Lifestyle and Food Habits Impact on Chronic Diseases: Roles of PPARs

 ,
,  ,
,  , ,
, , 
Abstract
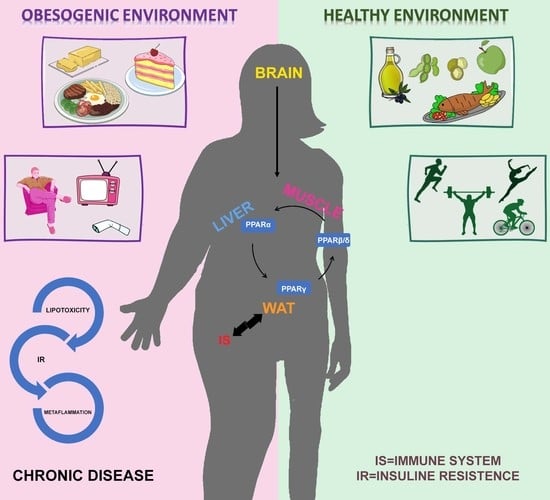
1. Introduction
2. PPARs and Metabolism
3. Systemic Low-Grade Inflammation
3.1. Adipose Tissue the Core of Metaflammation
3.2. Insulin Resistance and Inflammation
4. Chronic Diseases, from Lifestyle to PPARs
4.1. Polycystic Ovary Syndrome
4.2. Non-Alcoholic Fatty Liver Disease (NAFLD)
5. Conclusions
Funding
Conflicts of Interest
References
- Reaven, G.M. The Insulin Resistance Syndrome: Definition and Dietary Approaches to Treatment. Annu. Rev. Nutr. 2005, 25, 391–406. [Google Scholar] [CrossRef]
- Dandona, P.; Aljada, A.; Chaudhuri, A.; Mohanty, P.; Garg, R. Metabolic Syndrome: A Comprehensive Perspective Based on Interactions between Obesity, Diabetes, and Inflammation. Circulation 2005, 111, 1448–1454. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Kassi, E.; Pervanidou, P.; Kaltsas, G.; Chrousos, G. Metabolic syndrome: Definitions and controversies. BMC Med. 2011, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Cornier, M.-A.; Dabelea, D.; Hernandez, T.L.; Lindstrom, R.C.; Steig, A.J.; Stob, N.R.; Van Pelt, R.E.; Wang, H.; Eckel, R.H. The Metabolic Syndrome. Endocr. Rev. 2008, 29, 777–822. [Google Scholar] [CrossRef] [PubMed]
- Galland, L. Diet and Inflammation. Nutr. Clin. Pract. 2010, 25, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Kunnumakkara, A.B.; Kunnumakara, A.B.; Sundaram, C.; Harikumar, K.B.; Tharakan, S.T.; Lai, O.S.; Sung, B.; Aggarwal, B.B. Cancer is a preventable disease that requires major lifestyle changes. Pharm. Res. 2008, 25, 2097–2116. [Google Scholar] [CrossRef]
- Egger, G.; Dixon, J. Inflammatory effects of nutritional stimuli: Further support for the need for a big picture approach to tackling obesity and chronic disease. Obes. Rev. 2010, 11, 137–149. [Google Scholar] [CrossRef]
- Egger, G.; Dixon, J. Non-nutrient causes of low-grade, systemic inflammation: Support for a ‘canary in the mineshaft’ view of obesity in chronic disease: Inflammation and obesity. Obes. Rev. 2011, 12, 339–345. [Google Scholar] [CrossRef]
- Blaak, E.E. Carbohydrate quantity and quality and cardio-metabolic risk. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 289–293. [Google Scholar] [CrossRef]
- Kaartinen, N.E.; Knekt, P.; Kanerva, N.; Valsta, L.M.; Eriksson, J.G.; Rissanen, H.; Jääskeläinen, T.; Männistö, S. Dietary carbohydrate quantity and quality in relation to obesity: A pooled analysis of three Finnish population-based studies. Scand. J. Public Health 2016, 44, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Cornejo-Monthedoro, A.; Negreiros-Sánchez, I.; Del Águila, C.; Ysla-Marquillo, M.; Mayta-Tristán, P. Association between dietary glycemic load and metabolic syndrome in obese children and adolescents. Arch. Argent. Pediatr. 2017, 115, 323–330. [Google Scholar] [PubMed]
- Silva, K.C.; Neri Nobre, L.; Emanuelle de Castro Ferreira Vicente, S.; Lopes Moreira, L.; do Carmo Lessa, A.; Lamounier, J.A. Influence of glycemic index and glycemic load of the diet on the risk of overweight and adiposity in childhood. Rev. Paul. Pediatr. 2016, 34, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Milajerdi, A.; Saneei, P.; Larijani, B.; Esmaillzadeh, A. The effect of dietary glycemic index and glycemic load on inflammatory biomarkers: A systematic review and meta-analysis of randomized clinical trials. Am. J. Clin. Nutr. 2018, 107, 593–606. [Google Scholar] [CrossRef]
- Bell, S.J.; Sears, B. Low-glycemic-load diets: Impact on obesity and chronic diseases. Crit. Rev. Food Sci. Nutr. 2003, 43, 357–377. [Google Scholar] [CrossRef]
- Taylor, M.K.; Sullivan, D.K.; Swerdlow, R.H.; Vidoni, E.D.; Morris, J.K.; Mahnken, J.D.; Burns, J.M. A high-glycemic diet is associated with cerebral amyloid burden in cognitively normal older adults. Am. J. Clin. Nutr. 2017, 106, 1463–1470. [Google Scholar] [CrossRef]
- Hu, J.; La Vecchia, C.; Augustin, L.S.; Negri, E.; de Groh, M.; Morrison, H.; Mery, L. Canadian Cancer Registries Epidemiology Research Group Glycemic index, glycemic load and cancer risk. Ann. Oncol. 2013, 24, 245–251. [Google Scholar] [CrossRef]
- Jiménez-Gómez, Y.; López-Miranda, J.; Blanco-Colio, L.M.; Marín, C.; Pérez-Martínez, P.; Ruano, J.; Paniagua, J.A.; Rodríguez, F.; Egido, J.; Pérez-Jiménez, F. Olive oil and walnut breakfasts reduce the postprandial inflammatory response in mononuclear cells compared with a butter breakfast in healthy men. Atherosclerosis 2009, 204, e70–e76. [Google Scholar] [CrossRef]
- Mozaffarian, D. Trans fatty acids—Effects on systemic inflammation and endothelial function. Atheroscler. Suppl. 2006, 7, 29–32. [Google Scholar] [CrossRef]
