Excitatory Amino Acid Transporters (EAATs): Glutamate Transport and Beyond


Abstract
1. Glutamatergic Neurotransmission and Glutamate Transport: An Overview
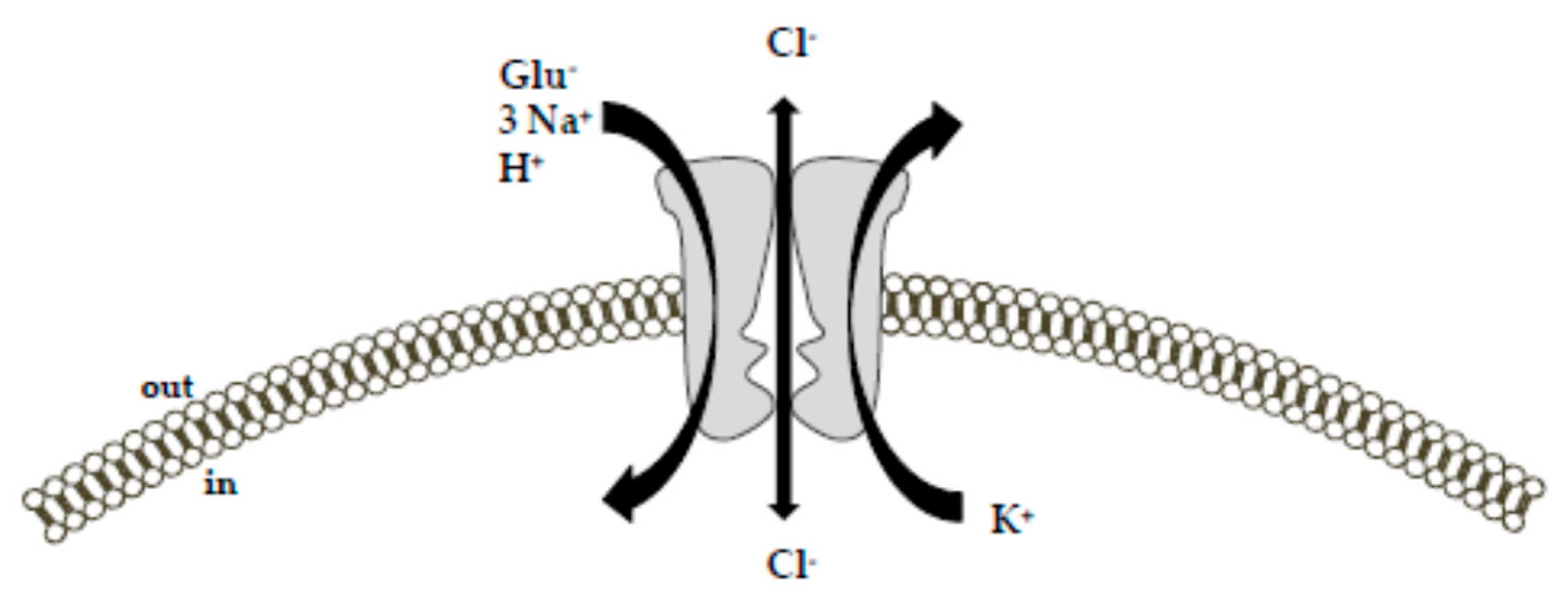
2. Na+-Dependent High-Affinity Glutamate Transporters
3. EAATs and the Maintenance of the Antioxidant Defenses
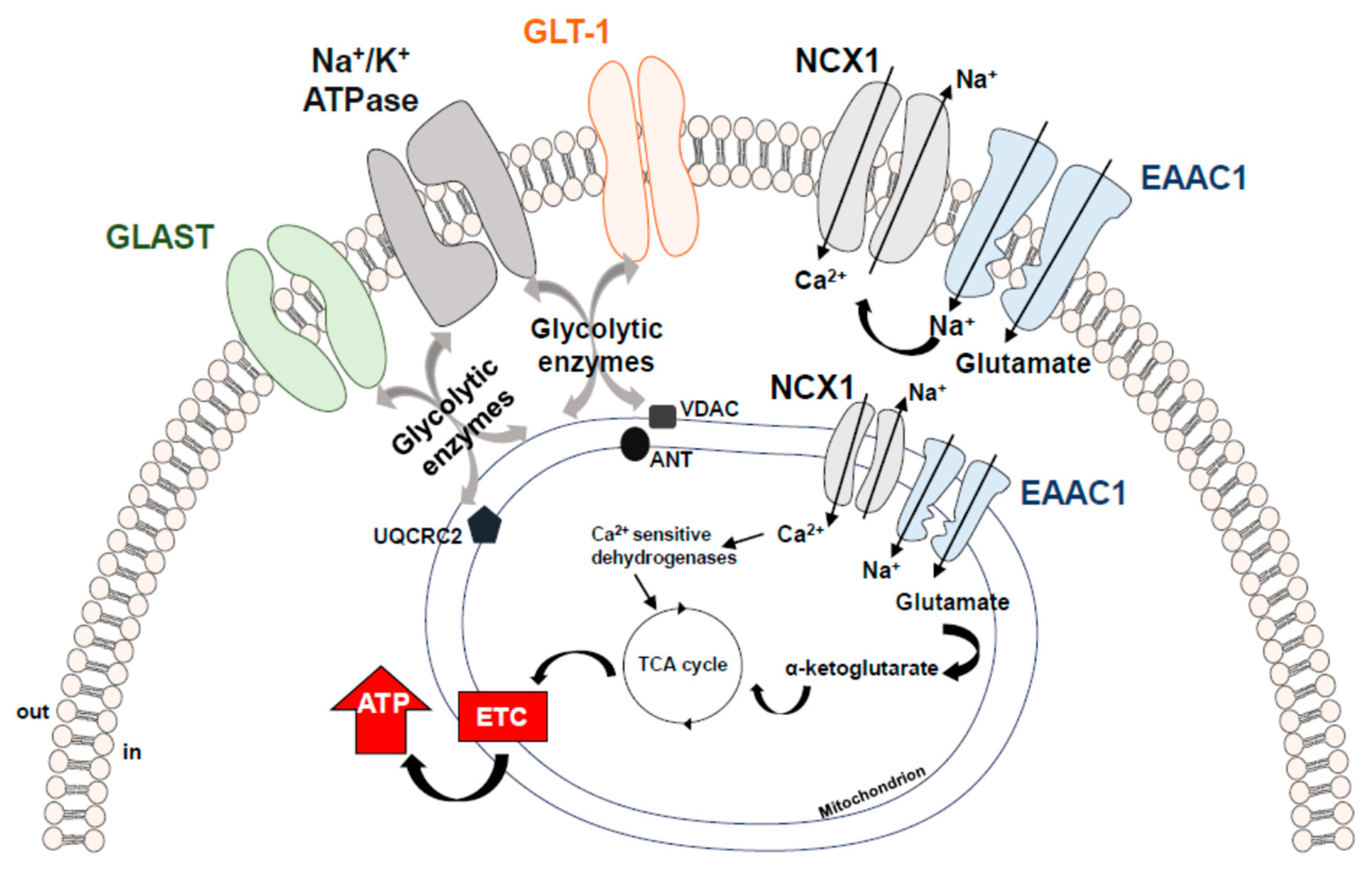
4. Metabolic Role of Glutamate and its Transport Systems
5. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AGCs | Aspartate/Glutamate Carriers |
| ANT | Adenine Nucleotide Translocator |
| AMPA | α-Amino-3-hydroxy-5-Methyl-4-Isoxazole Propionic acid |
| EAAC1 | Excitatory Amino Acid Carrier 1 |
| EAATs | Excitatory Amino Acid Transporters |
| GAPDH | Glyceraldehyde 3-Phosphate Dehydrogenase |
| GLAST | Glutamate Aspartate Transporter |
| GLT-1 | Glutamate Transporter-1 |
| GSH | γ-Glutamyl-Cysteinyl-Glycine |
| GSSG | Glutathione Disulfide |
| KA | Kainate |
| MPC | Mitochondrial Pyruvate Carrier |
| NMDA | N-Methyl-D-Aspartate |
| TCA | Tricarboxylic Acid |
| UQRC2 | Ubiquinol Cytochrome c Oxidoreductase Subunit Core 2 |
| VDAC | Voltage-Dependent Anion Channel |
| VGLUTs | Vescicular Glutamate Transporters |
References
- Gonzalez, J.; Barreto, G.E. In silico insights of L-glutamate: Structural features in vacuum and in complex with its receptor. J. Amino Acids 2013, 2013, 872058. [Google Scholar] [CrossRef] [PubMed]
- Magi, S.; Piccirillo, S.; Amoroso, S. The dual face of glutamate: From a neurotoxin to a potential survival factor-metabolic implications in health and disease. Cell. Mol. Life Sci. 2019, 76, 1473–1488. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; Levitz, J. Glutamatergic Signaling in the Central Nervous System: Ionotropic and Metabotropic Receptors in Concert. Neuron 2018, 98, 1080–1098. [Google Scholar] [CrossRef] [PubMed]
- Whetsell, W.O., Jr.; Shapira, N.A. Neuroexcitation, excitotoxicity and human neurological disease. Lab. Investig. 1993, 68, 372–387. [Google Scholar]
- Ribeiro, F.M.; Vieira, L.B.; Pires, R.G.; Olmo, R.P.; Ferguson, S.S. Metabotropic glutamate receptors and neurodegenerative diseases. Pharmacol. Res. 2017, 115, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Crupi, R.; Impellizzeri, D.; Cuzzocrea, S. Role of Metabotropic Glutamate Receptors in Neurological Disorders. Front. Mol. Neurosci. 2019, 12, 20. [Google Scholar] [CrossRef]
- Olloquequi, J.; Cornejo-Cordova, E.; Verdaguer, E.; Soriano, F.X.; Binvignat, O.; Auladell, C.; Camins, A. Excitotoxicity in the pathogenesis of neurological and psychiatric disorders: Therapeutic implications. J. Psychopharmacol. 2018, 32, 265–275. [Google Scholar] [CrossRef]
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural Transm. 2014, 121, 799–817. [Google Scholar] [CrossRef]
