DGKα in Neutrophil Biology and Its Implications for Respiratory Diseases



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
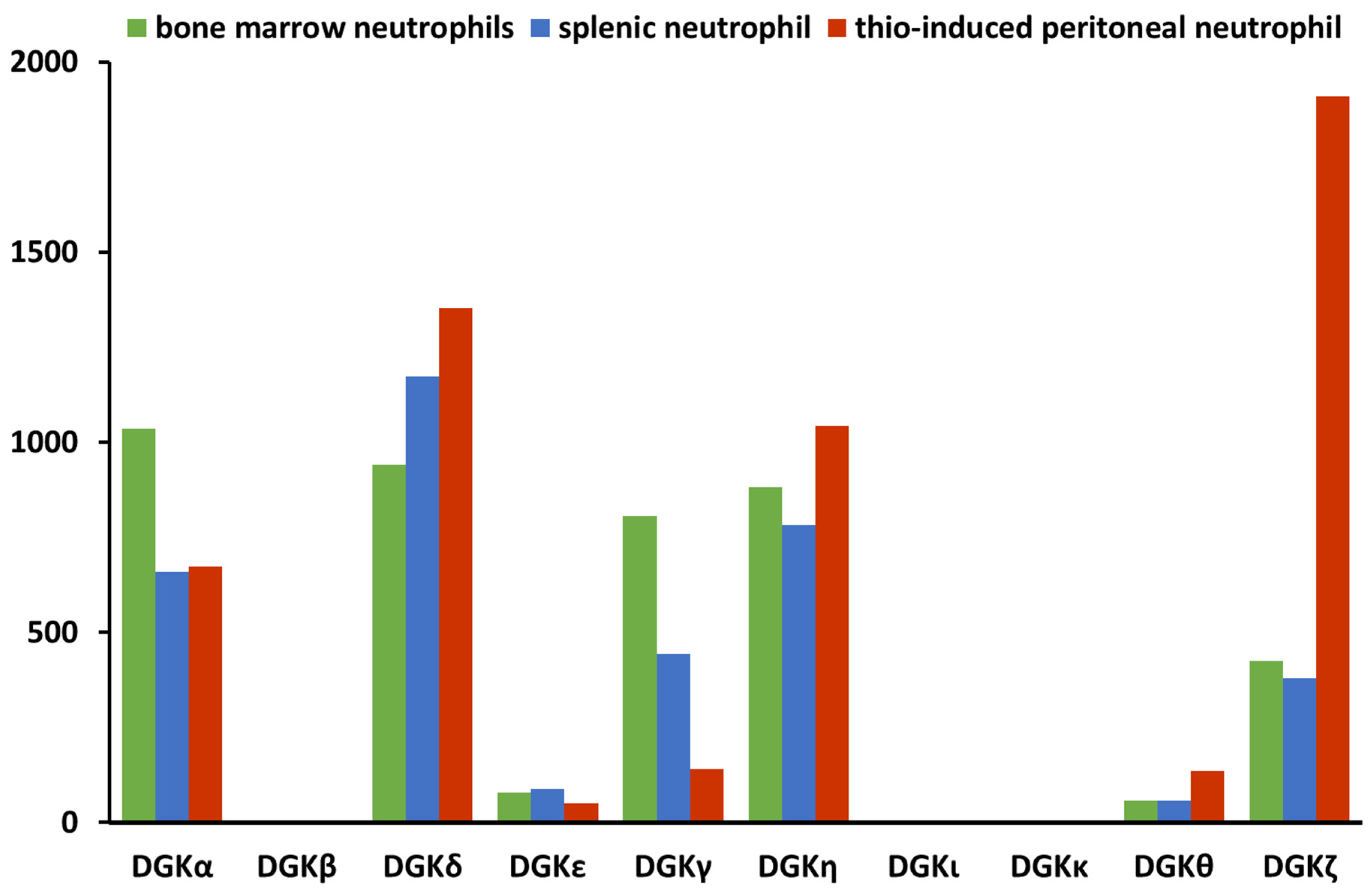
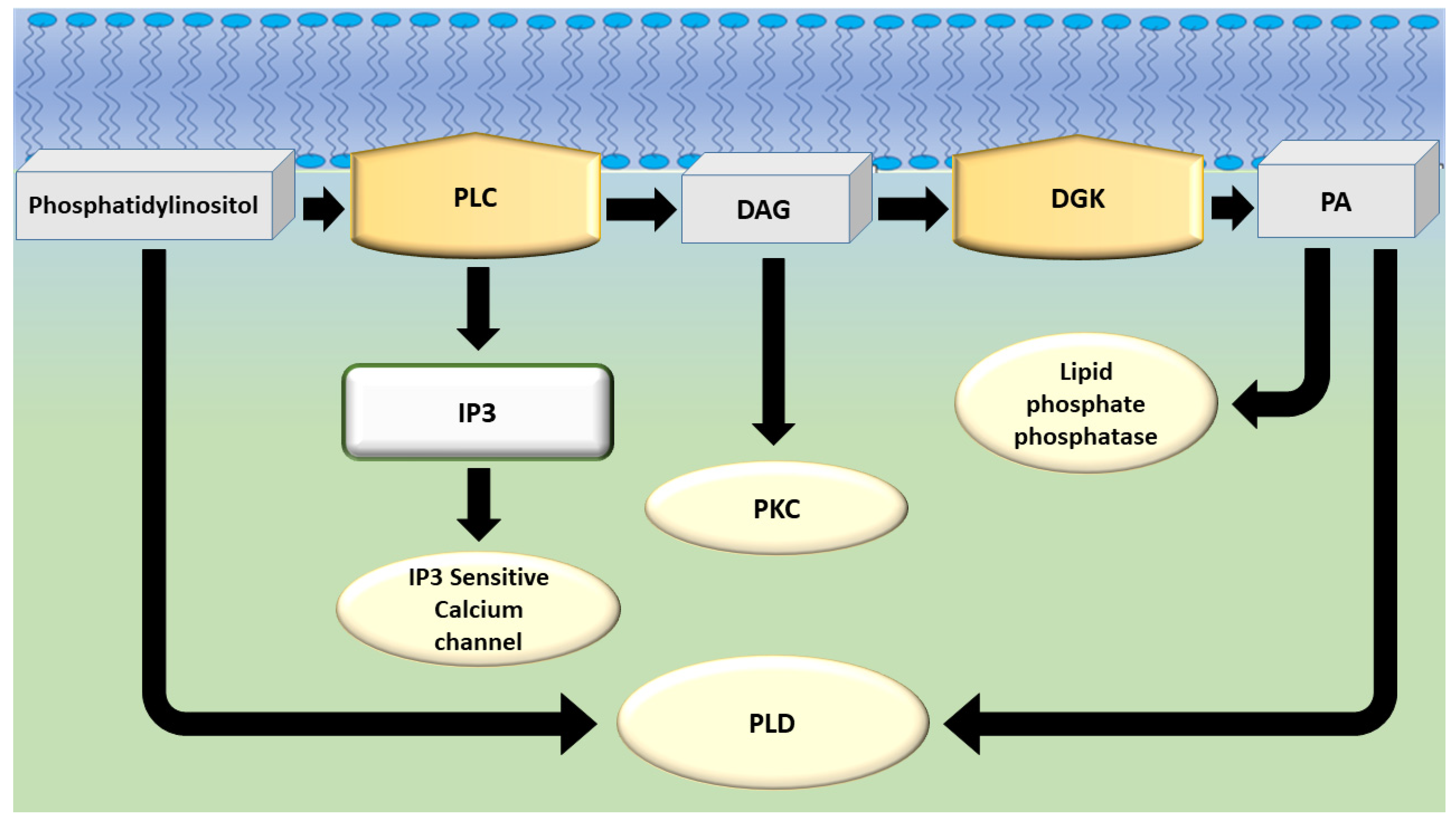
2. The Diacylglycerol Kinase Family
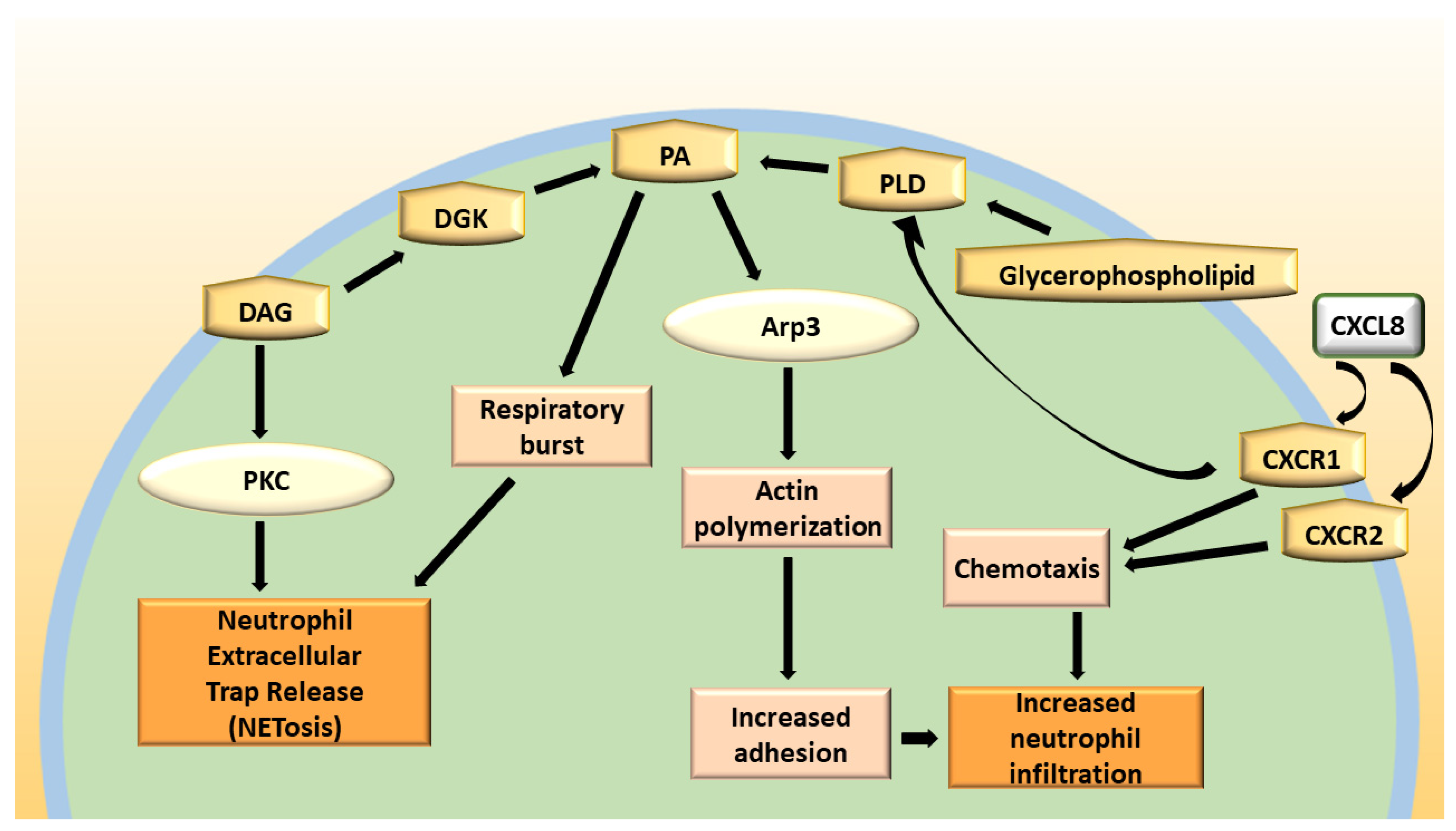
3. DGKα in Neutrophil Biology
4. DGKs in Respiratory Diseases
5. Discussion
Funding
Conflicts of Interest
Abbreviations
| Arp3 | Actin related protein 3 |
| CXCL8 | C-X-C motif chemokine ligand-8 |
| CXCR1 | C-X-C motif chemokine receptor-1 |
| CXCR2 | C-X-C motif chemokine receptor-2. |
| COPD | Chronic obstructive pulmonary disease |
| DAG | Diacylglycerol |
| DGK | Diacylglycerol kinase |
| IP3 | Inositol trisphosphate |
| NET | Neutrophil extracellular traps |
| PA | Phosphatidic acid |
| PKC | Protein kinase C |
| PLC | Phospholipase C |
| PLD | Phospholipase D |
| RCP | Rab-coupling protein |
| RICD | Restimulation-induced cell death |
| TCR | T cell receptor |
| XLP-1 | X-linked lymphoproliferative disease 1 |
References
- Mérida, I.; Avila-Flores, A.; Merino, E. Diacylglycerol kinases: At the hub of cell signalling. Biochem. J. 2008, 409, 1–18. [Google Scholar] [CrossRef]
- Topham, M.K. Signaling roles of diacylglycerol kinases. J. Cell. Biochem. 2006, 97, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Los, A.P.; van Baal, J.; de Widt, J.; Divecha, N.; van Blitterswijk, W.J. Structure-activity relationship of diacylglycerol kinase theta. Biochim. Biophys. Acta 2004, 1636, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Schaap, D.; van der Wal, J.; van Blitterswijk, W.J. Consensus sequences for ATP-binding sites in protein kinases do not apply to diacylglycerol kinases. Biochem. J. 1994, 304 Pt 2, 661–662. [Google Scholar] [CrossRef]
- Kazanietz, M.G. Targeting protein kinase C and “non-kinase” phorbol ester receptors: Emerging concepts and therapeutic implications. Biochim. Biophys. Acta 2005, 1754, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Sakane, F.; Kai, M.; Wada, I.; Imai, S.; Kanoh, H. The C-terminal part of diacylglycerol kinase alpha lacking zinc fingers serves as a catalytic domain. Biochem. J. 1996, 318 Pt 2, 583–590. [Google Scholar] [CrossRef]
- Shindo, M.; Irie, K.; Masuda, A.; Ohigashi, H.; Shirai, Y.; Miyasaka, K.; Saito, N. Synthesis and phorbol ester binding of the cysteine-rich domains of diacylglycerol kinase (DGK) isozymes. DGKgamma and DGKbeta are new targets of tumor-promoting phorbol esters. J. Biol. Chem. 2003, 278, 18448–18454. [Google Scholar] [CrossRef]
- Shindo, M.; Irie, K.; Ohigashi, H.; Kuriyama, M.; Saito, N. Diacylglycerol kinase gamma is one of the specific receptors of tumor-promoting phorbol esters. Biochem. Biophys. Res. Commun. 2001, 289, 451–456. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef]
- Sakane, F.; Yamada, K.; Imai, S.; Kanoh, H. Porcine 80-kDa diacylglycerol kinase is a calcium-binding and calcium/phospholipid-dependent enzyme and undergoes calcium-dependent translocation. J. Biol. Chem. 1991, 266, 7096–7100. [Google Scholar]
- Takahashi, M.; Yamamoto, T.; Sakai, H.; Sakane, F. Calcium negatively regulates an intramolecular interaction between the N-terminal recoverin homology and EF-hand motif domains and the C-terminal C1 and catalytic domains of diacylglycerol kinase α. Biochem. Biophys. Res. Commun. 2012, 423, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, D.; Suzuki, K.; Sakamoto, T.; Iwamoto, T.; Murata, T.; Sakane, F. Crystal structure and calcium-induced conformational changes of diacylglycerol kinase α EF-hand domains. Protein Sci. 2019, 28, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Kume, A.; Kawase, K.; Komenoi, S.; Usuki, T.; Takeshita, E.; Sakai, H.; Sakane, F. The Pleckstrin Homology Domain of Diacylglycerol Kinase η Strongly and Selectively Binds to Phosphatidylinositol 4,5-Bisphosphate. J. Biol. Chem. 2016, 291, 8150–8161. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Sakiyama, S.; Usuki, T.; Sakai, H.; Sakane, F. Diacylglycerol kinase δ1 transiently translocates to the plasma membrane in response to high glucose. Biochim. Biophys. Acta 2012, 1823, 2210–2216. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Sakane, F. Recent progress on type II diacylglycerol kinases: The physiological functions of diacylglycerol kinase δ, η and κ and their involvement in disease. J. Biochem. 2012, 152, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Hozumi, Y.; Tanaka, T.; Okada, M.; Nakano, T.; Suzuki, Y.; Iseki, K.; Kakehata, S.; Topham, M.K.; Goto, K. Role of the N-terminal hydrophobic residues of DGKε in targeting the endoplasmic reticulum. Biochim. Biophys. Acta 2014, 1842, 1440–1450. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, K.; Epand, R.M. Catalytic activity and acyl-chain selectivity of diacylglycerol kinase ɛ are modulated by residues in and near the lipoxygenase-like motif. J. Mol. Biol. 2012, 416, 619–628. [Google Scholar] [CrossRef]