- Mozaffarian, D.; Aro, A.; Willett, W.C. Health effects of trans-fatty acids: Experimental and observational evidence. Eur. J. Clin. Nutr. 2009, 63, S5–S21. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N. Endogenous pro-resolving and anti-inflammatory lipid mediators: A new pharmacologic genus. Br. J. Pharmacol. 2008, 153 (Suppl. 1), S200–S215. [Google Scholar] [CrossRef]
- Simopoulos, A.P. The Importance of the Omega-6/Omega-3 Fatty Acid Ratio in Cardiovascular Disease and Other Chronic Diseases. Exp. Biol. Med. 2008, 233, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. n−3 Polyunsaturated fatty acids, inflammation, and inflammatory diseases. Am. J. Clin. Nutr. 2006, 83, 1505S–1519S. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Liu, K.; Daviglus, M.L.; Jenny, N.S.; Mayer-Davis, E.; Jiang, R.; Steffen, L.; Siscovick, D.; Tsai, M.; Herrington, D. Associations of Dietary Long-Chain n-3 Polyunsaturated Fatty Acids and Fish With Biomarkers of Inflammation and Endothelial Activation (from the Multi-Ethnic Study of Atherosclerosis [MESA]). Am. J. Cardiol. 2009, 103, 1238–1243. [Google Scholar] [CrossRef] [PubMed]
- Huffman, K.M.; Samsa, G.P.; Slentz, C.A.; Duscha, B.D.; Johnson, J.L.; Bales, C.W.; Tanner, C.J.; Houmard, J.A.; Kraus, W.E. Response of high-sensitivity C-reactive protein to exercise training in an at-risk population. Am. Heart J. 2006, 152, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Petersen, A.M.W.; Pedersen, B.K. The anti-inflammatory effect of exercise. J. Appl. Physiol. 2005, 98, 1154–1162. [Google Scholar] [CrossRef]
- Roubenoff, R. Molecular Basis of Inflammation: Relationships between Catabolic Cytokines, Hormones, Energy Balance, and Muscle. J. Parenter. Enter. Nutr. 2008, 32, 630–632. [Google Scholar] [CrossRef]
- Handschin, C.; Spiegelman, B.M. The role of exercise and PGC1α in inflammation and chronic disease. Nature 2008, 454, 463–469. [Google Scholar] [CrossRef]
- Petrakis, D.; Vassilopoulou, L.; Mamoulakis, C.; Psycharakis, C.; Anifantaki, A.; Sifakis, S.; Docea, A.; Tsiaoussis, J.; Makrigiannakis, A.; Tsatsakis, A. Endocrine Disruptors Leading to Obesity and Related Diseases. Int. J. Environ. Res. Public Health 2017, 14, 1282. [Google Scholar] [CrossRef]
- Wahli, W.; Michalik, L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012, 23, 351–363. [Google Scholar] [CrossRef]
- Hong, F.; Pan, S.; Guo, Y.; Xu, P.; Zhai, Y. PPARs as Nuclear Receptors for Nutrient and Energy Metabolism. Molecules 2019, 24, 2545. [Google Scholar] [CrossRef] [PubMed]
- Schupp, M.; Lazar, M.A. Endogenous Ligands for Nuclear Receptors: Digging Deeper. J. Biol. Chem. 2010, 285, 40409–40415. [Google Scholar] [CrossRef]
- Bensinger, S.J.; Tontonoz, P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature 2008, 454, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Woller, A.; Duez, H.; Staels, B.; Lefranc, M. A Mathematical Model of the Liver Circadian Clock Linking Feeding and Fasting Cycles to Clock Function. Cell Rep. 2016, 17, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Caputo, T.; Gilardi, F.; Desvergne, B. From chronic overnutrition to metaflammation and insulin resistance: Adipose tissue and liver contributions. FEBS Lett. 2017, 591, 3061–3088. [Google Scholar] [CrossRef]
- Montagner, A.; Polizzi, A.; Fouché, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Régnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef]
- Dubois, V.; Eeckhoute, J.; Lefebvre, P.; Staels, B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J. Clin. Investig. 2017, 127, 1202–1214. [Google Scholar] [CrossRef]
- Lefebvre, P. Sorting out the roles of PPAR in energy metabolism and vascular homeostasis. J. Clin. Investig. 2006, 116, 571–580. [Google Scholar] [CrossRef]
- Jitrapakdee, S.; Slawik, M.; Medina-Gomez, G.; Campbell, M.; Wallace, J.C.; Sethi, J.K.; O’Rahilly, S.; Vidal-Puig, A.J. The Peroxisome Proliferator-activated Receptor-γ Regulates Murine Pyruvate Carboxylase Gene Expression in Vivo and in Vitro. J. Biol. Chem. 2005, 280, 27466–27476. [Google Scholar] [CrossRef]
- Patsouris, D.; Mandard, S.; Voshol, P.J.; Escher, P.; Tan, N.S.; Havekes, L.M.; Koenig, W.; März, W.; Tafuri, S.; Wahli, W.; et al. PPARα governs glycerol metabolism. J. Clin. Investig. 2004, 114, 94–103. [Google Scholar] [CrossRef]
- Sengupta, S.; Peterson, T.R.; Laplante, M.; Oh, S.; Sabatini, D.M. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature 2010, 468, 1100–1104. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Hirata, M.; Aoki, Y.; Iwase, M.; Takahashi, H.; Kim, M.; Li, Y.; Jheng, H.-F.; Nomura, W.; Takahashi, N.; et al. The hepatokine FGF21 is crucial for peroxisome proliferator-activated receptor-α agonist-induced amelioration of metabolic disorders in obese mice. J. Biol. Chem. 2017, 292, 9175–9190. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Lee, J.-Y.; Teraminami, A.; Kim, Y.-I.; Hirai, S.; Uemura, T.; Inoue, H.; Takahashi, N.; Kawada, T. Activation of peroxisome proliferator-activated receptor-alpha stimulates both differentiation and fatty acid oxidation in adipocytes. J. Lipid Res. 2011, 52, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Goto, T. A review of the studies on food-derived factors which regulate energy metabolism via the modulation of lipid-sensing nuclear receptors. Biosci. Biotechnol. Biochem. 2019, 83, 579–588. [Google Scholar] [CrossRef]
