Epidermolysis Bullosa-Associated Squamous Cell Carcinoma: From Pathogenesis to Therapeutic Perspectives

 , ,
, ,
Abstract
1. Introduction
2. SCC in the General Population
3. Wound Healing and SCC
4. Dystrophic EB
4.1. Clinical Features
4.2. DEB-SCC Genetics
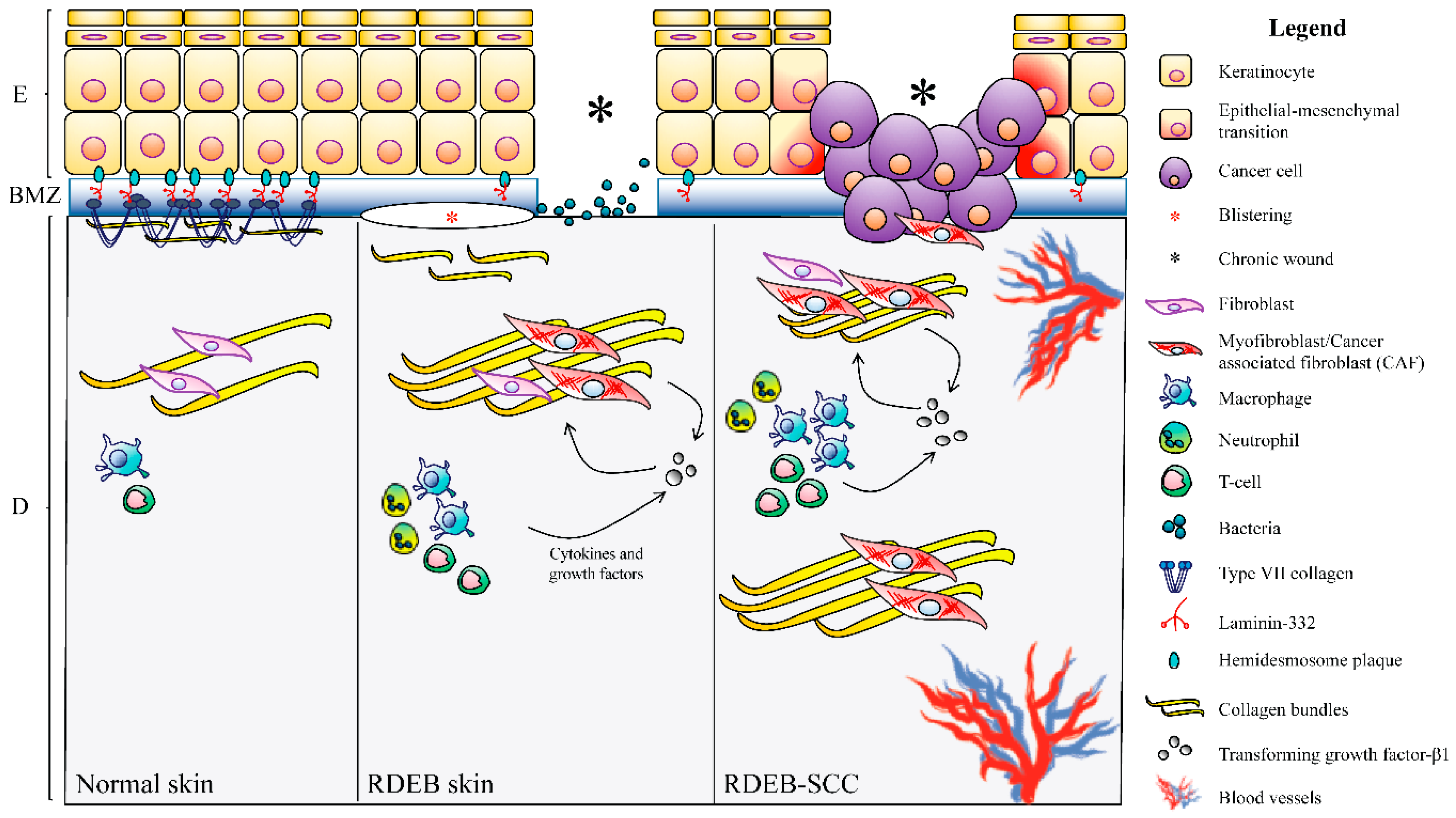
4.3. DEB-SCC Microenvironment
4.3.1. The Wound-Healing Process
4.3.2. Fibrosis
4.3.3. Intracellular Signaling
4.3.4. Inflammation
4.3.5. Microbial Infection
4.3.6. Immunity

4.4. Therapeutic Strategies: The Present and the Future
4.4.1. Current SCC Therapies
4.4.2. Therapeutic Perspectives
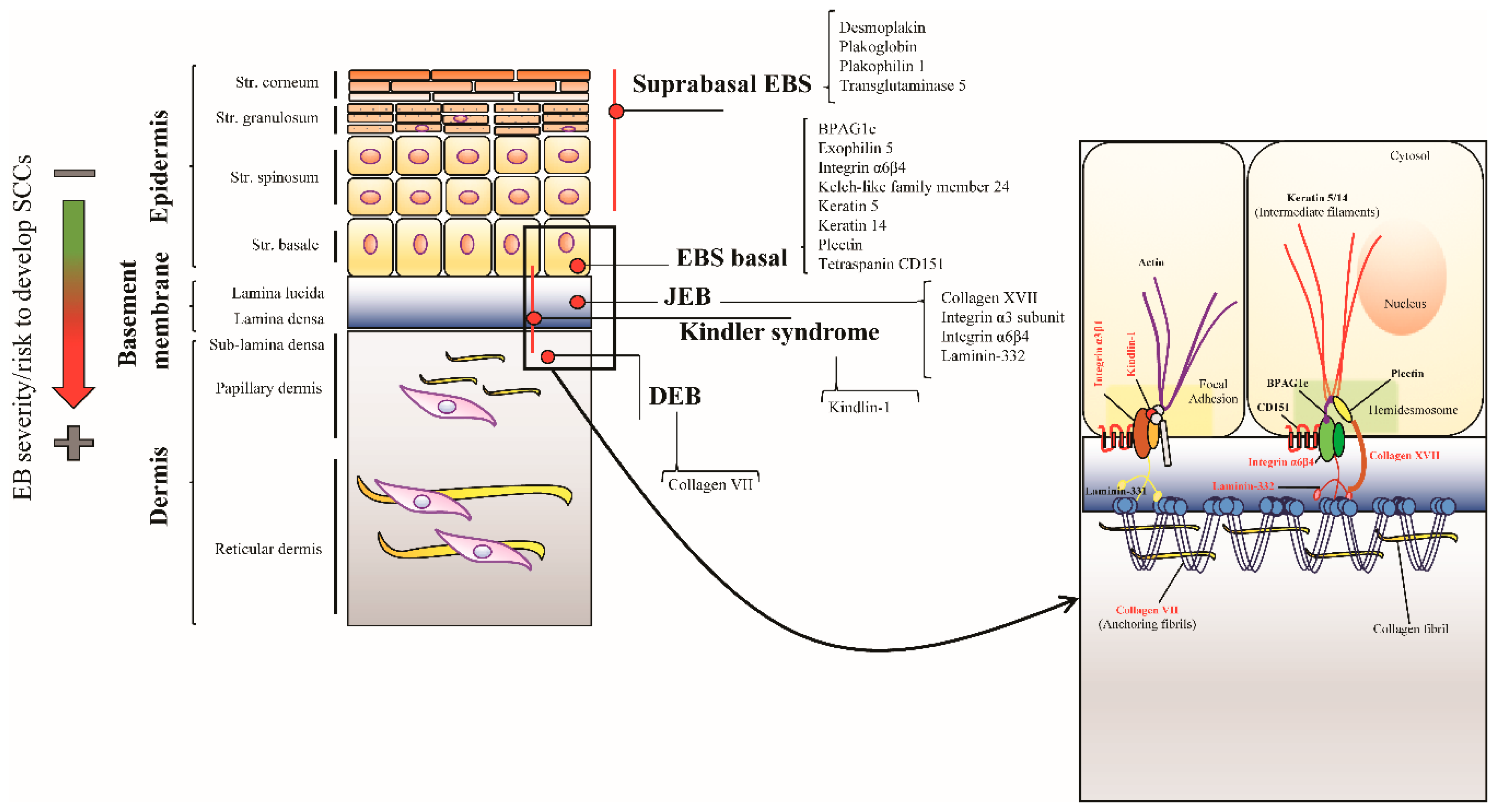
5. Junctional EB
5.1. Clinical Features
5.2. LM332 and COL17 in SCC in the General Population
5.3. LM332 and COL17 in SCC in JEB Patients
6. Kindler Syndrome
6.1. Clinical Features
6.2. Pathways Involved in KS-Related SCC Development
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EB | Epidermolysis bullosa |
| BMZ | Basement membrane zone |
| RDEB | Recessive dystrophic epidermolysis bullosa |
| JEB | Junctional epidermolysis bullosa |
| KS | Kindler syndrome |
| SCC | Squamous cell carcinoma |
| COL7 | Type VII collagen |
| ECM | Extracellular matrix |
| TNC | Tenascin-C |
| TGF-β1 | Transforming growth factor-β1 |
| DCN | Decorin |
| EMT | Epithelial-mesenchymal transition |
| IL-6 | Interleukin-6 |
| LM332 | Laminin-332 |
| COL17 | Type XVII collagen |
| LG45 | Laminin globular domains 4 and 5 |
| JEB-GI | Junctional epidermolysis bullosa, generalized intermediate subtype |
References
- Fine, J.D.; Bruckner-Tuderman, L.; Eady, R.A.; Bauer, E.A.; Bauer, J.W.; Has, C.; Heagerty, A.; Hintner, H.; Hovnanian, A.; Jonkman, M.F.; et al. Inherited epidermolysis bullosa: Updated recommendations on diagnosis and classification. J. Am. Acad. Dermatol. 2014, 70, 1103–1126. [Google Scholar] [CrossRef] [PubMed]
- Has, C.; Liu, L.; Bolling, M.C.; Charlesworth, A.V.; El Hachem, M.; Escámez, M.J.; Fuentes, I.; Büchel, S.; Hiremagalore, R.; Pohla-Gubo, G.; et al. Clinical practice guidelines for laboratory diagnosis of epidermolysis bullosa. Br. J. Dermatol. 2019, 15. [Google Scholar] [CrossRef] [PubMed]
- Chahal, H.S.; Rieger, K.E.; Sarin, K.Y. Incidence ratio of basal cell carcinoma to squamous cell carcinoma equalizes with age. J. Am. Acad. Dermatol. 2017, 76, 353–354. [Google Scholar] [CrossRef] [PubMed]
- Venables, Z.C.; Nijsten, T.; Wong, K.F.; Autier, P.; Broggio, J.; Deas, A.; Harwood, C.A.; Hollestein, L.M.; Langan, S.M.; Morgan, E.; et al. Epidemiology of basal and cutaneous squamous cell carcinoma in the U.K. 2013-15: A cohort study. Br. J. Dermatol. 2019, 181, 474–482. [Google Scholar] [CrossRef]
- Que, S.K.T.; Zwald, F.O.; Schmults, C.D. Cutaneous squamous cell carcinoma: Incidence, risk factors, diagnosis, and staging. J. Am. Acad. Dermatol. 2018, 78, 237–247. [Google Scholar] [CrossRef]
- Green, A.C.; Olsen, C.M. Cutaneous squamous cell carcinoma: An epidemiological review. Br. J. Dermatol. 2017, 177, 373–381. [Google Scholar] [CrossRef]
- Inman, G.J.; Wang, J.; Nagano, A.; Alexandrov, L.B.; Purdie, K.J.; Taylor, R.G.; Sherwood, V.; Thomson, J.; Hogan, S.; Spender, L.C.; et al. The genomic landscape of cutaneous SCC reveals drivers and a novel azathioprine associated mutational signature. Nat. Commun. 2018, 9, 3667. [Google Scholar] [CrossRef]
- Werner, R.N.; Stockfleth, E.; Connolly, S.M.; Correia, O.; Erdmann, R.; Foley, P.; Gupta, A.K.; Jacobs, A.; Kerl, H.; Lim, H.W.; et al. Evidence and consensus-based (S3) guidelines for the treatment of actinic keratosis – International League of Dermatological Societies in cooperation with the European Dermatology Forum–short version. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 2069–2079. [Google Scholar] [CrossRef]
- Werner, R.N.; Sammain, A.; Erdmann, R.; Hartmann, V.; Stockfleth, E.; Nast, A. The natural history of actinic keratosis: A systematic review. Br. J. Dermatol. 2013, 169, 502–518. [Google Scholar] [CrossRef]
- Chitsazzadeh, V.; Coarfa, C.; Drummond, J.A.; Nguyen, T.; Joseph, A.; Chilukuri, S.; Charpiot, E.; Adelmann, C.H.; Ching, G.; Nguyen, T.N.; et al. Cross-species identification of genomic drivers of squamous cell carcinoma development across preneoplastic intermediates. Nat. Commun. 2016, 7, 12601. [Google Scholar] [CrossRef]
- Albibas, A.A.; Rose-Zerilli, M.J.J.; Lai, C.; Pengelly, R.J.; Lockett, G.A.; Theaker, J.; Ennis, S.; Holloway, J.W.; Healy, E. Subclonal evolution of cancer-related gene mutations in p53 immunopositive patches in human skin. J. Investig. Dermatol. 2018, 138, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Skulsky, S.L.; O’Sullivan, B.; McArdle, O.; Leader, M.; Roche, M.; Conlon, P.J.; O’Neill, J.P. Review of high-risk features of cutaneous squamous cell carcinoma and discrepancies between the American Joint Committee on Cancer and NCCN clinical practice guidelines in oncology. Head Neck 2017, 39, 578–594. [Google Scholar] [CrossRef] [PubMed]
- Bazaliński, D.; Przybek-Mita, J.; Barańska, B.; Więch, P. Marjolin’s ulcer in chronic wounds - review of available literature. Contemp. Oncol. 2017, 21, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.S.; Jones, R.E.; Ransom, R.C.; Longaker, M.T.; Norton, J.A. The evolving relationship of wound healing and tumor stroma. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Su, S.; Workentine, M.; Agabalyan, N.; Cheng, M.; Gabriel, V.; Biernaskie, J. Transcriptional analysis reveals evidence of chronically impeded ECM turnover and epithelium-to-mesenchyme transition in scar tissue giving rise to Marjolin’s ulcer. J. Burn Care Res. 2017, 38, e14–e22. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar]
- Cianfarani, F.; Zambruno, G.; Castiglia, D.; Odorisio, T. Pathomechanisms of altered wound healing in recessive dystrophic epidermolysis bullosa. Am. J. Pathol. 2017, 187, 1445–1453. [Google Scholar] [CrossRef]