- Grewer, C.; Gameiro, A.; Rauen, T. SLC1 glutamate transporters. Pflug. Arch. Eur. J. Physiol. 2014, 466, 3–24. [Google Scholar] [CrossRef]
- Vandenberg, R.J.; Ryan, R.M. Mechanisms of glutamate transport. Physiol. Rev. 2013, 93, 1621–1657. [Google Scholar] [CrossRef]
- El Mestikawy, S.; Wallen-Mackenzie, A.; Fortin, G.M.; Descarries, L.; Trudeau, L.E. From glutamate co-release to vesicular synergy: Vesicular glutamate transporters. Nat. Rev. Neurosci. 2011, 12, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Fremeau, R.T., Jr.; Voglmaier, S.; Seal, R.P.; Edwards, R.H. VGLUTs define subsets of excitatory neurons and suggest novel roles for glutamate. Trends Neurosci. 2004, 27, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Danbolt, N.C.; Pines, G.; Kanner, B.I. Purification and reconstitution of the sodium- and potassium-coupled glutamate transport glycoprotein from rat brain. Biochemistry 1990, 29, 6734–6740. [Google Scholar] [CrossRef] [PubMed]
- Pines, G.; Danbolt, N.C.; Bjoras, M.; Zhang, Y.; Bendahan, A.; Eide, L.; Koepsell, H.; Storm-Mathisen, J.; Seeberg, E.; Kanner, B.I. Cloning and expression of a rat brain L-glutamate transporter. Nature 1992, 360, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Storck, T.; Schulte, S.; Hofmann, K.; Stoffel, W. Structure, expression, and functional analysis of a Na+-dependent glutamate/aspartate transporter from rat brain. Proc. Natl. Acad. Sci. USA 1992, 89, 10955–10959. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Smith, C.P.; Hediger, M.A. The elusive transporters with a high affinity for glutamate. Trends Neurosci. 1993, 16, 365–370. [Google Scholar] [CrossRef]
- Fairman, W.A.; Vandenberg, R.J.; Arriza, J.L.; Kavanaugh, M.P.; Amara, S.G. An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature 1995, 375, 599–603. [Google Scholar] [CrossRef]
- Arriza, J.L.; Eliasof, S.; Kavanaugh, M.P.; Amara, S.G. Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc. Natl. Acad. Sci. USA 1997, 94, 4155–4160. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Martin, L.; Levey, A.I.; Dykes-Hoberg, M.; Jin, L.; Wu, D.; Nash, N.; Kuncl, R.W. Localization of neuronal and glial glutamate transporters. Neuron 1994, 13, 713–725. [Google Scholar] [CrossRef]
- Mennerick, S.; Dhond, R.P.; Benz, A.; Xu, W.; Rothstein, J.D.; Danbolt, N.C.; Isenberg, K.E.; Zorumski, C.F. Neuronal expression of the glutamate transporter GLT-1 in hippocampal microcultures. J. Neurosci. 1998, 18, 4490–4499. [Google Scholar] [CrossRef]
- Kugler, P.; Schmitt, A. Glutamate transporter EAAC1 is expressed in neurons and glial cells in the rat nervous system. Glia 1999, 27, 129–142. [Google Scholar] [CrossRef]