- Coskun, T.; Bina, H.A.; Schneider, M.A.; Dunbar, J.D.; Hu, C.C.; Chen, Y.; Moller, D.E.; Kharitonenkov, A. Fibroblast Growth Factor 21 Corrects Obesity in Mice. Endocrinology 2008, 149, 6018–6027. [Google Scholar] [CrossRef]
- Fisher, F.M.; Kleiner, S.; Douris, N.; Fox, E.C.; Mepani, R.J.; Verdeguer, F.; Wu, J.; Kharitonenkov, A.; Flier, J.S.; Maratos-Flier, E.; et al. FGF21 regulates PGC-1 and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 2012, 26, 271–281. [Google Scholar] [CrossRef]
- Takahashi, H.; Goto, T.; Yamazaki, Y.; Kamakari, K.; Hirata, M.; Suzuki, H.; Shibata, D.; Nakata, R.; Inoue, H.; Takahashi, N.; et al. Metabolomics reveal 1-palmitoyl lysophosphatidylcholine production by peroxisome proliferator-activated receptor α. J. Lipid Res. 2015, 56, 254–265. [Google Scholar] [CrossRef]
- Takahashi, H.; Sanada, K.; Nagai, H.; Li, Y.; Aoki, Y.; Ara, T.; Seno, S.; Matsuda, H.; Yu, R.; Kawada, T.; et al. Over-expression of PPARα in obese mice adipose tissue improves insulin sensitivity. Biochem. Biophys. Res. Commun. 2017, 493, 108–114. [Google Scholar] [CrossRef]
- Tontonoz, P.; Spiegelman, B.M. Fat and Beyond: The Diverse Biology of PPARγ. Annu. Rev. Biochem. 2008, 77, 289–312. [Google Scholar] [CrossRef]
- Lehrke, M.; Lazar, M.A. The Many Faces of PPARγ. Cell 2005, 123, 993–999. [Google Scholar] [CrossRef]
- Rosen, E.D. C/EBPalpha induces adipogenesis through PPARgamma: A unified pathway. Genes Dev. 2002, 16, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Lefterova, M.I.; Haakonsson, A.K.; Lazar, M.A.; Mandrup, S. PPARγ and the global map of adipogenesis and beyond. Trends Endocrinol. Metab. 2014, 25, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Sakai, J.; Kajimura, S. Transcriptional and epigenetic control of brown and beige adipose cell fate and function. Nat. Rev. Mol. Cell Biol. 2016, 17, 480–495. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.M.; Moore, L.B.; Smith-Oliver, T.A.; Wilkison, W.O.; Willson, T.M.; Kliewer, S.A. An Antidiabetic Thiazolidinedione Is a High Affinity Ligand for Peroxisome Proliferator-activated Receptor γ (PPARγ). J. Biol. Chem. 1995, 270, 12953–12956. [Google Scholar] [CrossRef]
- Sharma, A.M.; Staels, B. Peroxisome Proliferator-Activated Receptor γ and Adipose Tissue—Understanding Obesity-Related Changes in Regulation of Lipid and Glucose Metabolism. J. Clin. Endocrinol. Metab. 2007, 92, 386–395. [Google Scholar] [CrossRef]
- Egger, G.; Dixon, J. Obesity and chronic disease: Always offender or often just accomplice? Br. J. Nutr. 2009, 102, 1238–1242. [Google Scholar] [CrossRef]
- Wildman, R.P. The Obese without Cardiometabolic Risk Factor Clustering and the Normal Weight with Cardiometabolic Risk Factor Clustering: Prevalence and Correlates of 2 Phenotypes Among the US Population (NHANES 1999-2004). Arch. Intern. Med. 2008, 168, 1617. [Google Scholar] [CrossRef]
- Hoffstedt, J.; Arner, E.; Wahrenberg, H.; Andersson, D.P.; Qvisth, V.; Löfgren, P.; Rydén, M.; Thörne, A.; Wirén, M.; Palmér, M.; et al. Regional impact of adipose tissue morphology on the metabolic profile in morbid obesity. Diabetologia 2010, 53, 2496–2503. [Google Scholar] [CrossRef]
- Gustafson, B.; Gogg, S.; Hedjazifar, S.; Jenndahl, L.; Hammarstedt, A.; Smith, U. Inflammation and impaired adipogenesis in hypertrophic obesity in man. Am. J. Physiol.-Endocrinol. Metab. 2009, 297, E999–E1003. [Google Scholar] [CrossRef]
- Lee, M.-J.; Wu, Y.; Fried, S.K. Adipose tissue remodeling in pathophysiology of obesity. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 371–376. [Google Scholar] [CrossRef]
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, B.; Hammarstedt, A.; Hedjazifar, S.; Smith, U. Restricted Adipogenesis in Hypertrophic Obesity: The Role of WISP2, WNT, and BMP4. Diabetes 2013, 62, 2997–3004. [Google Scholar] [CrossRef] [PubMed]
- Klöting, N.; Blüher, M. Adipocyte dysfunction, inflammation and metabolic syndrome. Rev. Endocr. Metab. Disord. 2014, 15, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Vishvanath, L.; Busbuso, N.C.; Hepler, C.; Shan, B.; Sharma, A.X.; Chen, S.; Yu, X.; An, Y.A.; Zhu, Y.; et al. De novo adipocyte differentiation from Pdgfrβ+ preadipocytes protects against pathologic visceral adipose expansion in obesity. Nat. Commun. 2018, 9, 890. [Google Scholar] [CrossRef] [PubMed]