- Montaudié, H.; Chiaverini, C.; Sbidian, E.; Charlesworth, A.; Lacour, J.P. Inherited epidermolysis bullosa and squamous cell carcinoma: A systematic review of 117 cases. Orphanet. J. Rare Dis. 2016, 11, 117. [Google Scholar] [CrossRef]
- Fine, J.D. Epidemiology of inherited epidermolysis bullosa based on incidence and prevalence estimates from the national epidermolysis bullosa registry. JAMA Dermatol. 2016, 152, 1231–1238. [Google Scholar] [CrossRef]
- Hernandez-Martín, A.; Aranegui, B.; Escámez, M.J.; de Lucas, R.; Vicente, A.; Rodríguez-Díaz, E.; Bernabeu-Wittel, J.; Gonzalez-Hermosa, R.; García-Patos, V.; Ginarte, M.; et al. Prevalence of dystrophic epidermolysis bullosa in Spain: A population-based study using the 3-source capture-recapture method. Evidence of a need for improvement in care. Actas Dermosifiliogr. 2013, 104, 890–896. [Google Scholar] [CrossRef]
- Horn, H.M.; Priestley, G.C.; Eady, R.A.; Tidman, M.J. The prevalence of epidermolysis bullosa in Scotland. Br. J. Dermatol. 1997, 136, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Fine, J.D.; Mellerio, J.E. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: Part I. Epithelial associated tissues. J. Am. Acad. Dermatol. 2009, 61, 367–384. [Google Scholar] [CrossRef] [PubMed]
- Fine, J.D.; Mellerio, J.E. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: Part II. Other organs. J. Am. Acad. Dermatol. 2009, 61, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Has, C.; Nyström, A.; Saeidian, A.H.; Bruckner-Tuderman, L.; Uitto, J. Epidermolysis bullosa: Molecular pathology of connective tissue components in the cutaneous basement membrane zone. Matrix Biol. 2018, 71–72. [Google Scholar] [CrossRef]
- Fine, J.D.; Johnson, L.B.; Weiner, M.; Li, K.P.; Suchindran, C. Epidermolysis bullosa and the risk of life-threatening cancers: The National EB Registry experience, 1986–2006. J. Am. Acad. Dermatol. 2009, 60, 203–211. [Google Scholar] [CrossRef]
- Mellerio, J.E.; Robertson, S.J.; Bernardis, C.; Diem, A.; Fine, J.D.; George, R.; Goldberg, D.; Halmos, G.B.; Harries, M.; Jonkman, M.F.; et al. Management of cutaneous squamous cell carcinoma in patients with epidermolysis bullosa: Best clinical practice guidelines. Br. J. Dermatol. 2016, 174, 56–67. [Google Scholar] [CrossRef]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef]
- Jonason, A.S.; Kunala, S.; Price, G.J.; Restifo, R.J.; Spinelli, H.M.; Persing, J.A.; Leffell, D.J.; Tarone, R.E.; Brash, D.E. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc. Natl. Acad. Sci. USA 1996, 93, 14025–14029. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Sans-DeSanNicolas, L.; Caratú, G.; Vidal-Cortés, O.; Sanchez-Redondo, S.; Ferrer, B.; Mancuso, F.; González-Sanchez, E.; Pérez-Alea, M.; McGrail, K.; Hernandez-Losa, J.; et al. Genetic profiles of squamous cell carcinomas associated with recessive dystrophic epidermolysis bullosa unveil NOTCH and TP53 mutations and an increased MYC expression. J. Investig. Dermatol. 2018, 138, 1423–1427. [Google Scholar] [CrossRef]
- Cho, R.J.; Alexandrov, L.B.; den Breems, N.Y.; Atanasova, V.S.; Farshchian, M.; Purdom, E.; Nguyen, T.N.; Coarfa, C.; Rajapakshe, K.; Prisco, M.; et al. APOBEC mutation drives early-onset squamous cell carcinomas in recessive dystrophic epidermolysis bullosa. Sci. Transl. Med. 2018, 10, eaas9668. [Google Scholar] [CrossRef] [PubMed]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y.; et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Hanna, G.J.; Laga, A.C.; Haddad, R.I.; Lorch, J.H.; Hammerman, P.S. Genomic analysis of metastatic cutaneous squamous cell carcinoma. Clin. Cancer. Res. 2015, 21, 1447–1456. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, M.; Wolfer, A.; Raj, K.; Kummer, J.A.; Mill, P.; van Noort, M.; Hui, C.C.; Clevers, H.; Dotto, G.P.; Radtke, F. Notch1 functions as a tumor suppressor in mouse skin. Nat. Genet. 2003, 33, 416–421. [Google Scholar] [CrossRef]
- Dainichi, T.; Nakano, Y.; Wakae, K.; Otsuka, M.; Muramatsu, M.; Kabashima, K. APOBEC3 regulates keratinocyte differentiation and expression of Notch3. Exp. Dermatol. 2019, 10. [Google Scholar] [CrossRef]
- Silvas, T.V.; Schiffer, C.A. APOBEC3s: DNA-editing human cytidine deaminases. Protein Sci. 2019, 28, 1552–1566. [Google Scholar] [CrossRef]
- Petljak, M.; Alexandrov, L.B.; Brammeld, J.S.; Price, S.; Wedge, D.C.; Grossmann, S.; Dawson, K.J.; Ju, Y.S.; Iorio, F.; Tubio, J.M.C.; et al. Characterizing mutational signatures in human cancer cell lines reveals episodic APOBEC mutagenesis. Cell 2019, 176, 1282–1294.e20. [Google Scholar] [CrossRef]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef]
- Pakshir, P.; Hinz, B. The big five in fibrosis: Macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol. 2018, 68–69, 81–93. [Google Scholar] [CrossRef]
- Hinz, B.; McCulloch, C.A.; Coelho, N.M. Mechanical regulation of myofibroblast phenoconversion and collagen contraction. Exp. Cell Res. 2019, 379, 119–128. [Google Scholar] [CrossRef]
- Nyström, A.; Bruckner-Tuderman, L. Injury- and inflammation-driven skin fibrosis: The paradigm of epidermolysis bullosa. Matrix Biol. 2018, 68, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, A.G.; Logli, E.; Cianfarani, F.; Teson, M.; Diociaiuti, A.; El Hachem, M.; Zambruno, G.; Castiglia, D.; Odorisio, T. miR-145-5p regulates fibrotic features of recessive dystrophic epidermolysis bullosa skin fibroblasts. Br. J. Dermatol. 2019, 28. [Google Scholar] [CrossRef]