- Zhu, J.; Chaki, M.; Lu, D.; Ren, C.; Wang, S.S.; Rauhauser, A.; Li, B.; Zimmerman, S.; Jun, B.; Du, Y.; et al. Loss of diacylglycerol kinase epsilon in mice causes endothelial distress and impairs glomerular Cox-2 and PGE2 production. Am. J. Physiol. Renal Physiol. 2016, 310, F895–F908. [Google Scholar] [CrossRef]
- Luo, B.; Prescott, S.M.; Topham, M.K. Protein kinase C alpha phosphorylates and negatively regulates diacylglycerol kinase zeta. J. Biol. Chem. 2003, 278, 39542–39547. [Google Scholar] [CrossRef]
- Los, A.P.; de Widt, J.; Topham, M.K.; van Blitterswijk, W.J.; Divecha, N. Protein kinase C inhibits binding of diacylglycerol kinase-zeta to the retinoblastoma protein. Biochim. Biophys. Acta 2007, 1773, 352–357. [Google Scholar] [CrossRef][Green Version]
- Luo, B.; Prescott, S.M.; Topham, M.K. Association of diacylglycerol kinase zeta with protein kinase C alpha: Spatial regulation of diacylglycerol signaling. J. Cell Biol. 2003, 160, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Houssa, B.; Schaap, D.; van der Wal, J.; Goto, K.; Kondo, H.; Yamakawa, A.; Shibata, M.; Takenawa, T.; van Blitterswijk, W.J. Cloning of a novel human diacylglycerol kinase (DGKtheta) containing three cysteine-rich domains, a proline-rich region, and a pleckstrin homology domain with an overlapping Ras-associating domain. J. Biol. Chem. 1997, 272, 10422–10428. [Google Scholar] [CrossRef] [PubMed]
- Topham, M.K.; Epand, R.M. Mammalian diacylglycerol kinases: Molecular interactions and biological functions of selected isoforms. Biochim. Biophys. Acta 2009, 1790, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Olenchock, B.A.; Guo, R.; Carpenter, J.H.; Jordan, M.; Topham, M.K.; Koretzky, G.A.; Zhong, X.P. Disruption of diacylglycerol metabolism impairs the induction of T cell anergy. Nat. Immunol. 2006, 7, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Shirai, Y.; Kouzuki, T.; Kakefuda, K.; Moriguchi, S.; Oyagi, A.; Horie, K.; Morita, S.Y.; Shimazawa, M.; Fukunaga, K.; Takeda, J.; et al. Essential role of neuron-enriched diacylglycerol kinase (DGK), DGKbeta in neurite spine formation, contributing to cognitive function. PLoS ONE 2010, 5, e11602. [Google Scholar] [CrossRef] [PubMed]
- Crotty, T.; Cai, J.; Sakane, F.; Taketomi, A.; Prescott, S.M.; Topham, M.K. Diacylglycerol kinase delta regulates protein kinase C and epidermal growth factor receptor signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 15485–15490. [Google Scholar] [CrossRef] [PubMed]
- Mannerås-Holm, L.; Schönke, M.; Brozinick, J.T.; Vetterli, L.; Bui, H.H.; Sanders, P.; Nascimento, E.B.M.; Björnholm, M.; Chibalin, A.V.; Zierath, J.R. Diacylglycerol kinase ε deficiency preserves glucose tolerance and modulates lipid metabolism in obese mice. J. Lipid Res. 2017, 58, 907–915. [Google Scholar] [CrossRef]
- Isozaki, T.; Komenoi, S.; Lu, Q.; Usuki, T.; Tomokata, S.; Matsutomo, D.; Sakai, H.; Bando, K.; Kiyonari, H.; Sakane, F. Deficiency of diacylglycerol kinase η induces lithium-sensitive mania-like behavior. J. Neurochem. 2016, 138, 448–456. [Google Scholar] [CrossRef]
- Regier, D.S.; Higbee, J.; Lund, K.M.; Sakane, F.; Prescott, S.M.; Topham, M.K. Diacylglycerol kinase iota regulates Ras guanyl-releasing protein 3 and inhibits Rap1 signaling. Proc. Natl. Acad. Sci. USA 2005, 102, 7595–7600. [Google Scholar] [CrossRef]
- Zhong, X.P.; Hainey, E.A.; Olenchock, B.A.; Jordan, M.S.; Maltzman, J.S.; Nichols, K.E.; Shen, H.; Koretzky, G.A. Enhanced T cell responses due to diacylglycerol kinase zeta deficiency. Nat. Immunol. 2003, 4, 882–890. [Google Scholar] [CrossRef]