- Domercq, M.; Sanchez-Gomez, M.V.; Areso, P.; Matute, C. Expression of glutamate transporters in rat optic nerve oligodendrocytes. Eur. J. Neurosci. 1999, 11, 2226–2236. [Google Scholar] [CrossRef] [PubMed]
- van Landeghem, F.K.; Stover, J.F.; Bechmann, I.; Bruck, W.; Unterberg, A.; Buhrer, C.; von Deimling, A. Early expression of glutamate transporter proteins in ramified microglia after controlled cortical impact injury in the rat. Glia 2001, 35, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Palos, T.P.; Ramachandran, B.; Boado, R.; Howard, B.D. Rat C6 and human astrocytic tumor cells express a neuronal type of glutamate transporter. Mol. Brain Res. 1996, 37, 297–303. [Google Scholar] [CrossRef]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Ryan, R.M.; Vandenberg, R.J. A channel in a transporter. Clin. Exp. Pharmacol. Physiol. 2005, 32, 1–6. [Google Scholar] [CrossRef]
- Owe, S.G.; Marcaggi, P.; Attwell, D. The ionic stoichiometry of the GLAST glutamate transporter in salamander retinal glia. J. Physiol. 2006, 577, 591–599. [Google Scholar] [CrossRef]
- Zerangue, N.; Kavanaugh, M.P. Flux coupling in a neuronal glutamate transporter. Nature 1996, 383, 634–637. [Google Scholar] [CrossRef]
- Kanai, Y.; Nussberger, S.; Romero, M.F.; Boron, W.F.; Hebert, S.C.; Hediger, M.A. Electrogenic properties of the epithelial and neuronal high affinity glutamate transporter. J. Biol. Chem. 1995, 270, 16561–16568. [Google Scholar] [CrossRef]
- Wadiche, J.I.; Amara, S.G.; Kavanaugh, M.P. Ion fluxes associated with excitatory amino acid transport. Neuron 1995, 15, 721–728. [Google Scholar] [CrossRef]
- Vandenberg, R.J.; Arriza, J.L.; Amara, S.G.; Kavanaugh, M.P. Constitutive ion fluxes and substrate binding domains of human glutamate transporters. J. Biol. Chem. 1995, 270, 17668–17671. [Google Scholar] [CrossRef]
- Billups, B.; Rossi, D.; Attwell, D. Anion conductance behavior of the glutamate uptake carrier in salamander retinal glial cells. J. Neurosci. 1996, 16, 6722–6731. [Google Scholar] [CrossRef]
- Sonders, M.S.; Amara, S.G. Channels in transporters. Curr. Opin. Neurobiol. 1996, 6, 294–302. [Google Scholar] [CrossRef]
- Vandenberg, R.J.; Huang, S.; Ryan, R.M. Slips, leaks and channels in glutamate transporters. Channels 2008, 2, 51–58. [Google Scholar] [CrossRef]
- Veruki, M.L.; Morkve, S.H.; Hartveit, E. Activation of a presynaptic glutamate transporter regulates synaptic transmission through electrical signaling. Nat. Neurosci. 2006, 9, 1388–1396. [Google Scholar] [CrossRef]
- Wersinger, E.; Schwab, Y.; Sahel, J.A.; Rendon, A.; Pow, D.V.; Picaud, S.; Roux, M.J. The glutamate transporter EAAT5 works as a presynaptic receptor in mouse rod bipolar cells. J. Physiol. 2006, 577, 221–234. [Google Scholar] [CrossRef]
