA Filament Active During ATPase Cycle. J. Am. Chem. Soc. 2014, 136, 14796–14800. [Google Scholar] [CrossRef]
- Yang, S.-H.; Zhou, R.; Campbell, J.; Chen, J.; Ha, T.; Paull, T.T. The SOSS1 single-stranded DNA binding complex promotes DNA end resection in concert with Exo1. EMBO J. 2013, 32, 126–139. [Google Scholar] [CrossRef]
- Ragunathan, K.; Liu, C.; Ha, T. RecA filament sliding on DNA facilitates homology search. eLife 2012, 1, e00067. [Google Scholar] [CrossRef]
- Qi, Z.; Greene, E.C. Visualizing recombination intermediates with single-stranded DNA curtains. Methods 2016, 105, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.C.; Liu, B.; Kowalczykowski, S.C. Imaging and energetics of single SSB-ssDNA molecules reveal intramolecular condensation and insight into RecOR function. eLife 2015, 4, e08646. [Google Scholar] [CrossRef] [PubMed]
- Galletto, R.; Amitani, I.; Baskin, R.J.; Kowalczykowski, S.C. Direct observation of individual RecA filaments assembling on single DNA molecules. Nature 2006, 443, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Joo, C.; Ha, T.; Kim, D. Molecular mechanism of sequence-dependent stability of RecA filament. Nucleic Acids Res. 2013, 41, 7738–7744. [Google Scholar] [CrossRef]
- Lu, C.-H.; Yeh, H.-Y.; Su, G.-C.; Ito, K.; Kurokawa, Y.; Iwasaki, H.; Chi, P.; Li, H.-W. Swi5–Sfr1 stimulates Rad51 recombinase filament assembly by modulating Rad51 dissociation. Proc. Natl. Acad. Sci. USA 2018, 115, E10059–E10068. [Google Scholar] [CrossRef]
- Gibb, B.; Silverstein, T.D.; Finkelstein, I.J.; Greene, E.C. Single-Stranded DNA Curtains for Real-Time Single-Molecule Visualization of Protein–Nucleic Acid Interactions. Anal. Chem. 2012, 84, 7607–7612. [Google Scholar] [CrossRef]
- Gibb, B.; Ye, L.F.; Kwon, Y.; Niu, H.; Sung, P.; Greene, E.C. Protein dynamics during presynaptic-complex assembly on individual single-stranded DNA molecules. Nat. Struct. Mol. Biol. 2014, 21, 893–900. [Google Scholar]
- Ma, C.J.; Gibb, B.; Kwon, Y.; Sung, P.; Greene, E.C. Protein dynamics of human RPA and RAD51 on ssDNA during assembly and disassembly of the RAD51 filament. Nucleic Acids Res. 2017, 45, 749–761. [Google Scholar] [CrossRef]
- Myler, L.R.; Gallardo, I.F.; Zhou, Y.; Gong, F.; Yang, S.-H.; Wold, M.S.; Miller, K.M.; Paull, T.T.; Finkelstein, I.J. Single-molecule imaging reveals the mechanism of Exo1 regulation by single-stranded DNA binding proteins. Proc. Natl. Acad. Sci. USA 2016, 113, E1170–E1179. [Google Scholar] [CrossRef]
- Kaniecki, K.; Kwon, Y.; Crickard, J.B.; Sung, P.; Greene, E.C. Yeast Srs2 Helicase Promotes Redistribution of Single-Stranded DNA-Bound RPA and Rad52 in Homologous Recombination Regulation. Cell Rep. 2017, 21, 570–577. [Google Scholar]
- Kaniecki, K.; De Tullio, L.; Gibb, B.; Kwon, Y.; Sung, P.; Greene, E.C. Dissociation of Rad51 Presynaptic Complexes and Heteroduplex DNA Joints by Tandem Assemblies of Srs2. Cell Rep. 2017, 21, 3166–3177. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, I.; Zhang, H.; Candelli, A.; Normanno, D.; Peterman, E.J.G.; Wuite, G.J.L.; Modesti, M. Human RAD52 Captures and Holds DNA Strands, Increases DNA Flexibility, and Prevents Melting of Duplex DNA: Implications for DNA Recombination. Cell Rep. 2017, 18, 2845–2853. [Google Scholar] [CrossRef] [PubMed]
- Crickard, J.B.; Kwon, Y.; Sung, P.; Greene, E.C. Dynamic interactions of the homologous pairing 2 (Hop2)–meiotic nuclear divisions 1 (Mnd1) protein complex with meiotic presynaptic filaments in budding yeast. J. Biol. Chem. 2019, 294, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Forget, A.L.; Kowalczykowski, S.C. Single-molecule imaging of DNA pairing by RecA reveals a three-dimensional homology search. Nature 2012, 482, 423–427. [Google Scholar] [CrossRef]
- Bianco, P.R.; Brewer, L.R.; Corzett, M.; Balhorn, R.; Yeh, Y.; Kowalczykowski, S.C.; Baskin, R.J. Processive translocation and DNA unwinding by individual RecBCD enzyme molecules. Nature 2001, 409, 374–378. [Google Scholar] [CrossRef]
- Singleton, M.R.; Dillingham, M.S.; Gaudier, M.; Kowalczykowski, S.C.; Wigley, D.B. Crystal structure of RecBCD enzyme reveals a machine for processing DNA breaks. Nature 2004, 432, 187–193. [Google Scholar] [CrossRef]
- Yang, L.; Handa, N.; Liu, B.; Dillingham, M.S.; Wigley, D.B.; Kowalczykowski, S.C. Alteration of χ recognition by RecBCD reveals a regulated molecular latch and suggests a channel-bypass mechanism for biological control. Proc. Natl. Acad. Sci. USA 2012, 109, 8907–8912. [Google Scholar] [CrossRef]