- De Chaffoy de Courcelles, D.; Roevens, P.; Van Belle, H.; Kennis, L.; Somers, Y.; De Clerck, F. The role of endogenously formed diacylglycerol in the propagation and termination of platelet activation. A biochemical and functional analysis using the novel diacylglycerol kinase inhibitor, R 59 949. J. Biol. Chem. 1989, 264, 3274–3285. [Google Scholar] [PubMed]
- De Chaffoy de Courcelles, D.C.; Roevens, P.; Van Belle, H. R 59 022, a diacylglycerol kinase inhibitor. Its effect on diacylglycerol and thrombin-induced C kinase activation in the intact platelet. J. Biol. Chem. 1985, 260, 15762–15770. [Google Scholar] [PubMed]
- Sato, M.; Liu, K.; Sasaki, S.; Kunii, N.; Sakai, H.; Mizuno, H.; Saga, H.; Sakane, F. Evaluations of the selectivities of the diacylglycerol kinase inhibitors r59022 and r59949 among diacylglycerol kinase isozymes using a new non-radioactive assay method. Pharmacology 2013, 92, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Sakane, F.; Kanoh, H.; Walsh, J.P. Selectivity of the diacylglycerol kinase inhibitor 3-[2-(4-[bis-(4-fluorophenyl)methylene]-1-piperidinyl)ethyl]-2, 3-dihydro-2-thioxo-4(1H)quinazolinone (R59949) among diacylglycerol kinase subtypes. Biochem. Pharmacol. 2000, 59, 763–772. [Google Scholar] [CrossRef]
- Ruffo, E.; Malacarne, V.; Larsen, S.E.; Das, R.; Patrussi, L.; Wülfing, C.; Biskup, C.; Kapnick, S.M.; Verbist, K.; Tedrick, P.; et al. Inhibition of diacylglycerol kinase α restores restimulation-induced cell death and reduces immunopathology in XLP-1. Sci. Transl. Med. 2016, 8, 321ra7. [Google Scholar] [CrossRef]
- Dominguez, C.L.; Floyd, D.H.; Xiao, A.; Mullins, G.R.; Kefas, B.A.; Xin, W.; Yacur, M.N.; Abounader, R.; Lee, J.K.; Wilson, G.M.; et al. Diacylglycerol kinase α is a critical signaling node and novel therapeutic target in glioblastoma and other cancers. Cancer Discov. 2013, 3, 782–797. [Google Scholar] [CrossRef]
- Boroda, S.; Niccum, M.; Raje, V.; Purow, B.W.; Harris, T.E. Dual activities of ritanserin and R59022 as DGKα inhibitors and serotonin receptor antagonists. Biochem. Pharmacol. 2017, 123, 29–39. [Google Scholar] [CrossRef]
- McCloud, R.L.; Franks, C.E.; Campbell, S.T.; Purow, B.W.; Harris, T.E.; Hsu, K.L. Deconstructing Lipid Kinase Inhibitors by Chemical Proteomics. Biochemistry 2018, 57, 231–236. [Google Scholar] [CrossRef]
- Franks, C.E.; Campbell, S.T.; Purow, B.W.; Harris, T.E.; Hsu, K.L. The Ligand Binding Landscape of Diacylglycerol Kinases. Cell Chem. Biol. 2017, 24, 870–880.e5. [Google Scholar] [CrossRef]
- Akhondzadeh, S.; Malek-Hosseini, M.; Ghoreishi, A.; Raznahan, M.; Rezazadeh, S.A. Effect of ritanserin, a 5HT2A/2C antagonist, on negative symptoms of schizophrenia: A double-blind randomized placebo-controlled study. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 1879–1883. [Google Scholar] [CrossRef]
- Akhondzadeh, S.; Mohajari, H.; Reza Mohammadi, M.; Amini, H. Ritanserin as an adjunct to lithium and haloperidol for the treatment of medication-naive patients with acute mania: A double blind and placebo controlled trial. BMC Psychiatry 2003, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Cornish, J.W.; Maany, I.; Fudala, P.J.; Ehrman, R.N.; Robbins, S.J.; O’Brien, C.P. A randomized, double-blind, placebo-controlled study of ritanserin pharmacotherapy for cocaine dependence. Drug Alcohol Depend. 2001, 61, 183–189. [Google Scholar] [CrossRef]
- Wiesbeck, G.A.; Weijers, H.G.; Chick, J.; Naranjo, C.A.; Boening, J. Ritanserin in relapse prevention in abstinent alcoholics: Results from a placebo-controlled double-blind international multicenter trial. Ritanserin in Alcoholism Work Group. Alcohol Clin. Exp. Res. 1999, 23, 230–235. [Google Scholar] [PubMed]
- Olin, R.; Klein, R.; Berg, P.A. A randomised double-blind 16-week study of ritanserin in fibromyalgia syndrome: Clinical outcome and analysis of autoantibodies to serotonin, gangliosides and phospholipids. Clin. Rheumatol. 1998, 17, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Duinkerke, S.J.; Botter, P.A.; Jansen, A.A.; van Dongen, P.A.; van Haaften, A.J.; Boom, A.J.; van Laarhoven, J.H.; Busard, H.L. Ritanserin, a selective 5-HT2/1C antagonist, and negative symptoms in schizophrenia. A placebo-controlled double-blind trial. Br. J. Psychiatry 1993, 163, 451–455. [Google Scholar] [CrossRef]