- Stump, M.; Guo, D.-F.; Lu, K.-T.; Mukohda, M.; Liu, X.; Rahmouni, K.; Sigmund, C.D. Effect of selective expression of dominant-negative PPARγ in pro-opiomelanocortin neurons on the control of energy balance. Physiol. Genom. 2016, 48, 491–501. [Google Scholar] [CrossRef]
- Long, L.; Toda, C.; Jeong, J.K.; Horvath, T.L.; Diano, S. PPARγ ablation sensitizes proopiomelanocortin neurons to leptin during high-fat feeding. J. Clin. Investig. 2014, 124, 4017–4027. [Google Scholar] [CrossRef]
- Kocalis, H.E.; Turney, M.K.; Printz, R.L.; Laryea, G.N.; Muglia, L.J.; Davies, S.S.; Stanwood, G.D.; McGuinness, O.P.; Niswender, K.D. Neuron-Specific Deletion of Peroxisome Proliferator-Activated Receptor Delta (PPARδ) in Mice Leads to Increased Susceptibility to Diet-Induced Obesity. PLoS ONE 2012, 7, e42981. [Google Scholar] [CrossRef]
- Wang, H.; Liu, H.; Liu, R.M. Gender difference in glutathione metabolism during aging in mice. Exp. Gerontol. 2003, 38, 507–517. [Google Scholar] [CrossRef]
- Tanaka, T.; Yamamoto, J.; Iwasaki, S.; Asaba, H.; Hamura, H.; Ikeda, Y.; Watanabe, M.; Magoori, K.; Ioka, R.X.; Tachibana, K.; et al. Activation of peroxisome proliferator-activated receptor induces fatty acid -oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl. Acad. Sci. USA 2003, 100, 15924–15929. [Google Scholar] [CrossRef]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauça, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef]
- Wang, Y.-X.; Zhang, C.-L.; Yu, R.T.; Cho, H.K.; Nelson, M.C.; Bayuga-Ocampo, C.R.; Ham, J.; Kang, H.; Evans, R.M. Regulation of Muscle Fiber Type and Running Endurance by PPARδ. PLoS Biol. 2004, 2, e294. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Waizenegger, W.; Lin, C.S.; Sorrentino, V.; He, M.-X.; Wall, C.E.; Li, H.; Liddle, C.; Yu, R.T.; Atkins, A.R.; et al. PPARδ Promotes Running Endurance by Preserving Glucose. Cell Metab. 2017, 25, 1186–1193. [Google Scholar] [CrossRef] [PubMed]
- Holst, D.; Luquet, S.; Nogueira, V.; Kristiansen, K.; Leverve, X.; Grimaldi, P.A. Nutritional regulation and role of peroxisome proliferator-activated receptor δ in fatty acid catabolism in skeletal muscle. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2003, 1633, 43–50. [Google Scholar] [CrossRef]
- Oliver, W.R.; Shenk, J.L.; Snaith, M.R.; Russell, C.S.; Plunket, K.D.; Bodkin, N.L.; Lewis, M.C.; Winegar, D.A.; Sznaidman, M.L.; Lambert, M.H.; et al. A selective peroxisome proliferator-activated receptor agonist promotes reverse cholesterol transport. Proc. Natl. Acad. Sci. USA 2001, 98, 5306–5311. [Google Scholar] [CrossRef]
- Luquet, S.; Lopez-Soriano, J.; Holst, D.; Fredenrich, A.; Melki, J.; Rassoulzadegan, M.; Grimaldi, P.A. Peroxisome proliferator-activated receptor δ controls muscle development and oxidative capability. FASEB J. 2003, 17, 2299–2301. [Google Scholar] [CrossRef]
- Watt, M.J.; Southgate, R.J.; Holmes, A.G.; Febbraio, M.A. Suppression of plasma free fatty acids upregulates peroxisome proliferator-activated receptor (PPAR) α and δ and PPAR coactivator 1α in human skeletal muscle, but not lipid regulatory genes. J. Mol. Endocrinol. 2004, 33, 533–544. [Google Scholar] [CrossRef]
- Mahoney, D.J.; Parise, G.; Melov, S.; Safdar, A.; Tarnopolsky, M.A. Analysis of global mRNA expression in human skeletal muscle during recovery from endurance exercise. FASEB J. 2005, 19, 1498–1500. [Google Scholar] [CrossRef]
- Baskin, K.K.; Winders, B.R.; Olson, E.N. Muscle as a “Mediator” of Systemic Metabolism. Cell Metab. 2015, 21, 237–248. [Google Scholar] [CrossRef]
- Pette, D.; Staron, R.S. Myosin isoforms, muscle fiber types, and transitions. Microsc. Res. Tech. 2000, 50, 500–509. [Google Scholar] [CrossRef]
- Bassel-Duby, R.; Olson, E.N. Signaling Pathways in Skeletal Muscle Remodeling. Annu. Rev. Biochem. 2006, 75, 19–37. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Zanuso, S.; Jimenez, A.; Pugliese, G.; Corigliano, G.; Balducci, S. Exercise for the management of type 2 diabetes: A review of the evidence. Acta Diabetol. 2010, 47, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Willis, L.H.; Slentz, C.A.; Bateman, L.A.; Shields, A.T.; Piner, L.W.; Bales, C.W.; Houmard, J.A.; Kraus, W.E. Effects of aerobic and/or resistance training on body mass and fat mass in overweight or obese adults. J. Appl. Physiol. 2012, 113, 1831–1837. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Febbraio, M.A. Muscles, exercise and obesity: Skeletal muscle as a secretory organ. Nat. Rev. Endocrinol. 2012, 8, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Weigert, C.; Lehmann, R.; Hartwig, S.; Lehr, S. The secretome of the working human skeletal muscle--a promising opportunity to combat the metabolic disaster? Proteom. Clin. Appl. 2014, 8, 5–18. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Steensberg, A.; Fischer, C.; Keller, C.; Keller, P.; Plomgaard, P.; Wolsk-Petersen, E.; Febbraio, M. The metabolic role of IL-6 produced during exercise: Is IL-6 an exercise factor? Proc. Nutr. Soc. 2004, 63, 263–267. [Google Scholar] [CrossRef]