- Odorisio, T.; Di Salvio, M.; Orecchia, A.; Di Zenzo, G.; Piccinni, E.; Cianfarani, F.; Travaglione, A.; Uva, P.; Bellei, B.; Conti, A.; et al. Monozygotic twins discordant for recessive dystrophic epidermolysis bullosa phenotype highlight the role of TGF-β signalling in modifying disease severity. Hum. Mol. Genet. 2014, 23, 3907–3922. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, A.; Loeckermann, S.; Kern, J.S.; Braun, A.; Bösl, M.R.; Bley, T.A.; Schumann, H.; von Elverfeldt, D.; Paul, D.; Erlacher, M.; et al. A hypomorphic mouse model of dystrophic epidermolysis bullosa reveals mechanisms of disease and response to fibroblast therapy. J. Clin. Investig. 2008, 118, 1669–1679. [Google Scholar] [CrossRef]
- Nyström, A.; Thriene, K.; Mittapalli, V.; Kern, J.S.; Kiritsi, D.; Dengjel, J.; Bruckner-Tuderman, L. Losartan ameliorates dystrophic epidermolysis bullosa and uncovers new disease mechanisms. EMBO Mol. Med. 2015, 7, 1211–1228. [Google Scholar] [CrossRef]
- Cianfarani, F.; De Domenico, E.; Nyström, A.; Mastroeni, S.; Abeni, D.; Baldini, E.; Ulisse, S.; Uva, P.; Bruckner-Tuderman, L.; Zambruno, G.; et al. Decorin counteracts disease progression in mice with recessive dystrophic epidermolysis bullosa. Matrix Biol. 2019, 81, 3–16. [Google Scholar] [CrossRef]
- Atanasova, V.S.; Russell, R.J.; Webster, T.G.; Cao, Q.; Agarwal, P.; Lim, Y.Z.; Krishnan, S.; Fuentes, I.; Guttmann-Gruber, C.; McGrath, J.A.; et al. Thrombospondin-1 is a major activator of TGF-β signaling in recessive dystrophic epidermolysis bullosa fibroblasts. J. Investig. Dermatol. 2019, 139, 1497–1505. [Google Scholar] [CrossRef]
- Zhang, W.; Ge, Y.; Cheng, Q.; Zhang, Q.; Fang, L.; Zheng, J. Decorin is a pivotal effector in the extracellular matrix and tumour microenvironment. Oncotarget 2018, 9, 5480–5491. [Google Scholar] [CrossRef]
- Ng, Y.Z.; Pourreyron, C.; Salas-Alanis, J.C.; Dayal, J.H.; Cepeda-Valdes, R.; Yan, W.; Wright, S.; Chen, M.; Fine, J.D.; Hogg, F.J.; et al. Fibroblast-derived dermal matrix drives development of aggressive cutaneous squamous cell carcinoma in patients with recessive dystrophic epidermolysis bullosa. Cancer Res. 2012, 72, 3522–3534. [Google Scholar] [CrossRef]
- Guerra, L.; Odorisio, T.; Zambruno, G.; Castiglia, D. Stromal microenvironment in type VII collagen-deficient skin: The ground for squamous cell carcinoma development. Matrix Biol. 2017, 63, 1–10. [Google Scholar] [CrossRef]
- Liu, T.; Zhou, L.; Li, D.; Andl, T.; Zhang, Y. Cancer-associated fibroblasts build and secure the tumor microenvironment. Front. Cell Dev. Biol. 2019, 7, 60. [Google Scholar] [CrossRef] [PubMed]
- Küttner, V.; Mack, C.; Rigbolt, K.T.; Kern, J.S.; Schilling, O.; Busch, H.; Bruckner-Tuderman, L.; Dengjel, J. Global remodelling of cellular microenvironment due to loss of collagen VII. Mol. Syst. Biol. 2013, 9, 657. [Google Scholar] [CrossRef]
- Chacón-Solano, E.; León, C.; Díaz, F.; García-García, F.; García, M.; Escámez, M.J.; Guerrero-Aspizua, S.; Conti, C.J.; Mencía, Á.; Martínez-Santamaría, L.; et al. Fibroblast activation and abnormal extracellular matrix remodelling as common hallmarks in three cancer-prone genodermatoses. Br. J. Dermatol. 2019, 181, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Föll, M.C.; Fahrner, M.; Gretzmeier, C.; Thoma, K.; Biniossek, M.L.; Kiritsi, D.; Meiss, F.; Schilling, O.; Nyström, A.; Kern, J.S. Identification of tissue damage, extracellular matrix remodeling and bacterial challenge as common mechanisms associated with high-risk cutaneous squamous cell carcinomas. Matrix Biol. 2018, 66, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Kiraly, O.; Gong, G.; Olipitz, W.; Muthupalani, S.; Engelward, B.P. Inflammation-induced cell proliferation potentiates DNA damage-induced mutations in vivo. PLoS Genet. 2015, 11, e1004901. [Google Scholar] [CrossRef] [PubMed]
- Lechner, A.; Schlößer, H.A.; Thelen, M.; Wennhold, K.; Rothschild, S.I.; Gilles, R.; Quaas, A.; Siefer, O.G.; Huebbers, C.U.; Cukuroglu, E.; et al. Tumor-associated B cells and humoral immune response in head and neck squamous cell carcinoma. Oncoimmunology 2019, 8, 1535293. [Google Scholar] [CrossRef] [PubMed]
- Thriene, K.; Grüning, B.A.; Bornert, O.; Erxleben, A.; Leppert, J.; Athanasiou, I.; Weber, E.; Kiritsi, D.; Nyström, A.; Reinheckel, T.; et al. Combinatorial omics analysis reveals perturbed lysosomal homeostasis in collagen VII-deficient keratinocytes. Mol. Cell. Proteomics. 2018, 17, 565–579. [Google Scholar] [CrossRef]
- Martins, V.L.; Vyas, J.J.; Chen, M.; Purdie, K.; Mein, C.A.; South, A.P.; Storey, A.; McGrath, J.A.; O’Toole, E.A. Increased invasive behaviour in cutaneous squamous cell carcinoma with loss of basement-membrane type VII collagen. J. Cell Sci. 2009, 122, 1788–1799. [Google Scholar] [CrossRef]
- Martins, V.L.; Caley, M.P.; Moore, K.; Szentpetery, Z.; Marsh, S.T.; Murrell, D.F.; Kim, M.H.; Avari, M.; McGrath, J.A.; Cerio, R.; et al. Suppression of TGFβ and angiogenesis by type VII collagen in cutaneous SCC. J. Natl. Cancer Inst. 2015, 108, djv293. [Google Scholar] [CrossRef]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-β-mediated epithelial-mesenchymal transition and cancer metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef]
- Mittapalli, V.R.; Madl, J.; Löffek, S.; Kiritsi, D.; Kern, J.S.; Römer, W.; Nyström, A.; Bruckner-Tuderman, L. Injury-driven stiffening of the dermis expedites skin carcinoma progression. Cancer Res. 2016, 76, 940–951. [Google Scholar] [CrossRef] [PubMed]
- Dayal, J.H.; Cole, C.L.; Pourreyron, C.; Watt, S.A.; Lim, Y.Z.; Salas-Alanis, J.C.; Murrell, D.F.; McGrath, J.A.; Stieger, B.; Jahoda, C.; et al. Type VII collagen regulates expression of OATP1B3, promotes front-to-rear polarity and increases structural organisation in 3D spheroid cultures of RDEB tumour keratinocytes. J. Cell Sci. 2014, 127, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Woess, K.; Kienzl, M.; Leb-Reichl, V.M.; Feinle, A.; Wimmer, M.; Zauner, R.; Wally, V.; Luetz-Meindl, U.; Mellerio, J.E.; et al. Extracellular vesicles as biomarkers for the detection of a tumor marker gene in epidermolysis bullosa-associated squamous cell carcinoma. J. Investig. Dermatol. 2018, 138, 1197–1200. [Google Scholar] [CrossRef] [PubMed]