- Bersani, G.; Grispini, A.; Marini, S.; Pasini, A.; Valducci, M.; Ciani, N. 5-HT2 antagonist ritanserin in neuroleptic-induced parkinsonism: A double-blind comparison with orphenadrine and placebo. Clin. Neuropharmacol. 1990, 13, 500–506. [Google Scholar] [CrossRef]
- Den Boer, J.A.; Westenberg, H.G. Serotonin function in panic disorder: A double blind placebo controlled study with fluvoxamine and ritanserin. Psychopharmacology 1990, 102, 85–94. [Google Scholar] [CrossRef]
- Hedner, T.; Persson, B. Experience with ketanserin and ritanserin in hypertensive patients. J. Cardiovasc. Pharmacol. 1988, 11 (Suppl. 1), S44–S48. [Google Scholar]
- Paiva, T.; Arriaga, F.; Wauquier, A.; Lara, E.; Largo, R.; Leitao, J.N. Effects of ritanserin on sleep disturbances of dysthymic patients. Psychopharmacology 1988, 96, 395–399. [Google Scholar] [CrossRef]
- Bressa, G.M.; Marini, S.; Gregori, S. Serotonin S2 receptors blockage and generalized anxiety disorders. A double-blind study on ritanserin and lorazepam. Int. J. Clin. Pharmacol. Res. 1987, 7, 111–119. [Google Scholar]
- Maertens de Noordhout, A.; Delwaide, P.J. Open pilot trial of ritanserin in parkinsonism. Clin. Neuropharmacol. 1986, 9, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Ceulemans, D.L.; Hoppenbrouwers, M.L.; Gelders, Y.G.; Reyntjens, A.J. The influence of ritanserin, a serotonin antagonist, in anxiety disorders: A double-blind placebo-controlled study versus lorazepam. Pharmacopsychiatry 1985, 18, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Olmez, I.; Love, S.; Xiao, A.; Manigat, L.; Randolph, P.; McKenna, B.D.; Neal, B.P.; Boroda, S.; Li, M.; Brenneman, B.; et al. Targeting the mesenchymal subtype in glioblastoma and other cancers via inhibition of diacylglycerol kinase alpha. Neuro Oncol. 2018, 20, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Velnati, S.; Ruffo, E.; Massarotti, A.; Talmon, M.; Varma, K.S.S.; Gesu, A.; Fresu, L.G.; Snow, A.L.; Bertoni, A.; Capello, D.; et al. Identification of a novel DGKα inhibitor for XLP-1 therapy by virtual screening. Eur. J. Med. Chem. 2019, 164, 378–390. [Google Scholar] [CrossRef]
- Liu, K.; Kunii, N.; Sakuma, M.; Yamaki, A.; Mizuno, S.; Sato, M.; Sakai, H.; Kado, S.; Kumagai, K.; Kojima, H.; et al. A novel diacylglycerol kinase α-selective inhibitor, CU-3, induces cancer cell apoptosis and enhances immune response. J. Lipid Res. 2016, 57, 368–379. [Google Scholar] [CrossRef]
- Gray, R.D.; Lucas, C.D.; MacKellar, A.; Li, F.; Hiersemenzel, K.; Haslett, C.; Davidson, D.J.; Rossi, A.G. Activation of conventional protein kinase C (PKC) is critical in the generation of human neutrophil extracellular traps. J. Inflamm. 2013, 10, 12. [Google Scholar] [CrossRef]
- Suire, S.; Lécureuil, C.; Anderson, K.E.; Damoulakis, G.; Niewczas, I.; Davidson, K.; Guillou, H.; Pan, D.; Clark, J.; Hawkins, P.T.; et al. GPCR activation of Ras and PI3Kc in neutrophils depends on PLCb2/b3 and the RasGEF RasGRP4. EMBO J. 2012, 31, 3118–3129. [Google Scholar] [CrossRef]
- Speranza, F.; Mahankali, M.; Henkels, K.M.; Gomez-Cambronero, J. The molecular basis of leukocyte adhesion involving phosphatidic acid and phospholipase D. J. Biol. Chem. 2014, 289, 28885–28897. [Google Scholar] [CrossRef]
- Tou, J.S.; Gill, J.S. Lysophosphatidic acid increases phosphatidic acid formation, phospholipase D activity and degranulation by human neutrophils. Cell Signal. 2005, 17, 77–82. [Google Scholar] [CrossRef]
- Erickson, R.W.; Langel-Peveri, P.; Traynor-Kaplan, A.E.; Heyworth, P.G.; Curnutte, J.T. Activation of human neutrophil NADPH oxidase by phosphatidic acid or diacylglycerol in a cell-free system. Activity of diacylglycerol is dependent on its conversion to phosphatidic acid. J. Biol. Chem. 1999, 274, 22243–22250. [Google Scholar] [CrossRef]
- Karathanassis, D.; Stahelin, R.V.; Bravo, J.; Perisic, O.; Pacold, C.M.; Cho, W.; Williams, R.L. Binding of the PX domain of p47(phox) to phosphatidylinositol 3,4-bisphosphate and phosphatidic acid is masked by an intramolecular interaction. EMBO J. 2002, 21, 5057–5068. [Google Scholar] [CrossRef]
- Batista, E.L.; Warbington, M.; Badwey, J.A.; Van Dyke, T.E. Differentiation of HL-60 cells to granulocytes involves regulation of select diacylglycerol kinases (DGKs). J. Cell. Biochem. 2005, 94, 774–793. [Google Scholar] [CrossRef]