- Muñoz-Cánoves, P.; Scheele, C.; Pedersen, B.K.; Serrano, A.L. Interleukin-6 myokine signaling in skeletal muscle: A double-edged sword? FEBS J. 2013, 280, 4131–4148. [Google Scholar] [CrossRef]
- Huang, Z.; Chen, X.; Chen, D. Myostatin: A novel insight into its role in metabolism, signal pathways, and expression regulation. Cell. Signal. 2011, 23, 1441–1446. [Google Scholar] [CrossRef]
- Mattijssen, F.; Kersten, S. Regulation of triglyceride metabolism by Angiopoietin-like proteins. Biochim. Biophys. Acta 2012, 1821, 782–789. [Google Scholar] [CrossRef]
- Rao, R.R.; Long, J.Z.; White, J.P.; Svensson, K.J.; Lou, J.; Lokurkar, I.; Jedrychowski, M.P.; Ruas, J.L.; Wrann, C.D.; Lo, J.C.; et al. Meteorin-like is a hormone that regulates immune-adipose interactions to increase beige fat thermogenesis. Cell 2014, 157, 1279–1291. [Google Scholar] [CrossRef] [PubMed]
- Sinha, M.; Jang, Y.C.; Oh, J.; Khong, D.; Wu, E.Y.; Manohar, R.; Miller, C.; Regalado, S.G.; Loffredo, F.S.; Pancoast, J.R.; et al. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science 2014, 344, 649–652. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.D.; Boström, P.; O’Sullivan, J.F.; Schinzel, R.T.; Lewis, G.D.; Dejam, A.; Lee, Y.-K.; Palma, M.J.; Calhoun, S.; Georgiadi, A.; et al. β-Aminoisobutyric acid induces browning of white fat and hepatic β-oxidation and is inversely correlated with cardiometabolic risk factors. Cell Metab. 2014, 19, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Schoonjans, K.; Peinado-Onsurbe, J.; Lefebvre, A.M.; Heyman, R.A.; Briggs, M.; Deeb, S.; Staels, B.; Auwerx, J. PPARalpha and PPARgamma activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. EMBO J. 1996, 15, 5336–5348. [Google Scholar] [CrossRef] [PubMed]
- Vu-Dac, N.; Gervois, P.; Jakel, H.; Nowak, M.; Bauge, E.; Dehondt, H.; Staels, B.; Pennacchio, L.A.; Rubin, E.M.; Fruchart-Najib, J.; et al. Apolipoprotein A5, a crucial determinant of plasma triglyceride levels, is highly responsive to peroxisome proliferator-activated receptor alpha activators. J. Biol. Chem. 2003, 278, 17982–17985. [Google Scholar] [CrossRef] [PubMed]
- Berthou, L.; Duverger, N.; Emmanuel, F.; Langouët, S.; Auwerx, J.; Guillouzo, A.; Fruchart, J.C.; Rubin, E.; Denèfle, P.; Staels, B.; et al. Opposite regulation of human versus mouse apolipoprotein A-I by fibrates in human apolipoprotein A-I transgenic mice. J. Clin. Investig. 1996, 97, 2408–2416. [Google Scholar] [CrossRef]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef]
- Gross, B.; Pawlak, M.; Lefebvre, P.; Staels, B. PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat. Rev. Endocrinol. 2017, 13, 36–49. [Google Scholar] [CrossRef]
- Derosa, G.; Sahebkar, A.; Maffioli, P. The role of various peroxisome proliferator-activated receptors and their ligands in clinical practice. J. Cell. Physiol. 2018, 233, 153–161. [Google Scholar] [CrossRef]
- Peeters, A.; Baes, M. Role of PPAR in Hepatic Carbohydrate Metabolism. PPAR Res. 2010, 2010, 1–12. [Google Scholar] [CrossRef]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Hatano, B.; Zhao, M.; Yen, C.-C.; Kang, K.; Reilly, S.M.; Gangl, M.R.; Gorgun, C.; Balschi, J.A.; Ntambi, J.M.; et al. Role of Peroxisome Proliferator-activated Receptor δ/β in Hepatic Metabolic Regulation. J. Biol. Chem. 2011, 286, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Rubins, H.B.; Robins, S.J.; Collins, D. The veterans affairs high-density lipoprotein intervention trial: Baseline characteristics of normocholesterolemic men with coronary artery disease and low levels of high-density lipoprotein cholesterol. Am. J. Cardiol. 1996, 78, 572–575. [Google Scholar] [CrossRef]
- Manninen, V.; Tenkanen, L.; Koskinen, P.; Huttunen, J.K.; Mänttäri, M.; Heinonen, O.P.; Frick, M.H. Joint effects of serum triglyceride and LDL cholesterol and HDL cholesterol concentrations on coronary heart disease risk in the Helsinki Heart Study. Implications for treatment. Circulation 1992, 85, 37–45. [Google Scholar] [CrossRef]
- Vrins, C.L.J.; van der Velde, A.E.; van den Oever, K.; Levels, J.H.M.; Huet, S.; Oude Elferink, R.P.J.; Kuipers, F.; Groen, A.K. Peroxisome proliferator-activated receptor delta activation leads to increased transintestinal cholesterol efflux. J. Lipid Res. 2009, 50, 2046–2054. [Google Scholar] [CrossRef]
- Lee, T.-W.; Bai, K.-J.; Lee, T.-I.; Chao, T.-F.; Kao, Y.-H.; Chen, Y.-J. PPARs modulate cardiac metabolism and mitochondrial function in diabetes. J. Biomed. Sci. 2017, 24, 5. [Google Scholar] [CrossRef]
- Duan, S.Z.; Ivashchenko, C.Y.; Russell, M.W.; Milstone, D.S.; Mortensen, R.M. Cardiomyocyte-Specific Knockout and Agonist of Peroxisome Proliferator–Activated Receptor-γ Both Induce Cardiac Hypertrophy in Mice. Circ. Res. 2005, 97, 372–379. [Google Scholar] [CrossRef]