- Küttner, V.; Mack, C.; Gretzmeier, C.; Bruckner-Tuderman, L.; Dengjel, J. Loss of collagen VII is associated with reduced transglutaminase 2 abundance and activity. J. Investig. Dermatol. 2014, 134, 2381–2389. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.W.; Lee, S.H. The roles of autophagy in cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Shi, S.; Wu, B.; Zhang, J.; Li, Y.; Wu, X.; Zhang, J.; Wang, K.; Zhao, B.; Cai, W.; et al. Autophagy protein LC3 regulates the fibrosis of hypertrophic scar by controlling Bcl-xL in dermal fibroblasts. Oncotarget 2017, 8, 93757–93770. [Google Scholar] [CrossRef]
- Vanden Oever, M.; Muldoon, D.; Mathews, W.; McElmurry, R.; Tolar, J. miR-29 Regulates Type VII collagen in recessive dystrophic epidermolysis bullosa. J. Investig. Dermatol. 2016, 136, 2013–2021. [Google Scholar] [CrossRef]
- Bi, S.; Chai, L.; Yuan, X.; Cao, C.; Li, S. MicroRNA-98 inhibits the cell proliferation of human hypertrophic scar fibroblasts via targeting Col1A1. Biol. Res. 2017, 50, 22. [Google Scholar] [CrossRef]
- García-Sancha, N.; Corchado-Cobos, R.; Pérez-Losada, J.; Cañueto, J. MicroRNA dysregulation in cutaneous squamous cell carcinoma. Int. J. Mol. Sci. 2019, 20, 2181. [Google Scholar] [CrossRef]
- Liao, Y.; Ivanova, L.; Zhu, H.; Plumer, T.; Hamby, C.; Mehta, B.; Gevertz, A.; Christiano, A.M.; McGrath, J.A.; Cairo, M.S. Cord blood-derived stem cells suppress fibrosis and may prevent malignant progression in recessive dystrophic epidermolysis bullosa. Stem Cells 2018, 36, 1839–1850. [Google Scholar] [CrossRef]
- Esposito, S.; Guez, S.; Orenti, A.; Tadini, G.; Scuvera, G.; Corti, L.; Scala, A.; Biganzoli, E.; Berti, E.; Principi, N. Autoimmunity and cytokine imbalance in inherited epidermolysis bullosa. Int. J. Mol. Sci. 2016, 17, 1625. [Google Scholar] [CrossRef] [PubMed]
- Pedroza, M.; To, S.; Assassi, S.; Wu, M.; Tweardy, D.; Agarwal, S.K. Role of STAT3 in skin fibrosis and transforming growth factor beta signalling. Rheumatology 2018, 57, 1838–1850. [Google Scholar] [CrossRef] [PubMed]
- Karakasheva, T.A.; Lin, E.W.; Tang, Q.; Qiao, E.; Waldron, T.J.; Soni, M.; Klein-Szanto, A.J.; Sahu, V.; Basu, D.; Ohashi, S.; et al. IL-6 mediates cross-talk between tumor cells and activated fibroblasts in the tumor microenvironment. Cancer Res. 2018, 78, 4957–4970. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, M.M.; France, T.J.; Teknos, T.N.; Kumar, P. Interleukin-6 role in head and neck squamous cell carcinoma progression. World J. Otorhinolaryngol. Head Neck Surg. 2016, 2, 90–97. [Google Scholar] [CrossRef]
- Qiao, Y.; Zhang, C.; Li, A.; Wang, D.; Luo, Z.; Ping, Y.; Zhou, B.; Liu, S.; Li, H.; Yue, D.; et al. IL6 derived from cancer-associated fibroblasts promotes chemoresistance via CXCR7 in esophageal squamous cell carcinoma. Oncogene 2018, 37, 873–883. [Google Scholar] [CrossRef]
- Wu, X.; Tao, P.; Zhou, Q.; Li, J.; Yu, Z.; Wang, X.; Li, J.; Li, C.; Yan, M.; Zhu, Z.; et al. IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget 2017, 8, 20741–20750. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Kang, R.; Zhang, Q.; Zeh, H.J., 3rd; Lotze, M.T.; Tang, D. HMGB1 in cancer: Good, bad, or both? Clin. Cancer Res. 2013, 19, 4046–4057. [Google Scholar] [CrossRef]
- Tripathi, A.; Shrinet, K.; Kumar, A. HMGB1 protein as a novel target for cancer. Toxicol. Rep. 2019, 6, 253–261. [Google Scholar] [CrossRef]
- Sun, Y.; Tu, Y.; He, L.I.; Ji, C.; Cheng, B.O. High mobility group box 1 regulates tumor metastasis in cutaneous squamous cell carcinoma via the PI3K/AKT and MAPK signaling pathways. Oncol. Lett. 2016, 11, 59–62. [Google Scholar] [CrossRef][Green Version]
- Petrof, G.; Abdul-Wahab, A.; Proudfoot, L.; Pramanik, R.; Mellerio, J.E.; McGrath, J.A. Serum levels of high mobility group box 1 correlate with disease severity in recessive dystrophic epidermolysis bullosa. Exp. Dermatol. 2013, 22, 433–435. [Google Scholar] [CrossRef] [PubMed]
- Tamai, K.; Yamazaki, T.; Chino, T.; Ishii, M.; Otsuru, S.; Kikuchi, Y.; Iinuma, S.; Saga, K.; Nimura, K.; Shimbo, T.; et al. PDGFRalpha-positive cells in bone marrow are mobilized by high mobility group box 1 (HMGB1) to regenerate injured epithelia. Proc. Natl. Acad. Sci. USA 2011, 108, 6609–6614. [Google Scholar] [CrossRef] [PubMed]
- Hoste, E.; Arwert, E.N.; Lal, R.; South, A.P.; Salas-Alanis, J.C.; Murrell, D.F.; Donati, G.; Watt, F.M. Innate sensing of microbial products promotes wound-induced skin cancer. Nat. Commun. 2015, 6, 5932. [Google Scholar] [CrossRef] [PubMed]
- Nyström, A.; Bornert, O.; Kühl, T.; Gretzmeier, C.; Thriene, K.; Dengjel, J.; Pfister-Wartha, A.; Kiritsi, D.; Bruckner-Tuderman, L. Impaired lymphoid extracellular matrix impedes antibacterial immunity in epidermolysis bullosa. Proc. Natl. Acad. Sci. USA. 2018, 115, E705–E714. [Google Scholar] [CrossRef] [PubMed]
- Van der Kooi-Pol, M.M.; de Vogel, C.P.; Westerhout-Pluister, G.N.; Veenstra-Kyuchukova, Y.K.; Duipmans, J.C.; Glasner, C.; Buist, G.; Elsinga, G.S.; Westra, H.; Bonarius, H.P.J.; et al. High anti-staphylococcal antibody titers in patients with epidermolysis bullosa relate to long-term colonization with alternating types of Staphylococcus aureus. J. Investig. Dermatol. 2013, 133, 847–850. [Google Scholar] [CrossRef]
- Vindenes, H.; Bjerknes, R. Microbial colonization of large wounds. Burns 1995, 21, 575–579. [Google Scholar] [CrossRef]
- Purdie, K.J.; Pourreyron, C.; Fassihi, H.; Cepeda-Valdes, R.; Frew, J.W.; Volz, A.; Weissenborn, S.J.; Pfister, H.; Proby, C.M.; Bruckner-Tuderman, L.; et al. No evidence that human papillomavirus is responsible for the aggressive nature of recessive dystrophic epidermolysis bullosa-associated squamous cell carcinoma. J. Investig. Dermatol. 2010, 130, 2853–2855. [Google Scholar] [CrossRef]