- Yamada, K.; Sakane, F.; Imai, S.; Tsushima, S.; Murakami, T.; Kanoh, H. Regulatory role of diacylglycerol kinase gamma in macrophage differentiation of leukemia cells. Biochem. Biophys. Res. Commun. 2003, 305, 101–107. [Google Scholar] [CrossRef]
- Yamamoto, M.; Tanaka, T.; Hozumi, Y.; Saino-Saito, S.; Nakano, T.; Tajima, K.; Kato, T.; Goto, K. Expression of mRNAs for the diacylglycerol kinase family in immune cellsduring an inflammatory reaction. Biomed. Res. 2014, 35, 61–68. [Google Scholar] [CrossRef]
- Oyaizu, K.; Kantarci, A.; Maeda, H.; Batista, E.L.; Hasturk, H.; Murayama, Y.; Badwey, J.A.; Van Dyke, T.E. Identification of mRNAs for the various diacylglycerol kinase isoforms in neutrophils from patients with localized aggressive periodontitis. J. Periodontal Res. 2003, 38, 488–495. [Google Scholar] [CrossRef]
- Heng, T.S.; Painter, M.W.; Consortium, I.G.P. The Immunological Genome Project: Networks of gene expression in immune cells. Nat. Immunol. 2008, 9, 1091–1094. [Google Scholar] [CrossRef]
- Perkins, R.S.; Lindsay, M.A.; Barnes, P.J.; Giembycz, M.A. Early signalling events implicated in leukotriene B4-induced activation of the NADPH oxidase in eosinophils: Role of Ca2+, protein kinase C and phospholipases C and D. Biochem. J. 1995, 310 Pt 3, 795–806. [Google Scholar] [CrossRef]
- Reali, E.; Spisani, S.; Gavioli, R.; Lanza, F.; Moretti, S.; Traniello, S. IL-8 enhances antibody-dependent cellular cytotoxicity in human neutrophils. Immunol. Cell Biol. 1995, 73, 234–238. [Google Scholar] [CrossRef]
- Muid, R.E.; Penfield, A.; Dale, M.M. The diacylglycerol kinase inhibitor, R59022, enhances the superoxide generation from human neutrophils induced by stimulation of fMet-Leu-Phe, IgG and C3b receptors. Biochem. Biophys. Res. Commun. 1987, 143, 630–637. [Google Scholar] [CrossRef]
- Gomez-Cambronero, J.; Molski, T.F.; Becker, E.L.; Sha’afi, R.I. The diacylglycerol kinase inhibitor R59022 potentiates superoxide production but not secretion induced by fMet-Leu-Phe: Effects of leupeptin and the protein kinase C inhibitor H-7. Biochem. Biophys. Res. Commun. 1987, 148, 38–46. [Google Scholar] [CrossRef]
- Hurttia, H.; Leino, L. Subcellular localization of diacylglycerol kinase activity in stimulated and unstimulated human peripheral blood lymphocytes and neutrophils. Biochem. Mol. Biol. Int. 1996, 40, 579–585. [Google Scholar] [CrossRef]
- Tao, W.; Molski, T.F.; Sha’afi, R.I. Arachidonic acid release in rabbit neutrophils. Biochem. J. 1989, 257, 633–637. [Google Scholar] [CrossRef]
- Ohtsuka, T.; Hiura, M.; Yoshida, K.; Okamura, N.; Ishibashi, S. A diacylglycerol kinase inhibitor, R 59 022, potentiates superoxide anion production and 46-kDa protein phosphorylation in guinea pig polymorphonuclear leukocytes. J. Biol. Chem. 1990, 265, 15418–15423. [Google Scholar]
- Boonen, G.J.; de Koster, B.M.; VanSteveninck, J.; Elferink, J.G. Neutrophil chemotaxis induced by the diacylglycerol kinase inhibitor R59022. Biochim. Biophys. Acta 1993, 1178, 97–102. [Google Scholar] [CrossRef]
- Silva, L.M.; Brenchley, L.; Moutsopoulos, N.M. Primary immunodeficiencies reveal the essential role of tissue neutrophils in periodontitis. Immunol. Rev. 2019, 287, 226–235. [Google Scholar] [CrossRef]
- Tyagi, S.R.; Uhlinger, D.J.; Lambeth, J.D.; Champagne, C.; Van Dyke, T.E. Altered diacylglycerol level and metabolism in neutrophils from patients with localized juvenile periodontitis. Infect. Immun. 1992, 60, 2481–2487. [Google Scholar]
- Leino, L.; Hurttia, H.; Peltonen, E. Diacylglycerol in peripheral blood neutrophils from patients with localized juvenile periodontitis. J. Periodontal Res. 1994, 29, 334–338. [Google Scholar] [CrossRef]
- Hurttia, H.M.; Pelto, L.M.; Leino, L. Evidence of an association between functional abnormalities and defective diacylglycerol kinase activity in peripheral blood neutrophils from patients with localized juvenile periodontitis. J. Periodontal Res. 1997, 32, 401–407. [Google Scholar] [CrossRef]
- Hurttia, H.; Saarinen, K.; Leino, L. Increased adhesion of peripheral blood neutrophils from patients with localized juvenile periodontitis. J. Periodontal Res. 1998, 33, 292–297. [Google Scholar] [CrossRef]
- Gronert, K.; Kantarci, A.; Levy, B.D.; Clish, C.B.; Odparlik, S.; Hasturk, H.; Badwey, J.A.; Colgan, S.P.; Van Dyke, T.E.; Serhan, C.N. A molecular defect in intracellular lipid signaling in human neutrophils in localized aggressive periodontal tissue damage. J. Immunol. 2004, 172, 1856–1861. [Google Scholar] [CrossRef]