- Vázquez-Carrera, M. Unraveling the Effects of PPARβ/δ on Insulin Resistance and Cardiovascular Disease. Trends Endocrinol. Metab. 2016, 27, 319–334. [Google Scholar] [CrossRef]
- Wan, J.; Jiang, L.; Lü, Q.; Ke, L.; Li, X.; Tong, N. Activation of PPARδ up-regulates fatty acid oxidation and energy uncoupling genes of mitochondria and reduces palmitate-induced apoptosis in pancreatic β-cells. Biochem. Biophys. Res. Commun. 2010, 391, 1567–1572. [Google Scholar] [CrossRef]
- Li, L.; Li, T.; Zhang, Y.; Pan, Z.; Wu, B.; Huang, X.; Zhang, Y.; Mei, Y.; Ge, L.; Shen, G.; et al. Peroxisome proliferator-activated receptorβ/δ activation is essential for modulating p-Foxo1/Foxo1 status in functional insulin-positive cell differentiation. Cell Death Dis. 2015, 6, e1715. [Google Scholar] [CrossRef]
- Cohen, G.; Riahi, Y.; Shamni, O.; Guichardant, M.; Chatgilialoglu, C.; Ferreri, C.; Kaiser, N.; Sasson, S. Role of Lipid Peroxidation and PPAR-δ in Amplifying Glucose-Stimulated Insulin Secretion. Diabetes 2011, 60, 2830–2842. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.-H.; Hancock, C.R.; Terada, S.; Higashida, K.; Holloszy, J.O.; Han, D.-H. PPARβ Is Essential for Maintaining Normal Levels of PGC-1α and Mitochondria and for the Increase in Muscle Mitochondria Induced by Exercise. Cell Metab. 2017, 25, 1176–1185. [Google Scholar] [CrossRef] [PubMed]
- Schnuck, J.K.; Sunderland, K.L.; Gannon, N.P.; Kuennen, M.R.; Vaughan, R.A. Leucine stimulates PPARβ/δ-dependent mitochondrial biogenesis and oxidative metabolism with enhanced GLUT4 content and glucose uptake in myotubes. Biochimie 2016, 128–129, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Bernal-Mizrachi, C.; Han, D.H.; Coleman, T.; Sambandam, N.; LaRiviere, L.L.; Holloszy, J.O.; Semenkovich, C.F.; Kelly, D.P. A potential link between muscle peroxisome proliferator- activated receptor-α signaling and obesity-related diabetes. Cell Metab. 2005, 1, 133–144. [Google Scholar] [CrossRef]
- Karimian Azari, E.; Leitner, C.; Jaggi, T.; Langhans, W.; Mansouri, A. Possible Role of Intestinal Fatty Acid Oxidation in the Eating-Inhibitory Effect of the PPAR-α Agonist Wy-14643 in High-Fat Diet Fed Rats. PLoS ONE 2013, 8, e74869. [Google Scholar] [CrossRef]
- Nozu, T.; Miyagishi, S.; Nozu, R.; Takakusaki, K.; Okumura, T. Pioglitazone improves visceral sensation and colonic permeability in a rat model of irritable bowel syndrome. J. Pharmacol. Sci. 2019, 139, 46–49. [Google Scholar] [CrossRef]
- Régnier, M.; Polizzi, A.; Lippi, Y.; Fouché, E.; Michel, G.; Lukowicz, C.; Smati, S.; Marrot, A.; Lasserre, F.; Naylies, C.; et al. Insights into the role of hepatocyte PPARα activity in response to fasting. Mol. Cell. Endocrinol. 2018, 471, 75–88. [Google Scholar] [CrossRef]
- Sanderson, L.M.; Boekschoten, M.V.; Desvergne, B.; Müller, M.; Kersten, S. Transcriptional profiling reveals divergent roles of PPARα and PPARβ/δ in regulation of gene expression in mouse liver. Physiol. Genom. 2010, 41, 42–52. [Google Scholar] [CrossRef]
- Morán-Salvador, E.; López-Parra, M.; García-Alonso, V.; Titos, E.; Martínez-Clemente, M.; González-Périz, A.; López-Vicario, C.; Barak, Y.; Arroyo, V.; Clària, J. Role for PPARγ in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J. 2011, 25, 2538–2550. [Google Scholar] [CrossRef]
- Hoekstra, M.; Kruijt, J.K.; Van Eck, M.; van Berkel, T.J.C. Specific Gene Expression of ATP-binding Cassette Transporters and Nuclear Hormone Receptors in Rat Liver Parenchymal, Endothelial, and Kupffer Cells. J. Biol. Chem. 2003, 278, 25448–25453. [Google Scholar] [CrossRef]
- Tontonoz, P.; Hu, E.; Graves, R.A.; Budavari, A.I.; Spiegelman, B.M. mPPAR gamma 2: Tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994, 8, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Zhang, J.; Li, H.; Liu, J.; He, L.; Zhang, J.; Zhai, Y. Inhibition of adipocyte differentiation and adipogenesis by the traditional Chinese herb Sibiraea Angustata. Exp. Biol. Med. 2010, 235, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Teng, C.T.; Li, Y.; Stockton, P.; Foley, J. Fasting Induces the Expression of PGC-1α and ERR Isoforms in the Outer Stripe of the Outer Medulla (OSOM) of the Mouse Kidney. PLoS ONE 2011, 6, e26961. [Google Scholar] [CrossRef] [PubMed]
- Dressel, U.; Allen, T.L.; Pippal, J.B.; Rohde, P.R.; Lau, P.; Muscat, G.E.O. The Peroxisome Proliferator-Activated Receptor β/δ Agonist, GW501516, Regulates the Expression of Genes Involved in Lipid Catabolism and Energy Uncoupling in Skeletal Muscle Cells. Mol. Endocrinol. 2003, 17, 2477–2493. [Google Scholar] [CrossRef]
- Wang, Y.-X.; Lee, C.-H.; Tiep, S.; Yu, R.T.; Ham, J.; Kang, H.; Evans, R.M. Peroxisome-Proliferator-Activated Receptor δ Activates Fat Metabolism to Prevent Obesity. Cell 2003, 113, 159–170. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef]