- Gameiro, A.; Braams, S.; Rauen, T.; Grewer, C. The discovery of slowness: Low-capacity transport and slow anion channel gating by the glutamate transporter EAAT5. Biophys. J. 2011, 100, 2623–2632. [Google Scholar] [CrossRef][Green Version]
- Had-Aissouni, L. Toward a new role for plasma membrane sodium-dependent glutamate transporters of astrocytes: Maintenance of antioxidant defenses beyond extracellular glutamate clearance. Amino Acids 2012, 42, 181–197. [Google Scholar] [CrossRef]
- Chen, Y.; Swanson, R.A. The glutamate transporters EAAT2 and EAAT3 mediate cysteine uptake in cortical neuron cultures. J. Neurochem. 2003, 84, 1332–1339. [Google Scholar] [CrossRef]
- Cho, Y.; Bannai, S. Uptake of glutamate and cysteine in C-6 glioma cells and in cultured astrocytes. J. Neurochem. 1990, 55, 2091–2097. [Google Scholar] [CrossRef]
- Shanker, G.; Allen, J.W.; Mutkus, L.A.; Aschner, M. Methylmercury inhibits cysteine uptake in cultured primary astrocytes, but not in neurons. Brain Res. 2001, 914, 159–165. [Google Scholar] [CrossRef]
- Bender, A.S.; Reichelt, W.; Norenberg, M.D. Characterization of cystine uptake in cultured astrocytes. Neurochem. Int. 2000, 37, 269–276. [Google Scholar] [CrossRef]
- Hayes, D.; Wiessner, M.; Rauen, T.; McBean, G.J. Transport of L-[14C]cystine and L-[14C]cysteine by subtypes of high affinity glutamate transporters over-expressed in HEK cells. Neurochem. Int. 2005, 46, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Shiiya, A.; Kimata, M.; Maebara, K.; Tamba, M.; Sakakura, Y.; Makino, N.; Sugiyama, F.; Yagami, K.; Moriguchi, T.; et al. Redox imbalance in cystine/glutamate transporter-deficient mice. J. Biol. Chem. 2005, 280, 37423–37429. [Google Scholar] [CrossRef]
- Zerangue, N.; Kavanaugh, M.P. Interaction of L-cysteine with a human excitatory amino acid transporter. J. Physiol. 1996, 493, 419–423. [Google Scholar] [CrossRef]
- Himi, T.; Ikeda, M.; Yasuhara, T.; Nishida, M.; Morita, I. Role of neuronal glutamate transporter in the cysteine uptake and intracellular glutathione levels in cultured cortical neurons. J. Neural Transm. 2003, 110, 1337–1348. [Google Scholar] [CrossRef]
- Harada, T.; Harada, C.; Nakamura, K.; Quah, H.M.; Okumura, A.; Namekata, K.; Saeki, T.; Aihara, M.; Yoshida, H.; Mitani, A.; et al. The potential role of glutamate transporters in the pathogenesis of normal tension glaucoma. J. Clin. Investig. 2007, 117, 1763–1770. [Google Scholar] [CrossRef]
- Aoyama, K.; Suh, S.W.; Hamby, A.M.; Liu, J.; Chan, W.Y.; Chen, Y.; Swanson, R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 2006, 9, 119–126. [Google Scholar] [CrossRef]