- Bottomley, M.J.; Thomson, J.; Harwood, C.; Leigh, I. The role of the immune system in cutaneous squamous cell carcinoma. Int. J. Mol. Sci. 2019, 20, 2009. [Google Scholar] [CrossRef]
- Riihilä, P.; Viiklepp, K.; Nissinen, L.; Farshchian, M.; Kallajoki, M.; Kivisaari, A.; Meri, S.; Peltonen, J.; Peltonen, S.; Kähäri, V.M. Tumour-cell-derived complement components C1r and C1s promote growth of cutaneous squamous cell carcinoma. Br. J. Dermatol. 2019, 3. [Google Scholar] [CrossRef]
- Kim, M.; Li, M.; Intong, L.R.; Tran, K.; Melbourne, W.; Marucci, D.; Bucci, J.; de Souza, P.; Mallesara, G.; Murrell, D.F. Use of cetuximab as an adjuvant agent to radiotherapy and surgery in recessive dystrophic epidermolysis bullosa with squamous cell carcinoma. Br. J. Dermatol. 2013, 169, 208–210. [Google Scholar] [CrossRef]
- Arnold, A.W.; Bruckner-Tuderman, L.; Zuger, C.; Itin, P.H. Cetuximab therapy of metastasizing cutaneous squamous cell carcinoma in a patient with severe recessive dystrophic epidermolysis bullosa. Dermatology 2009, 219, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Medek, K.; Koelblinger, P.; Koller, J.; Diem, A.; Ude-Schoder, K.; Bauer, J.W.; Laimer, M. Wound healing deficits in severe generalized recessive dystrophic epidermolysis bullosa along anticancer treatment with cetuximab. J. Dtsch. Dermatol. Ges. 2019, 17, 448–450. [Google Scholar] [CrossRef] [PubMed]
- Reimer, A.; Lu, S.; He, Y.; Bruckner-Tuderman, L.; Technau-Hafsi, K.; Meiss, F.; Has, C.; von Bubnoff, D. Combined anti-inflammatory and low-dose antiproliferative therapy for squamous cell carcinomas in recessive dystrophic epidermolysis bullosa. J. Eur. Acad. Dermatol. Venereol. 2019, 2. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Li, M.; Intong-Wheeler, L.R.A.; Tran, K.; Marucci, D.; Murrell, D.F. Epidemiology and outcome of squamous cell carcinoma in epidermolysis bullosa in Australia and New Zealand. Acta Derm. Venereol. 2018, 98, 70–76. [Google Scholar] [CrossRef]
- Tamai, K.; Yamazaki, S.; Wang, X.; Nishida, M.; Shimbo, T.; Kikuchi, Y.; Bruckner-Tuderman, L.; Uitto, J.; Katayama, I.; Kaneda, Y. 179 Systemic administration of HMGB1 peptide drastically improves survival of the RDEB model mice by mobilizing multipotent stem/progenitor cells from bone marrow. J. Investig. Dermatol. 2017, 137, S223. [Google Scholar] [CrossRef]
- Bellomo, F.; Medina, D.L.; De Leo, E.; Panarella, A.; Emma, F. High-content drug screening for rare diseases. J. Inherit. Metab. Dis. 2017, 40, 601–607. [Google Scholar] [CrossRef]
- Osborn, M.J.; Newby, G.A.; McElroy, A.N.; Knipping, F.; Nielsen, S.C.; Riddle, M.J.; Xia, L.; Chen, W.; Eide, C.R.; Webber, B.R.; et al. Base editor correction of COL7A1 in recessive dystrophic epidermolysis bullosa patient-derived fibroblasts and iPSCs. J. Investig. Dermatol. 2019, 19. [Google Scholar] [CrossRef]
- Lwin, S.M.; Syed, F.; Di, W.L.; Kadiyirire, T.; Liu, L.; Guy, A.; Petrova, A.; Abdul-Wahab, A.; Reid, F.; Phillips, R.; et al. Safety and early efficacy outcomes for lentiviral fibroblast gene therapy in recessive dystrophic epidermolysis bullosa. JCI Insight 2019, 4, 126243. [Google Scholar] [CrossRef]
- Gaucher, S.; Lwin, S.M.; Titeux, M.; Abdul-Wahab, A.; Pironon, N.; Izmiryan, A.; Miskinyte, S.; Ganier, C.; Duchatelet, S.; Mellerio, J.E.; et al. EBGene trial: Patient pre-selection outcomes for the European GENEGRAFT ex vivo phase I/II gene therapy trial for recessive dystrophic epidermolysis bullosa. Br. J. Dermatol. 2019, 26. [Google Scholar] [CrossRef]
- Mencía, Á.; Chamorro, C.; Bonafont, J.; Duarte, B.; Holguin, A.; Illera, N.; Llames, S.G.; Escámez, M.J.; Hausser, I.; Del Río, M.; et al. Deletion of a pathogenic mutation-containing exon of COL7A1 allows clonal gene editing correction of RDEB patient epidermal stem cells. Mol. Ther. Nucleic Acids. 2018, 11, 68–78. [Google Scholar] [CrossRef]
- Bonafont, J.; Mencía, Á.; García, M.; Torres, R.; Rodríguez, S.; Carretero, M.; Chacón-Solano, E.; Modamio-Høybjør, S.; Marinas, L.; León, C.; et al. Clinically relevant correction of recessive dystrophic epidermolysis bullosa by dual sgRNA CRISPR/Cas9-mediated gene editing. Mol Ther. 2019, 27, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Droz-Georget Lathion, S.; Rochat, A.; Knott, G.; Recchia, A.; Martinet, D.; Benmohammed, S.; Grasset, N.; Zaffalon, A.; Besuchet Schmutz, N.; Savioz-Dayer, E.; et al. A single epidermal stem cell strategy for safe ex vivo gene therapy. EMBO Mol. Med. 2015, 7, 380–393. [Google Scholar] [CrossRef] [PubMed]
- Siprashvili, Z.; Nguyen, N.T.; Gorell, E.S.; Loutit, K.; Khuu, P.; Furukawa, L.K.; Lorenz, H.P.; Leung, T.H.; Keene, D.R.; Rieger, K.E.; et al. Safety and wound outcomes following genetically corrected autologous epidermal grafts in patients with recessive dystrophic epidermolysis bullosa. JAMA 2016, 316, 1808–1817. [Google Scholar] [CrossRef] [PubMed]
- Eichstadt, S.; Barriga, M.; Ponakala, A.; Teng, C.; Nguyen, N.T.; Siprashvili, Z.; Nazaroff, J.; Gorell, E.S.; Chiou, A.S.; Taylor, L.; et al. Phase 1/2a clinical trial of gene-corrected autologous cell therapy for recessive dystrophic epidermolysis bullosa. JCI Insight 2019, 4, 130554. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, T.; Rothoeft, T.; Teig, N.; Bauer, J.W.; Pellegrini, G.; De Rosa, L.; Scaglione, D.; Reichelt, J.; Klausegger, A.; Kneisz, D.; et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature 2017, 551, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.S.; Loeckermann, S.; Fritsch, A.; Hausser, I.; Roth, W.; Magin, T.M.; Mack, C.; Müller, M.L.; Paul, O.; Ruther, P.; et al. Mechanisms of fibroblast cell therapy for dystrophic epidermolysis bullosa: High stability of collagen VII favors long-term skin integrity. Mol. Ther. 2009, 17, 1605–1615. [Google Scholar] [CrossRef]