- Woods, J.A.; Wilund, K.R.; Martin, S.A.; Kistler, B.M. Exercise, inflammation and aging. Aging Dis. 2012, 3, 130–140. [Google Scholar]
- Gabriely, I.; Ma, X.H.; Yang, X.M.; Rossetti, L.; Barzilai, N. Leptin Resistance During Aging Is Independent of Fat Mass. Diabetes 2002, 51, 1016–1021. [Google Scholar] [CrossRef]
- Villareal, D.T.; Apovian, C.M.; Kushner, R.F.; Klein, S. Obesity in Older Adults: Technical Review and Position Statement of the American Society for Nutrition and NAASO, the Obesity Society. Obes. Res. 2005, 13, 1849–1863. [Google Scholar] [CrossRef]
- Kotsis, V.; Stabouli, S.; Papakatsika, S.; Rizos, Z.; Parati, G. Mechanisms of obesity-induced hypertension. Hypertens. Res. 2010, 33, 386–393. [Google Scholar] [CrossRef]
- Eckel, R.H.; Kahn, S.E.; Ferrannini, E.; Goldfine, A.B.; Nathan, D.M.; Schwartz, M.W.; Smith, R.J.; Smith, S.R. Obesity and Type 2 Diabetes: What Can Be Unified and What Needs to Be Individualized? Diabetes Care 2011, 34, 1424–1430. [Google Scholar] [CrossRef] [PubMed]
- Poirier, P.; Giles, T.D.; Bray, G.A.; Hong, Y.; Stern, J.S.; Pi-Sunyer, F.X.; Eckel, R.H. Obesity and Cardiovascular Disease: Pathophysiology, Evaluation, and Effect of Weight Loss: An Update of the 1997 American Heart Association Scientific Statement on Obesity and Heart Disease From the Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Circulation 2006, 113, 898–918. [Google Scholar] [PubMed]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Prattichizzo, F.; De Nigris, V.; La Sala, L.; Procopio, A.D.; Olivieri, F.; Ceriello, A. “Inflammaging” as a Druggable Target: A Senescence-Associated Secretory Phenotype—Centered View of Type 2 Diabetes. Oxidative Med. Cell. Longev. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.R.; Justin Milner, J.; Makowski, L. The inflammation highway: Metabolism accelerates inflammatory traffic in obesity. Immunol. Rev. 2012, 249, 218–238. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Masuda, S.; Matsuo, Y.; Sasaki, Y.; Yamakage, H.; Muranaka, K.; Wada, H.; Hasegawa, K.; Tsukahara, T.; Shimatsu, A.; et al. Hyperglycemia and Inflammatory Property of Circulating Monocytes are Associated with Inflammatory Property of Carotid Plaques in Patients Undergoing Carotid Endarterectomy. J. Atheroscler. Thromb. 2016, 23, 1212–1221. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, X.; Madhankumar, A.B.; Miller, P.A.; Duck, K.A.; Hafenstein, S.; Rizk, E.; Slagle-Webb, B.; Sheehan, J.M.; Connor, J.R.; Yang, Q.X. MRI contrast agent for targeting glioma: Interleukin-13 labeled liposome encapsulating gadolinium-DTPA. Neuro-Oncology 2016, 18, 691–699. [Google Scholar] [CrossRef]
- Dror, E.; Dalmas, E.; Meier, D.T.; Wueest, S.; Thévenet, J.; Thienel, C.; Timper, K.; Nordmann, T.M.; Traub, S.; Schulze, F.; et al. Postprandial macrophage-derived IL-1β stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat. Immunol. 2017, 18, 283–292. [Google Scholar] [CrossRef]
- Esposito, K.; Nappo, F.; Marfella, R.; Giugliano, G.; Giugliano, F.; Ciotola, M.; Quagliaro, L.; Ceriello, A.; Giugliano, D. Inflammatory Cytokine Concentrations Are Acutely Increased by Hyperglycemia in Humans: Role of Oxidative Stress. Circulation 2002, 106, 2067–2072. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef]
- Dasu, M.R.; Ramirez, S.; Isseroff, R.R. Toll-like receptors and diabetes: A therapeutic perspective. Clin. Sci. 2012, 122, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.; Dasgupta, S.; Kundu, R.; Maitra, S.; Das, G.; Mukhopadhyay, S.; Ray, S.; Majumdar, S.S.; Bhattacharya, S. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat. Med. 2012, 18, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Sohn, K.H.; Rhee, S.H.; Hwang, D. Saturated Fatty Acids, but Not Unsaturated Fatty Acids, Induce the Expression of Cyclooxygenase-2 Mediated through Toll-like Receptor 4. J. Biol. Chem. 2001, 276, 16683–16689. [Google Scholar] [CrossRef] [PubMed]
- Drew, B.G.; Hamidi, H.; Zhou, Z.; Villanueva, C.J.; Krum, S.A.; Calkin, A.C.; Parks, B.W.; Ribas, V.; Kalajian, N.Y.; Phun, J.; et al. Estrogen receptor (ER)α-regulated lipocalin 2 expression in adipose tissue links obesity with breast cancer progression. J. Biol. Chem. 2015, 290, 5566–5581. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Cellular Senescence: A Translational Perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C. Obesity in geroscience—Is cellular senescence the culprit? Nat. Rev. Endocrinol. 2017, 13, 76–78. [Google Scholar] [CrossRef]
- Jung, U.; Choi, M.-S. Obesity and Its Metabolic Complications: The Role of Adipokines and the Relationship between Obesity, Inflammation, Insulin Resistance, Dyslipidemia and Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2014, 15, 6184–6223. [Google Scholar] [CrossRef]