- Berman, A.E.; Chan, W.Y.; Brennan, A.M.; Reyes, R.C.; Adler, B.L.; Suh, S.W.; Kauppinen, T.M.; Edling, Y.; Swanson, R.A. N-acetylcysteine prevents loss of dopaminergic neurons in the EAAC1-/- mouse. Ann. Neurol. 2011, 69, 509–520. [Google Scholar] [CrossRef]
- Li, L.; Zuo, Z. Glutamate transporter type 3 knockout reduces brain tolerance to focal brain ischemia in mice. J. Cereb. Blood Flow Metab. 2011, 31, 1283–1292. [Google Scholar] [CrossRef]
- McKenna, M.C. The glutamate-glutamine cycle is not stoichiometric: Fates of glutamate in brain. J. Neurosci. Res. 2007, 85, 3347–3358. [Google Scholar] [CrossRef] [PubMed]
- Panov, A.; Schonfeld, P.; Dikalov, S.; Hemendinger, R.; Bonkovsky, H.L.; Brooks, B.R. The neuromediator glutamate, through specific substrate interactions, enhances mitochondrial ATP production and reactive oxygen species generation in nonsynaptic brain mitochondria. J. Biol. Chem. 2009, 284, 14448–14456. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Hertz, E. Cataplerotic TCA cycle flux determined as glutamate-sustained oxygen consumption in primary cultures of astrocytes. Neurochem. Int. 2003, 43, 355–361. [Google Scholar] [CrossRef]
- McKenna, M.C. Glutamate pays its own way in astrocytes. Front. Endocrinol. 2013, 4, 191. [Google Scholar] [CrossRef]
- McKenna, M.C.; Tildon, J.T.; Stevenson, J.H.; Boatright, R.; Huang, S. Regulation of energy metabolism in synaptic terminals and cultured rat brain astrocytes: Differences revealed using aminooxyacetate. Dev. Neurosci. 1993, 15, 320–329. [Google Scholar] [CrossRef]
- Tildon, J.T.; Roeder, L.M.; Stevenson, J.H. Substrate oxidation by isolated rat brain mitochondria and synaptosomes. J. Neurosci. Res. 1985, 14, 207–215. [Google Scholar] [CrossRef]
- Olstad, E.; Qu, H.; Sonnewald, U. Glutamate is preferred over glutamine for intermediary metabolism in cultured cerebellar neurons. J. Cereb. Blood Flow Metab. 2007, 27, 811–820. [Google Scholar] [CrossRef]
- Divakaruni, A.S.; Wallace, M.; Buren, C.; Martyniuk, K.; Andreyev, A.Y.; Li, E.; Fields, J.A.; Cordes, T.; Reynolds, I.J.; Bloodgood, B.L.; et al. Inhibition of the mitochondrial pyruvate carrier protects from excitotoxic neuronal death. J. Cell. Biol. 2017, 216, 1091–1105. [Google Scholar] [CrossRef]
- Fendt, S.M.; Verstreken, P. Neurons eat glutamate to stay alive. J. Cell. Biol. 2017, 216, 863–865. [Google Scholar] [CrossRef]
- Magi, S.; Lariccia, V.; Castaldo, P.; Arcangeli, S.; Nasti, A.A.; Giordano, A.; Amoroso, S. Physical and functional interaction of NCX1 and EAAC1 transporters leading to glutamate-enhanced ATP production in brain mitochondria. PLoS ONE 2012, 7, e34015. [Google Scholar] [CrossRef]