- Kühl, T.; Mezger, M.; Hausser, I.; Handgretinger, R.; Bruckner-Tunderman, L.; Nyström, A. High local concentrations of intradermal MSCs restore skin integrity and facilitate wound healing in dystrophic epidermolysis bullosa. Mol. Ther. 2015, 23, 1368–1379. [Google Scholar] [CrossRef]
- Tolar, J.; Ishida-Yamamoto, A.; Riddle, M.; McElmurry, R.T.; Osborn, M.; Xia, L.; Lund, T.; Slattery, C.; Uitto, J.; Christiano, A.M.; et al. Amelioration of epidermolysis bullosa by transfer of wild-type bone marrow cells. Blood 2009, 113, 1167–1174. [Google Scholar] [CrossRef]
- Iinuma, S.; Aikawa, E.; Tamai, K.; Fujita, R.; Kikuchi, Y.; Chino, T.; Kikuta, J.; McGrath, J.A.; Uitto, J.; Ishii, M.; et al. Transplanted bone marrow-derived circulating PDGFRα+ cells restore type VII collagen in recessive dystrophic epidermolysis bullosa mouse skin graft. J. Immunol. 2015, 194, 1996–2003. [Google Scholar] [CrossRef]
- Liao, Y.; Itoh, M.; Yang, A.; Zhu, H.; Roberts, S.; Highet, A.M.; Latshaw, S.; Mitchell, K.; van de Ven, C.; Christiano, A.; et al. Human cord blood-derived unrestricted somatic stem cells promote wound healing and have therapeutic potential for patients with recessive dystrophic epidermolysis bullosa. Cell Transplant. 2014, 23, 303–317. [Google Scholar] [CrossRef]
- Liao, Y.; Ivanova, L.; Zhu, H.; Yahr, A.; Ayello, J.; van de Ven, C.; Rashad, A.; Uitto, J.; Christiano, A.M.; Cairo, M.S. Rescue of the mucocutaneous manifestations by human cord blood derived nonhematopoietic stem cells in a mouse model of recessive dystrophic epidermolysis bullosa. Stem Cells. 2015, 33, 1807–1817. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, M.K.; Otero-Vinas, M.; Falanga, V. Transforming growth factor beta (TGF-β) isoforms in wound healing and fibrosis. Wound Repair Regen. 2016, 24, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.; Gammon, L.; Liu, L.; Mellerio, J.E.; Dopping-Hepenstal, P.J.; Pacy, J.; Elia, G.; Jeffery, R.; Leigh, I.M.; Navsaria, H.; et al. Potential of fibroblast cell therapy for recessive dystrophic epidermolysis bullosa. J. Investig. Dermatol. 2008, 128, 2179–2189. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.E.; Ishida-Yamamoto, A.; McGrath, J.A.; Hordinsky, M.; Keene, D.R.; Woodley, D.T.; Chen, M.; Riddle, M.J.; Osborn, M.J.; Lund, T.; et al. Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. N. Engl. J. Med. 2010, 363, 629–639. [Google Scholar] [CrossRef]
- Petrof, G.; Martinez-Queipo, M.; Mellerio, J.E.; Kemp, P.; McGrath, J.A. Fibroblast cell therapy enhances initial healing in recessive dystrophic epidermolysis bullosa wounds: Results of a randomized, vehicle-controlled trial. Br. J. Dermatol. 2013, 169, 1025–1033. [Google Scholar] [CrossRef]
- Petrof, G.; Lwin, S.M.; Martinez-Queipo, M.; Abdul-Wahab, A.; Tso, S.; Mellerio, J.E.; Slaper-Cortenbach, I.; Boelens, J.J.; Tolar, J.; Veys, P.; et al. Potential of systemic allogenic mesenchymal stromal cell therapy for children with recessive dystrophic epidermolysis bullosa. J. Investig. Dermatol. 2015, 135, 2319–2321. [Google Scholar] [CrossRef]
- Ebens, C.L.; McGrath, J.A.; Tamai, K.; Hovnanian, A.; Wagner, J.E.; Riddle, M.J.; Keene, D.R.; DeFor, T.E.; Tryon, R.; Chen, M.; et al. Bone marrow transplant with post-transplant cyclophosphamide for recessive dystrophic epidermolysis bullosa expands the related donor pool and permits tolerance of nonhaematopoietic cellular grafts. Br. J. Dermatol. 2019, 6. [Google Scholar] [CrossRef]
- Remington, J.; Wang, X.; Hou, Y.; Zhou, H.; Burnett, J.; Muirhead, T.; Uitto, J.; Keene, D.R.; Woodley, D.T.; Chen, M. Injection of recombinant human type VII collagen corrects the disease phenotype in a murine model of dystrophic epidermolysis bullosa. Mol. Ther. 2009, 17, 26–33. [Google Scholar] [CrossRef]
- Bruckner-Tuderman, L. Can type VII collagen injections cure dystrophic epidermolysis bullosa? Mol. Ther. 2009, 17, 6–7. [Google Scholar] [CrossRef]
- Bremer, J.; Bornert, O.; Nyström, A.; Gostynski, A.; Jonkman, M.F.; Aartsma-Rus, A.; van den Akker, P.C.; Pasmooij, A.M. Antisense oligonucleotide-mediated exon skipping as a systemic therapeutic approach for recessive dystrophic epidermolysis bullosa. Mol. Ther. Nucleic Acids. 2016, 5, e379. [Google Scholar] [CrossRef]
- Turczynski, S.; Titeux, M.; Tonasso, L.; Décha, A.; Ishida-Yamamoto, A.; Hovnanian, A. Targeted exon skipping restores Type VII collagen expression and anchoring fibril formation in an in vivo RDEB model. J. Investig. Dermatol. 2016, 136, 2387–2395. [Google Scholar] [CrossRef] [PubMed]
- Woodley, D.T.; Cogan, J.; Hou, Y.; Lyu, C.; Marinkovich, M.P.; Keene, D.; Chen, M. Gentamicin induces functional type VII collagen in recessive dystrophic epidermolysis bullosa patients. J. Clin. Investig. 2017, 127, 3028–3038. [Google Scholar] [CrossRef] [PubMed]
- Atanasova, V.S.; Jiang, Q.; Prisco, M.; Gruber, C.; Piñón Hofbauer, J.; Chen, M.; Has, C.; Bruckner-Tuderman, L.; McGrath, J.A.; Uitto, J.; et al. Amlexanox enhances premature termination codon read-through in COL7A1 and expression of full length type VII collagen: Potential therapy for recessive dystrophic epidermolysis bullosa. J. Investig. Dermatol. 2017, 137, 1842–1849. [Google Scholar] [CrossRef] [PubMed]
- Watt, S.A.; Pourreyron, C.; Purdie, K.; Hogan, C.; Cole, C.L.; Foster, N.; Pratt, N.; Bourdon, J.C.; Appleyard, V.; Murray, K.; et al. Integrative mRNA profiling comparing cultured primary cells with clinical samples reveals PLK1 and C20orf20 as therapeutic targets in cutaneous squamous cell carcinoma. Oncogene 2011, 30, 4666–4677. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, Q.; Wang, X. PLK1, a potential target for cancer therapy. Transl. Oncol. 2017, 10, 22–32. [Google Scholar] [CrossRef]