- Liu, B.; Baskin, R.J.; Kowalczykowski, S.C. DNA unwinding heterogeneity by RecBCD results from static molecules able to equilibrate. Nature 2013, 500, 482–485. [Google Scholar] [CrossRef]
- Prasad, T.K.; Yeykal, C.C.; Greene, E.C. Visualizing the Assembly of Human Rad51 Filaments on Double-stranded DNA. J. Mol. Biol. 2006, 363, 713–728. [Google Scholar] [CrossRef]
- Yeykal, C.C.; Greene, E.C. Visualizing the Behavior of Human Rad51 at the Single-Molecule Level. Cell Cycle 2006, 5, 1033–1038. [Google Scholar] [CrossRef]
- Qi, Z.; Redding, S.; Lee, J.Y.; Gibb, B.; Kwon, Y.; Niu, H.; Gaines, W.A.; Sung, P.; Greene, E.C. DNA Sequence Alignment by Microhomology Sampling during Homologous Recombination. Cell 2015, 160, 856–869. [Google Scholar] [CrossRef] [PubMed]
- Rad, B.; Forget, A.L.; Baskin, R.J.; Kowalczykowski, S.C. Single-molecule visualization of RecQ helicase reveals DNA melting, nucleation, and assembly are required for processive DNA unwinding. Proc. Natl. Acad. Sci. USA 2015, 112, E6852–E6861. [Google Scholar] [CrossRef] [PubMed]
- McKinney, S.A.; Déclais, A.-C.; Lilley, D.M.J.; Ha, T. Structural dynamics of individual Holliday junctions. Nat. Struct. Biol. 2003, 10, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, D.R.; Dhakal, S. Single-Molecule Imaging Reveals Conformational Manipulation of Holliday Junction DNA by the Junction Processing Protein RuvA. Biochemistry 2018, 57, 3616–3624. [Google Scholar] [CrossRef] [PubMed]
- Karymov, M.; Daniel, D.; Sankey, O.F.; Lyubchenko, Y.L. Holliday junction dynamics and branch migration: Single-molecule analysis. Proc. Natl. Acad. Sci. USA 2005, 102, 8186–8191. [Google Scholar] [CrossRef]
- Joo, C.; McKinney, S.A.; Lilley, D.M.J.; Ha, T. Exploring Rare Conformational Species and Ionic Effects in DNA Holliday Junctions Using Single-molecule Spectroscopy. J. Mol. Biol. 2004, 341, 739–751. [Google Scholar] [CrossRef]
- Hohng, S.; Zhou, R.; Nahas, M.K.; Yu, J.; Schulten, K.; Lilley, D.M.J.; Ha, T. Fluorescence-Force Spectroscopy Maps Two-Dimensional Reaction Landscape of the Holliday Junction. Science 2007, 318, 279–283. [Google Scholar] [CrossRef]
- Lahiri, S.; Li, Y.; Hingorani, M.M.; Mukerji, I. MutSγ-Induced DNA Conformational Changes Provide Insights into Its Role in Meiotic Recombination. Biophys. J. 2018, 115, 2087–2101. [Google Scholar] [CrossRef]
- Zhao, D.; Mukerji, I.; Etson, C. Establishing a Single-Molecule FRET System for Studying DNA-Protein Interactions. Biophys. J. 2019, 116, 505a. [Google Scholar] [CrossRef]
- Iwasa, T.; Han, Y.-W.; Hiramatsu, R.; Yokota, H.; Nakao, K.; Yokokawa, R.; Ono, T.; Harada, Y. Synergistic effect of ATP for RuvA–RuvB–Holliday junction DNA complex formation. Sci. Rep. 2015, 5, 18177. [Google Scholar] [CrossRef]
- Zhou, R.; Yang, O.; Déclais, A.-C.; Jin, H.; Gwon, G.H.; Freeman, A.D.J.; Cho, Y.; Lilley, D.M.J.; Ha, T. Junction resolving enzymes use multivalency to keep the Holliday junction dynamic. Nat. Chem. Biol. 2019, 15, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Goyal, N.; Rossi, M.J.; Mazina, O.M.; Chi, Y.; Moritz, R.L.; Clurman, B.E.; Mazin, A.V. RAD54 N-terminal domain is a DNA sensor that couples ATP hydrolysis with branch migration of Holliday junctions. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Panyutin, I.G.; Hsieh, P. The kinetics of spontaneous DNA branch migration. Proc. Natl. Acad. Sci. USA 1994, 91, 2021–2025. [Google Scholar] [CrossRef] [PubMed]
- Hiom, K.; West, S.C. Branch migration during homologous recombination: Assembly of a RuvAB-holliday junction complex in vitro. Cell 1995, 80, 787–793. [Google Scholar] [CrossRef][Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gibbs, D.R.; Dhakal, S. Homologous Recombination under the Single-Molecule Fluorescence Microscope. Int. J. Mol. Sci. 2019, 20, 6102. https://doi.org/10.3390/ijms20236102
Gibbs DR, Dhakal S. Homologous Recombination under the Single-Molecule Fluorescence Microscope. International Journal of Molecular Sciences. 2019; 20(23):6102. https://doi.org/10.3390/ijms20236102
Chicago/Turabian StyleGibbs, Dalton R., and Soma Dhakal. 2019. "Homologous Recombination under the Single-Molecule Fluorescence Microscope" International Journal of Molecular Sciences 20, no. 23: 6102. https://doi.org/10.3390/ijms20236102
APA StyleGibbs, D. R., & Dhakal, S. (2019). Homologous Recombination under the Single-Molecule Fluorescence Microscope. International Journal of Molecular Sciences, 20(23), 6102. https://doi.org/10.3390/ijms20236102