- Palmieri, L.; Pardo, B.; Lasorsa, F.M.; del Arco, A.; Kobayashi, K.; Iijima, M.; Runswick, M.J.; Walker, J.E.; Saheki, T.; Satrustegui, J.; et al. Citrin and aralar1 are Ca2+-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 2001, 20, 5060–5069. [Google Scholar] [CrossRef] [PubMed]
- Fiermonte, G.; Palmieri, L.; Todisco, S.; Agrimi, G.; Palmieri, F.; Walker, J.E. Identification of the mitochondrial glutamate transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution of two human isoforms. J. Biol. Chem. 2002, 277, 19289–19294. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Pozos, E.; Chi-Castaneda, D.; Ortega, A. Glial Glutamate Transporters as Signaling Molecules. Adv. Neurobiol. 2017, 16, 185–198. [Google Scholar] [PubMed]
- Ralphe, J.C.; Segar, J.L.; Schutte, B.C.; Scholz, T.D. Localization and function of the brain excitatory amino acid transporter type 1 in cardiac mitochondria. J. Mol. Cell. Cardiol. 2004, 37, 33–41. [Google Scholar] [CrossRef]
- Ralphe, J.C.; Bedell, K.; Segar, J.L.; Scholz, T.D. Correlation between myocardial malate/aspartate shuttle activity and EAAT1 protein expression in hyper- and hypothyroidism. Am. J. Physiol. Heart. Circ. Physiol. 2005, 288, H2521–H2526. [Google Scholar] [CrossRef]
- Annunziato, L.; Pignataro, G.; Di Renzo, G.F. Pharmacology of brain Na+/Ca2+ exchanger: From molecular biology to therapeutic perspectives. Pharmacol. Rev. 2004, 56, 633–654. [Google Scholar] [CrossRef]
- Blaustein, M.P.; Lederer, W.J. Sodium/calcium exchange: Its physiological implications. Physiol. Rev. 1999, 79, 763–854. [Google Scholar] [CrossRef]
- Philipson, K.D.; Nicoll, D.A. Sodium-calcium exchange: A molecular perspective. Annu. Rev. Physiol. 2000, 62, 111–133. [Google Scholar] [CrossRef]
- Quednau, B.D.; Nicoll, D.A.; Philipson, K.D. Tissue specificity and alternative splicing of the Na+/Ca2+ exchanger isoforms NCX1, NCX2, and NCX3 in rat. Am. J. Physiol. 1997, 272, C1250–C1261. [Google Scholar] [CrossRef]
- Giladi, M.; Lee, S.Y.; Ariely, Y.; Teldan, Y.; Granit, R.; Strulovich, R.; Haitin, Y.; Chung, K.Y.; Khananshvili, D. Structure-based dynamic arrays in regulatory domains of sodium-calcium exchanger (NCX) isoforms. Sci. Rep. 2017, 7, 993. [Google Scholar] [CrossRef]
- Magi, S.; Arcangeli, S.; Castaldo, P.; Nasti, A.A.; Berrino, L.; Piegari, E.; Bernardini, R.; Amoroso, S.; Lariccia, V. Glutamate-induced ATP synthesis: Relationship between plasma membrane Na+/Ca2+ exchanger and excitatory amino acid transporters in brain and heart cell models. Mol. Pharmacol. 2013, 84, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Maiolino, M.; Castaldo, P.; Lariccia, V.; Piccirillo, S.; Amoroso, S.; Magi, S. Essential role of the Na+-Ca2+ exchanger (NCX) in glutamate-enhanced cell survival in cardiac cells exposed to hypoxia/reoxygenation. Sci. Rep. 2017, 7, 13073. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, S.; Castaldo, P.; Macri, M.L.; Amoroso, S.; Magi, S. Glutamate as a potential “survival factor” in an in vitro model of neuronal hypoxia/reoxygenation injury: Leading role of the Na+/Ca2+ exchanger. Cell. Death Dis. 2018, 9, 731. [Google Scholar] [CrossRef] [PubMed]