- Atanasova, V.S.; Pourreyron, C.; Farshchian, M.; Lawler, M.; Brown, C.A., 4th; Watt, S.A.; Wright, S.; Warkala, M.; Guttmann-Gruber, C.; Hofbauer, J.P.; et al. Identification of rigosertib for the treatment of recessive dystrophic epidermolysis bullosa-associated squamous cell carcinoma. Clin. Cancer Res. 2019, 25, 3384–3391. [Google Scholar] [CrossRef]
- Yuen, W.Y.; Lemmink, H.H.; van Dijk-Bos, K.K.; Sinke, R.J.; Jonkman, M.F. Herlitz junctional epidermolysis bullosa: Diagnostic features, mutational profile, incidence and population carrier frequency in the Netherlands. Br. J. Dermatol. 2011, 165, 1314–1322. [Google Scholar] [CrossRef]
- Yuen, W.Y.; Duipmans, J.C.; Molenbuur, B.; Herpertz, I.; Mandema, J.M.; Jonkman, M.F. Long-term follow-up of patients with Herlitz-type junctional epidermolysis bullosa. Br. J. Dermatol. 2012, 167, 374–382. [Google Scholar] [CrossRef]
- Yuen, W.Y.; Jonkman, M.F. Risk of squamous cell carcinoma in junctional epidermolysis bullosa, non-Herlitz type: Report of 7 cases and a review of the literature. J. Am. Acad. Dermatol. 2011, 65, 780–789. [Google Scholar] [CrossRef]
- Rousselle, P.; Beck, K. Laminin 332 processing impacts cellular behaviour. Cell. Adh. Migr. 2013, 7, 122–134. [Google Scholar] [CrossRef]
- Senyürek, I.; Kempf, W.E.; Klein, G.; Maurer, A.; Kalbacher, H.; Schäfer, L.; Wanke, I.; Christ, C.; Stevanovic, S.; Schaller, M.; et al. Processing of laminin α chains generates peptides involved in wound healing and host defense. J. Innate Immun. 2014, 6, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.E.; Carter, W.G. Laminin 5 deposition regulates keratinocyte polarization and persistent migration. J. Cell Sci. 2004, 117, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.; Rousselle, P.; Nokelainen, P.; Tallapragada, S.; Nguyen, N.T.; Fincher, E.F.; Marinkovich, M.P. Targeting a tumor-specific laminin domain critical for human carcinogenesis. Cancer Res. 2008, 68, 2885–2894. [Google Scholar] [CrossRef] [PubMed]
- Dajee, M.; Lazarov, M.; Zhang, J.Y.; Cai, T.; Green, C.L.; Russell, A.J.; Marinkovich, M.P.; Tao, S.; Lin, Q.; Kubo, Y.; et al. NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature 2003, 421, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Waterman, E.A.; Sakai, N.; Nguyen, N.T.; Horst, B.A.; Veitch, D.P.; Dey, C.N.; Ortiz-Urda, S.; Khavari, P.A.; Marinkovich, M.P. A laminin-collagen complex drives human epidermal carcinogenesis through phosphoinositol-3-kinase activation. Cancer Res. 2007, 67, 4264–4270. [Google Scholar] [CrossRef] [PubMed]
- Marinkovich, M.P. Tumor microenvironment: Laminin 332 in squamous-cell carcinoma. Nat. Rev. Cancer 2007, 7, 370–380. [Google Scholar] [CrossRef]
- Momota, Y.; Suzuki, N.; Kasuya, Y.; Kobayashi, T.; Mizoguchi, M.; Yokoyama, F.; Nomizu, M.; Shinkai, H.; Iwasaki, T.; Utani, A. Laminin alpha3 LG4 module induces keratinocyte migration: Involvement of matrix metalloproteinase-9. J. Recept. Signal Transduct. Res. 2005, 25, 1–17. [Google Scholar] [CrossRef]
- Cavaco, A.C.M.; Rezaei, M.; Caliandro, M.F.; Lima, A.M.; Stehling, M.; Dhayat, S.A.; Haier, J.; Brakebusch, C.; Eble, J.A. The interaction between Laminin-332 and α3β1 integrin determines differentiation and maintenance of CAFs, and supports invasion of pancreatic duct adenocarcinoma cells. Cancers 2018, 11, 14. [Google Scholar] [CrossRef]
- Guess, C.M.; Quaranta, V. Defining the role of laminin-332 in carcinoma. Matrix Biol. 2009, 28, 445–455. [Google Scholar] [CrossRef]
- Chen, J.; Wang, W.; Wei, J.; Zhou, D.; Zhao, X.; Song, W.; Sun, Q.; Huang, P.; Zheng, S. Overexpression of β3 chains of laminin-332 is associated with clinicopathologic features and decreased survival in patients with pancreatic adenocarcinoma. Appl. Immunohistochem. Mol. Morphol. 2015, 23, 516–521. [Google Scholar] [CrossRef]
- Carpenter, P.M.; Ziogas, A.; Markham, E.M.; Cantillep, A.S.; Yan, R.; Anton-Culver, H. Laminin 332 expression and prognosis in breast cancer. Hum. Pathol. 2018, 82, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Galiger, C.; Löffek, S.; Stemmler, M.P.; Kroeger, J.K.; Mittapalli, V.R.; Fauth, L.; Esser, P.R.; Kern, J.S.; Meiss, F.; Laßmann, S.; et al. Targeting of cell surface proteolysis of collagen XVII impedes squamous cell carcinoma progression. Mol. Ther. 2018, 26, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Di Zenzo, G.; El Hachem, M.; Diociaiuti, A.; Boldrini, R.; Calabresi, V.; Cianfarani, F.; Fortugno, P.; Piccinni, E.; Zambruno, G.; Castiglia, D. A truncating mutation in the laminin-332α chain highlights the role of the LG45 proteolytic domain in regulating keratinocyte adhesion and migration. Br. J. Dermatol. 2014, 170, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, E.A.; Marinkovich, M.P.; Hoeffler, W.K.; Furthmayr, H.; Woodley, D.T. Laminin-5 inhibits human keratinocyte migration. Exp. Cell Res. 1997, 233, 330–339. [Google Scholar] [CrossRef]
- Tasanen, K.; Tunggal, L.; Chometon, G.; Bruckner-Tuderman, L.; Aumailley, M. Keratinocytes from patients lacking collagen XVII display a migratory phenotype. Am. J. Pathol. 2004, 164, 2027–2038. [Google Scholar] [CrossRef][Green Version]
- Löffek, S.; Hurskainen, T.; Jackow, J.; Sigloch, F.C.; Schilling, O.; Tasanen, K.; Bruckner-Tuderman, L.; Franzke, F.C. Transmembrane collagen XVII modulates integrin dependent keratinocyte migration via PI3K/Rac1 signaling. PLoS ONE 2014, 9, e87263. [Google Scholar] [CrossRef]
- Has, C.; Castiglia, D.; del Rio, M.; Diez, M.G.; Piccinni, E.; Kiritsi, D.; Kohlhase, J.; Itin, P.; Martin, L.; Fischer, J.; et al. Kindler syndrome: Extension of FERMT1 mutational spectrum and natural history. Hum. Mutat. 2011, 32, 1204–1212. [Google Scholar] [CrossRef]
- Souldi, H.; Bajja, M.Y.; Mahtar, M. Kindler syndrome complicated by invasive squamous cell carcinoma of the palate. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2018, 135, 59–61. [Google Scholar] [CrossRef]