- Batista, E.L.; Kantarci, A.I.; Hasturk, H.; Van Dyke, T.E. Alternative Splicing Generates a Diacylglycerol Kinase α (DGKα) Transcript That Acts as a Dominant Negative Modulator of Superoxide Production in Localized Aggressive Periodontitis. J. Periodontol. 2014, 85, 934–943. [Google Scholar] [CrossRef]
- Kettritz, R. How anti-neutrophil cytoplasmic autoantibodies activate neutrophils. Clin. Exp. Immunol. 2012, 169, 220–228. [Google Scholar] [CrossRef]
- Jarrot, P.A.; Kaplanski, G. Pathogenesis of ANCA-associated vasculitis: An update. Autoimmun. Rev. 2016, 15, 704–713. [Google Scholar] [CrossRef]
- Williams, J.M.; Pettitt, T.R.; Powell, W.; Grove, J.; Savage, C.O.; Wakelam, M.J. Antineutrophil cytoplasm antibody-stimulated neutrophil adhesion depends on diacylglycerol kinase-catalyzed phosphatidic acid formation. J. Am. Soc. Nephrol. 2007, 18, 1112–1120. [Google Scholar] [CrossRef]
- Alonso, R.; Mazzeo, C.; Rodriguez, M.C.; Marsh, M.; Fraile-Ramos, A.; Calvo, V.; Avila-Flores, A.; Merida, I.; Izquierdo, M. Diacylglycerol kinase α regulates the formation and polarisation of mature multivesicular bodies involved in the secretion of Fas ligand-containing exosomes in T lymphocytes. Cell Death Differ. 2011, 18, 1161–1173. [Google Scholar] [CrossRef]
- Rainero, E.; Caswell, P.T.; Muller, P.A.; Grindlay, J.; McCaffrey, M.W.; Zhang, Q.; Wakelam, M.J.; Vousden, K.H.; Graziani, A.; Norman, J.C. Diacylglycerol kinase α controls RCP-dependent integrin trafficking to promote invasive migration. J. Cell Biol. 2012, 196, 277–295. [Google Scholar] [CrossRef]
- Xie, S.; Naslavsky, N.; Caplan, S. Diacylglycerol kinase α regulates tubular recycling endosome biogenesis and major histocompatibility complex class I recycling. J. Biol. Chem. 2014, 289, 31914–31926. [Google Scholar] [CrossRef]
- Rincón, E.; Sáez de Guinoa, J.; Gharbi, S.I.; Sorzano, C.O.; Carrasco, Y.R.; Mérida, I. Translocation dynamics of sorting nexin 27 in activated T cells. J. Cell Sci. 2011, 124, 776–788. [Google Scholar] [CrossRef]
- Nagaya, H.; Wada, I.; Jia, Y.J.; Kanoh, H. Diacylglycerol kinase delta suppresses ER-to-Golgi traffic via its SAM and PH domains. Mol. Biol Cell 2002, 13, 302–316. [Google Scholar] [CrossRef]
- Goldschmidt, H.L.; Tu-Sekine, B.; Volk, L.; Anggono, V.; Huganir, R.L.; Raben, D.M. DGKθ Catalytic Activity Is Required for Efficient Recycling of Presynaptic Vesicles at Excitatory Synapses. Cell Rep. 2016, 14, 200–207. [Google Scholar] [CrossRef]
- Holden, N.J.; Savage, C.O.; Young, S.P.; Wakelam, M.J.; Harper, L.; Williams, J.M. A dual role for diacylglycerol kinase generated phosphatidic acid in autoantibody-induced neutrophil exocytosis. Mol. Med. 2011, 17, 1242–1252. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Brightling, C.; Pedersen, S.E.; Reddel, H.K. Asthma. Lancet 2018, 391, 783–800. [Google Scholar] [CrossRef]
- Kulkarni, N.S.; Hollins, F.; Sutcliffe, A.; Saunders, R.; Shah, S.; Siddiqui, S.; Gupta, S.; Haldar, P.; Green, R.; Pavord, I.; et al. Eosinophil protein in airway macrophages: A novel biomarker of eosinophilic inflammation in patients with asthma. J. Allergy Clin. Immunol. 2010, 126, 61–69.e63. [Google Scholar] [CrossRef] [PubMed]
- Varricchi, G.; Bagnasco, D.; Borriello, F.; Heffler, E.; Canonica, G.W. Interleukin-5 pathway inhibition in the treatment of eosinophilic respiratory disorders: Evidence and unmet needs. Curr. Opin. Allergy Clin. Immunol. 2016, 16, 186–200. [Google Scholar] [CrossRef]
- Del Giacco, S.R.; Bakirtas, A.; Bel, E.; Custovic, A.; Diamant, Z.; Hamelmann, E.; Heffler, E.; Kalayci, Ö.; Saglani, S.; Sergejeva, S.; et al. Allergy in severe asthma. Allergy 2017, 72, 207–220. [Google Scholar] [CrossRef]
- Green, R.H.; Brightling, C.E.; Woltmann, G.; Parker, D.; Wardlaw, A.J.; Pavord, I.D. Analysis of induced sputum in adults with asthma: Identification of subgroup with isolated sputum neutrophilia and poor response to inhaled corticosteroids. Thorax 2002, 57, 875–879. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Hammad, H.; Fahy, J.V. The Cytokines of Asthma. Immunity 2019, 50, 975–991. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Q.; Ma, Q.; Zhang, Y.; Li, Z.; Wang, C. DGKα DNA vaccine relieves airway allergic inflammation in asthma model possibly via induction of T cell anergy. Int. J. Clin. Exp. Pathol. 2013, 6, 2404–2411. [Google Scholar]