- Gegelashvili, G.; Bjerrum, O.J. Glutamate transport system as a key constituent of glutamosome: Molecular pathology and pharmacological modulation in chronic pain. Neuropharmacology 2019, 153, 53–62. [Google Scholar] [CrossRef]
- Genda, E.N.; Jackson, J.G.; Sheldon, A.L.; Locke, S.F.; Greco, T.M.; O′Donnell, J.C.; Spruce, L.A.; Xiao, R.; Guo, W.; Putt, M.; et al. Co-compartmentalization of the astroglial glutamate transporter, GLT-1, with glycolytic enzymes and mitochondria. J. Neurosci. 2011, 31, 18275–18288. [Google Scholar] [CrossRef]
- Bauer, D.E.; Jackson, J.G.; Genda, E.N.; Montoya, M.M.; Yudkoff, M.; Robinson, M.B. The glutamate transporter, GLAST, participates in a macromolecular complex that supports glutamate metabolism. Neurochem. Int. 2012, 61, 566–574. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Glutamate Transporters Subtype | Rodent Homologue | Cell Type | DISTRIBUTION |
|---|---|---|---|
| EAAT1 | GLAST | Astrocytes, oligodendrocytes [2,19] | Cerebellum, cortex, spinal cord |
| EAAT2 | GLT-1 | Astrocytes [2,20] | Through the brain and spinal cord |
| EAAT3 | EAAC1 | Mostly neurons. Also found in cells of glial origin (i.e., oligodendrocytes, glioma cells) [2,21,22,23] | Hippocampus, striatum, cerebellum |
| EAAT4 | EAAT4 | Purkinje cells [2,24] | Cerebellum |
| EAAT5 | EAAT5 | Photoreceptor and bipolar cells [2,25] | Retina |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magi, S.; Piccirillo, S.; Amoroso, S.; Lariccia, V. Excitatory Amino Acid Transporters (EAATs): Glutamate Transport and Beyond. Int. J. Mol. Sci. 2019, 20, 5674. https://doi.org/10.3390/ijms20225674
Magi S, Piccirillo S, Amoroso S, Lariccia V. Excitatory Amino Acid Transporters (EAATs): Glutamate Transport and Beyond. International Journal of Molecular Sciences. 2019; 20(22):5674. https://doi.org/10.3390/ijms20225674
Chicago/Turabian StyleMagi, Simona, Silvia Piccirillo, Salvatore Amoroso, and Vincenzo Lariccia. 2019. "Excitatory Amino Acid Transporters (EAATs): Glutamate Transport and Beyond" International Journal of Molecular Sciences 20, no. 22: 5674. https://doi.org/10.3390/ijms20225674
APA StyleMagi, S., Piccirillo, S., Amoroso, S., & Lariccia, V. (2019). Excitatory Amino Acid Transporters (EAATs): Glutamate Transport and Beyond. International Journal of Molecular Sciences, 20(22), 5674. https://doi.org/10.3390/ijms20225674