- Saleva, M.; Has, C.; He, Y.; Vassileva, S.; Balabanova, M.; Miteva, L. Natural history of Kindler syndrome and propensity for skin cancer - case report and literature review. J. Dtsch. Dermatol. Ges. 2018, 16, 338–341. [Google Scholar] [CrossRef]
- Guerrero-Aspizua, S.; Conti, C.J.; Escamez, M.J.; Castiglia, D.; Zambruno, G.; Youssefian, L.; Vahidnezhad, H.; Requena, L.; Itin, P.; Tadini, G.; et al. Assessment of the risk and characterization of non-melanoma skin cancer in Kindler syndrome: Study of a series of 91 patients. Orphanet. J. Rare Dis. 2019, 14, 183. [Google Scholar] [CrossRef]
- Emmert, H.; Patel, H.; Brunton, V.G. Kindlin-1 protects cells from oxidative damage through activation of ERK signalling. Free Radic. Biol. Med. 2017, 108, 896–903. [Google Scholar] [CrossRef] [PubMed]
- Emmert, H.; Culley, J.; Brunton, V.G. Inhibition of cyclin-dependent kinase activity exacerbates H2 O2 -induced DNA damage in Kindler syndrome keratinocytes. Exp. Dermatol. 2019, 28, 1074–1078. [Google Scholar] [CrossRef] [PubMed]
- Piccinni, E.; Di Zenzo, G.; Maurelli, R.; Dellambra, E.; Teson, M.; Has, C.; Zambruno, G.; Castiglia, D. Induction of senescence pathways in Kindler syndrome primary keratinocytes. Br. J. Dermatol. 2013, 168, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef] [PubMed]
- Abbadie, C.; Pluquet, O.; Pourtier, A. Epithelial cell senescence: An adaptive response to pre-carcinogenic stresses? Cell. Mol. Life Sci. 2017, 74, 4471–4509. [Google Scholar] [CrossRef]
- Michael, M.; Begum, R.; Chan, G.K.; Whitewood, A.J.; Matthews, D.R.; Goult, B.T.; McGrath, J.A.; Parsons, M. Kindlin-1 regulates epidermal growth factor receptor signalling. J. Investig. Dermatol. 2019, 139, 369–379. [Google Scholar] [CrossRef]
- Abu-Humaidan, A.H.; Ananthoju, N.; Mohanty, T.; Sonesson, A.; Alberius, P.; Schmidtchen, A.; Garred, P.; Sørensen, O.E. The epidermal growth factor receptor is a regulator of epidermal complement component expression and complement activation. J. Immunol. 2014, 192, 3355–3364. [Google Scholar] [CrossRef]
- Campbell, P.; Morton, P.E.; Takeichi, T.; Salam, A.; Roberts, N.; Proudfoot, L.E.; Mellerio, J.E.; Aminu, K.; Wellington, C.; Patil, S.N.; et al. Epithelial inflammation resulting from an inherited loss-of-function mutation in EGFR. J. Investig. Dermatol. 2014, 134, 2570–2578. [Google Scholar] [CrossRef]
- Heinemann, A.; He, Y.; Zimina, E.; Boerries, M.; Busch, H.; Chmel, N.; Kurz, T.; Bruckner-Tuderman, L.; Has, C. Induction of phenotype modifying cytokines by FERMT1 mutations. Hum. Mutat. 2011, 32, 397–406. [Google Scholar] [CrossRef]
- Rognoni, E.; Widmaier, M.; Jakobson, M.; Ruppert, R.; Ussar, S.; Katsougkri, D.; Böttcher, R.T.; Lai-Cheong, J.E.; Rifkin, D.B.; McGrath, J.A.; et al. Kindlin-1 controls Wnt and TGF-β availability to regulate cutaneous stem cell proliferation. Nat. Med. 2014, 20, 350–359. [Google Scholar] [CrossRef]
- Lu, S.; Hess, M.E.; Reimer, A.; Castiglia, D.; He, Y.; Rafei-Shamsabadi, D.A.; Bubnoff, D.v.; Meiss, F.; Boerries, M.; Has, C. 302 Molecular and mutational signatures of squamous cell carcinomas in epidermolysis bullosa. J. Investig. Dermatol. 2019, 139, S266. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Significantly Mutated Genes | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference | Disease Model (Sample Size) | CASP8 | NOTCH1 | TP53 | CDKN2A | FAT1 | ARID2 | HRAS | KMT2B | ARHGEF6 | FAM114A2 | LRRC8A | PHF13 | SPTBN4 | NOTCH2 | SMARCA4 | EGFR | NF2 | NOTCH4 | PRDM9 | Other Genes |
| Cho R.J. et al. Sci. Transl. Med. 2018 (PMID: 30135250) [31] | RDEB SCCs (n = 31) | 38.7 | 54.8 | 45 | 32.2 | 22.5 | 12.9 | 12.9 | 19.3 | ||||||||||||
| Sans-DeSanNicolas L. et al. J. Invest. Dermatol. 2018 (PMID: 29291383) [30] | RDEB SCC1 (n = 1) | + | VAF 20% | VAF 15.9% | |||||||||||||||||
| RDEB SCC2 (n = 1) | + | + | + | + | + | VAF 36.47% | VAF 9.02% | ||||||||||||||
| Inman G.J. et al. Nat. Commun. 2018 (PMID: 30202019) [7] | non-EB SCCs (n = 40) | 75 | 70 | 45 | 22.5 | 50 | ATP1A1, CACNA1C, CLCN3, CRY1, FLNB, GLIS3, GRHL2, HERC6, LCLAT1, MAP3K9, MAPK1IP1L, PTEN, SF3B1, TMEM51, TRAPPC9, VSP41, WHSC1. | ||||||||||||||
| Li Y.Y. et al. Clin. Cancer. Res. 2015 (PMID: 25589618) [33] | metastatic SCCs (n = 29) | N/P | 48 | 79 | 45 | N/P | N/P | N/P | N/P | N/P | N/P | N/P | 28 | 14 | 17 | N/P | |||||
| Pickering C.R. et al. Clin. Cancer. Res. 2014 (PMID: 25303977) [32] | aggressive UV-induced SCCs (n = 39) | 23.1 | 59 | 94.9 | 43.6 | 43.6 | 20.5 | 51.3 | AJUBA, BBS9, BF2D, COBLL1, DCLK1, DCLRE1A, FBX021, KMT2C, OPN3, PARD3, PEG10, RASA1, RBM46, SEC31A, SNX25, ZNF644. | ||||||||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Condorelli, A.G.; Dellambra, E.; Logli, E.; Zambruno, G.; Castiglia, D. Epidermolysis Bullosa-Associated Squamous Cell Carcinoma: From Pathogenesis to Therapeutic Perspectives. Int. J. Mol. Sci. 2019, 20, 5707. https://doi.org/10.3390/ijms20225707
Condorelli AG, Dellambra E, Logli E, Zambruno G, Castiglia D. Epidermolysis Bullosa-Associated Squamous Cell Carcinoma: From Pathogenesis to Therapeutic Perspectives. International Journal of Molecular Sciences. 2019; 20(22):5707. https://doi.org/10.3390/ijms20225707
Chicago/Turabian StyleCondorelli, Angelo Giuseppe, Elena Dellambra, Elena Logli, Giovanna Zambruno, and Daniele Castiglia. 2019. "Epidermolysis Bullosa-Associated Squamous Cell Carcinoma: From Pathogenesis to Therapeutic Perspectives" International Journal of Molecular Sciences 20, no. 22: 5707. https://doi.org/10.3390/ijms20225707
APA StyleCondorelli, A. G., Dellambra, E., Logli, E., Zambruno, G., & Castiglia, D. (2019). Epidermolysis Bullosa-Associated Squamous Cell Carcinoma: From Pathogenesis to Therapeutic Perspectives. International Journal of Molecular Sciences, 20(22), 5707. https://doi.org/10.3390/ijms20225707