- Martínez-Moreno, M.; García-Liévana, J.; Soutar, D.; Torres-Ayuso, P.; Andrada, E.; Zhong, X.P.; Koretzky, G.A.; Mérida, I.; Ávila-Flores, A. FoxO-dependent regulation of diacylglycerol kinase α gene expression. Mol. Cell. Biol. 2012, 32, 4168–4180. [Google Scholar] [CrossRef]
- Mérida, I.; Andrada, E.; Gharbi, S.I.; Ávila-Flores, A. Redundant and specialized roles for diacylglycerol kinases α and ζ in the control of T cell functions. Sci. Signal. 2015, 8, re6. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, P.; Krishna, S.; Wang, J.; Lin, X.; Huang, H.; Xie, D.; Gorentla, B.; Huang, R.; Gao, J.; et al. Unexpected positive control of NFκB and miR-155 by DGKα and ζ ensures effector and memory CD8+ T cell differentiation. Oncotarget 2016, 7, 33744–33764. [Google Scholar] [CrossRef]
- Snow, A.L.; Pandiyan, P.; Zheng, L.; Krummey, S.M.; Lenardo, M.J. The power and the promise of restimulation-induced cell death in human immune diseases. Immunol. Rev. 2010, 236, 68–82. [Google Scholar] [CrossRef]
- Shin, J.; O’Brien, T.F.; Grayson, J.M.; Zhong, X.P. Differential regulation of primary and memory CD8 T cell immune responses by diacylglycerol kinases. J. Immunol. 2012, 188, 2111–2117. [Google Scholar] [CrossRef] [PubMed]
- Mérida, I.; Avila-Flores, A.; García, J.; Merino, E.; Almena, M.; Torres-Ayuso, P. Diacylglycerol kinase alpha, from negative modulation of T cell activation to control of cancer progression. Adv. Enzym. Regul. 2009, 49, 174–188. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, M.A.; Jones, D.R.; Izquierdo, M.; Mérida, I. Role of diacylglycerol kinase alpha in the attenuation of receptor signaling. J. Cell Biol. 2001, 153, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, M.A.; Pradet-Balade, B.; Jones, D.R.; Martínez-A, C.; Stone, J.C.; Garcia-Sanz, J.A.; Mérida, I. T cell activation in vivo targets diacylglycerol kinase alpha to the membrane: A novel mechanism for Ras attenuation. J. Immunol. 2003, 170, 2877–2883. [Google Scholar] [CrossRef]
- Chauveau, A.; Le Floc’h, A.; Bantilan, N.S.; Koretzky, G.A.; Huse, M. Diacylglycerol kinase α establishes T cell polarity by shaping diacylglycerol accumulation at the immunological synapse. Sci. Signal. 2014, 7, ra82. [Google Scholar] [CrossRef]
- Carrasco, S.; Mérida, I. Diacylglycerol, when simplicity becomes complex. Trends Biochem. Sci. 2007, 32, 27–36. [Google Scholar] [CrossRef]
- Stace, C.L.; Ktistakis, N.T. Phosphatidic acid- and phosphatidylserine-binding proteins. Biochim. Biophys. Acta 2006, 1761, 913–926. [Google Scholar] [CrossRef]
- Kooijman, E.E.; Chupin, V.; de Kruijff, B.; Burger, K.N. Modulation of membrane curvature by phosphatidic acid and lysophosphatidic acid. Traffic 2003, 4, 162–174. [Google Scholar] [CrossRef]
- Damaj, B.B.; McColl, S.R.; Neote, K.; Hébert, C.A.; Naccache, P.H. Diverging signal transduction pathways activated by interleukin 8 (IL-8) and related chemokines in human neutrophils. IL-8 and Gro-alpha differentially stimulate calcium influx through IL-8 receptors A and B. J. Biol. Chem. 1996, 271, 20540–20544. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Wolf, M.; Qin, S.; Mackay, C.R.; Baggiolini, M. Different functions for the interleukin 8 receptors (IL-8R) of human neutrophil leukocytes: NADPH oxidase and phospholipase D are activated through IL-8R1 but not IL-8R2. Proc. Natl. Acad. Sci. USA 1996, 93, 6682–6686. [Google Scholar] [CrossRef] [PubMed]
- Biernacki, W.A.; Kharitonov, S.A.; Barnes, P.J. Increased leukotriene B4 and 8-isoprostane in exhaled breath condensate of patients with exacerbations of COPD. Thorax 2003, 58, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Sapey, E.; Stockley, J.A.; Greenwood, H.; Ahmad, A.; Bayley, D.; Lord, J.M.; Insall, R.H.; Stockley, R.A. Behavioral and structural differences in migrating peripheral neutrophils from patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 183, 1176–1186. [Google Scholar] [CrossRef]
- Iyer, S.S.; Kusner, D.J. Assay of phospholipase D activity in cell-free systems. Methods Mol. Biol. 2006, 332, 281–298. [Google Scholar] [CrossRef]
- Lehman, N.; Di Fulvio, M.; McCray, N.; Campos, I.; Tabatabaian, F.; Gomez-Cambronero, J. Phagocyte cell migration is mediated by phospholipases PLD1 and PLD2. Blood 2006, 108, 3564–3572. [Google Scholar] [CrossRef]
- Antonescu, C.N.; Danuser, G.; Schmid, S.L. Phosphatidic acid plays a regulatory role in clathrin-mediated endocytosis. Mol. Biol. Cell 2010, 21, 2944–2952. [Google Scholar] [CrossRef]
- Cai, K.; Sewer, M.B. cAMP-stimulated transcription of DGKθ requires steroidogenic factor 1 and sterol regulatory element binding protein 1. J. Lipid Res. 2013, 54, 2121–2132. [Google Scholar] [CrossRef]
- Cai, K.; Lucki, N.C.; Sewer, M.B. Silencing diacylglycerol kinase-theta expression reduces steroid hormone biosynthesis and cholesterol metabolism in human adrenocortical cells. Biochim. Biophys. Acta 2014, 1841, 552–562. [Google Scholar] [CrossRef]
- Xu, X.; Jin, T. The Novel Functions of the PLC/PKC/PKD Signaling Axis in G Protein-Coupled Receptor-Mediated Chemotaxis of Neutrophils. J. Immunol. Res. 2015, 2015, 817604. [Google Scholar] [CrossRef]
- Kenny, E.F.; Herzig, A.; Krüger, R.; Muth, A.; Mondal, S.; Thompson, P.R.; Brinkmann, V.; Bernuth, H.V.; Zychlinsky, A. Diverse stimuli engage different neutrophil extracellular trap pathways. eLife 2017, 6, e24437. [Google Scholar] [CrossRef] [PubMed]
- Tatsiy, O.; McDonald, P.P. Physiological Stimuli Induce PAD4-Dependent, ROS-Independent NETosis, With Early and Late Events Controlled by Discrete Signaling Pathways. Front. Immunol. 2018, 9, 2036. [Google Scholar] [CrossRef] [PubMed]
- American Thoracic Society. Idiopathic pulmonary fibrosis: Diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am. J. Respir. Crit. Care Med. 2000, 161, 646–664. [Google Scholar] [CrossRef] [PubMed]
- Hewlett, J.C.; Kropski, J.A.; Blackwell, T.S. Idiopathic pulmonary fibrosis: Epithelial-mesenchymal interactions and emerging therapeutic targets. Matrix Biol. 2018, 71–72, 112–127. [Google Scholar] [CrossRef] [PubMed]
- Suryadevara, V.; Huang, L.; Kim, S.J.; Cheresh, S.; Shaaya, M.; Bandela, M.; Fu, P.; Feghali-Bostwick, C.A.; Di Paolo, G.; Kamp, D.W.; et al. Role of phospholipase D in bleomycin-induced mitochondrial reactive oxygen species generation, mitochondrial DNA damage and pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L175–L187. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.; Kumar, R.K.; Iyer, A.; Boswell, S.; Gerarduzzi, C.; Dadhania, V.P.; Herbert, Z.; Joshi, N.; Luyendyk, J.P.; Humphreys, B.D.; et al. Targeting Phospholipase D4 Attenuates Kidney Fibrosis. J. Am. Soc. Nephrol. 2017, 28, 3579–3589. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Lu, W.; Schmidt Paustian, A.M.; Ge, M.Q.; Koziol-White, C.J.; Flayer, C.H.; Killingbeck, S.S.; Wang, N.; Dong, X.; Riese, M.J.; et al. Diacylglycerol kinase ζ promotes allergic airway inflammation and airway hyperresponsiveness through distinct mechanisms. Sci. Signal. 2019, 12, eaax3332. [Google Scholar] [CrossRef]
- Malerba, M.; Ricciardolo, F.; Radaeli, A.; Torregiani, C.; Ceriani, L.; Mori, E.; Bontempelli, M.; Tantucci, C.; Grassi, V. Neutrophilic inflammation and IL-8 levels in induced sputum of alpha-1-antitrypsin PiMZ subjects. Thorax 2006, 61, 129–133. [Google Scholar] [CrossRef]
- Hazari, Y.M.; Bashir, A.; Habib, M.; Bashir, S.; Habib, H.; Qasim, M.A.; Shah, N.N.; Haq, E.; Teckman, J.; Fazili, K.M. Alpha-1-antitrypsin deficiency: Genetic variations, clinical manifestations and therapeutic interventions. Mutat. Res. 2017, 773, 14–25. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baldanzi, G.; Malerba, M. DGKα in Neutrophil Biology and Its Implications for Respiratory Diseases. Int. J. Mol. Sci. 2019, 20, 5673. https://doi.org/10.3390/ijms20225673
Baldanzi G, Malerba M. DGKα in Neutrophil Biology and Its Implications for Respiratory Diseases. International Journal of Molecular Sciences. 2019; 20(22):5673. https://doi.org/10.3390/ijms20225673
Chicago/Turabian StyleBaldanzi, Gianluca, and Mario Malerba. 2019. "DGKα in Neutrophil Biology and Its Implications for Respiratory Diseases" International Journal of Molecular Sciences 20, no. 22: 5673. https://doi.org/10.3390/ijms20225673
APA StyleBaldanzi, G., & Malerba, M. (2019). DGKα in Neutrophil Biology and Its Implications for Respiratory Diseases. International Journal of Molecular Sciences, 20(22), 5673. https://doi.org/10.3390/ijms20225673