- Corbi, G.; Polito, R.; Monaco, M.L.; Cacciatore, F.; Scioli, M.; Ferrara, N.; Daniele, A.; Nigro, E. Adiponectin Expression and Genotypes in Italian People with Severe Obesity Undergone a Hypocaloric Diet and Physical Exercise Program. Nutrients 2019, 11, 2195. [Google Scholar] [CrossRef]
- Nguyen, M.T.A.; Favelyukis, S.; Nguyen, A.-K.; Reichart, D.; Scott, P.A.; Jenn, A.; Liu-Bryan, R.; Glass, C.K.; Neels, J.G.; Olefsky, J.M. A Subpopulation of Macrophages Infiltrates Hypertrophic Adipose Tissue and Is Activated by Free Fatty Acids via Toll-like Receptors 2 and 4 and JNK-dependent Pathways. J. Biol. Chem. 2007, 282, 35279–35292. [Google Scholar] [CrossRef]
- Nguyen, M.T.A.; Satoh, H.; Favelyukis, S.; Babendure, J.L.; Imamura, T.; Sbodio, J.I.; Zalevsky, J.; Dahiyat, B.I.; Chi, N.-W.; Olefsky, J.M. JNK and Tumor Necrosis Factor-α Mediate Free Fatty Acid-induced Insulin Resistance in 3T3-L1 Adipocytes. J. Biol. Chem. 2005, 280, 35361–35371. [Google Scholar] [CrossRef]
- Yu, C.; Chen, Y.; Cline, G.W.; Zhang, D.; Zong, H.; Wang, Y.; Bergeron, R.; Kim, J.K.; Cushman, S.W.; Cooney, G.J.; et al. Mechanism by Which Fatty Acids Inhibit Insulin Activation of Insulin Receptor Substrate-1 (IRS-1)-associated Phosphatidylinositol 3-Kinase Activity in Muscle. J. Biol. Chem. 2002, 277, 50230–50236. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.; Rhee, S.H. Receptor-mediated signaling pathways: Potential targets of modulation by dietary fatty acids. Am. J. Clin. Nutr. 1999, 70, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Olefsky, J.M. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. 2012, 15, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Wong, V.; Guan, X.; Oprescu, A.I.; Giacca, A. Salicylate prevents hepatic insulin resistance caused by short-term elevation of free fatty acids in vivo. J. Endocrinol. 2007, 195, 323–331. [Google Scholar] [CrossRef]
- Summers, S. Ceramides in insulin resistance and lipotoxicity. Prog. Lipid Res. 2006, 45, 42–72. [Google Scholar] [CrossRef]
- Serhan, C.N.; Brain, S.D.; Buckley, C.D.; Gilroy, D.W.; Haslett, C.; O’Neill, L.A.J.; Perretti, M.; Rossi, A.G.; Wallace, J.L. Resolution of inflammation: State of the art, definitions and terms. FASEB J. 2007, 21, 325–332. [Google Scholar] [CrossRef]
- Serhan, C.N. Resolution Phase of Inflammation: Novel Endogenous Anti-Inflammatory and Proresolving Lipid Mediators and Pathways. Annu. Rev. Immunol. 2007, 25, 101–137. [Google Scholar] [CrossRef]
- Clària, J.; Dalli, J.; Yacoubian, S.; Gao, F.; Serhan, C.N. Resolvin D1 and Resolvin D2 Govern Local Inflammatory Tone in Obese Fat. J. Immunol. 2012, 189, 2597–2605. [Google Scholar] [CrossRef]
- Kuk, J.L.; Saunders, T.J.; Davidson, L.E.; Ross, R. Age-related changes in total and regional fat distribution. Ageing Res. Rev. 2009, 8, 339–348. [Google Scholar] [CrossRef]
- Mau, T.; Yung, R. Adipose tissue inflammation in aging. Exp. Gerontol. 2018, 105, 27–31. [Google Scholar] [CrossRef]
- Fox, C.S.; Massaro, J.M.; Hoffmann, U.; Pou, K.M.; Maurovich-Horvat, P.; Liu, C.-Y.; Vasan, R.S.; Murabito, J.M.; Meigs, J.B.; Cupples, L.A.; et al. Abdominal visceral and subcutaneous adipose tissue compartments: Association with metabolic risk factors in the Framingham Heart Study. Circulation 2007, 116, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Hausman, D.B.; DiGirolamo, M.; Bartness, T.J.; Hausman, G.J.; Martin, R.J. The biology of white adipocyte proliferation. Obes. Rev. 2001, 2, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kim, J.; Osborne, O.; Oh, D.Y.; Sasik, R.; Schenk, S.; Chen, A.; Chung, H.; Murphy, A.; Watkins, S.M.; et al. Increased Adipocyte O2 Consumption Triggers HIF-1α, Causing Inflammation and Insulin Resistance in Obesity. Cell 2014, 157, 1339–1352. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U. Endoplasmic Reticulum Stress Links Obesity, Insulin Action, and Type 2 Diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef]
- Murano, I.; Barbatelli, G.; Parisani, V.; Latini, C.; Muzzonigro, G.; Castellucci, M.; Cinti, S. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J. Lipid Res. 2008, 49, 1562–1568. [Google Scholar] [CrossRef]
- Zamarron, B.F.; Mergian, T.A.; Cho, K.W.; Martinez-Santibanez, G.; Luan, D.; Singer, K.; DelProposto, J.L.; Geletka, L.M.; Muir, L.A.; Lumeng, C.N. Macrophage Proliferation Sustains Adipose Tissue Inflammation in Formerly Obese Mice. Diabetes 2017, 66, 392–406. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Evans, G.; Tonne, J.M.; Verzosa, G.C.; Stout, M.B.; Mazula, D.L.; Palmer, A.K.; Baker, D.J.; Jensen, M.D.; et al. Exercise Prevents Diet-Induced Cellular Senescence in Adipose Tissue. Diabetes 2016, 65, 1606–1615. [Google Scholar] [CrossRef]
- Huang, D.; Zhao, Q.; Liu, H.; Guo, Y.; Xu, H. PPAR-α Agonist WY-14643 Inhibits LPS-Induced Inflammation in Synovial Fibroblasts via NF-κB Pathway. J. Mol. Neurosci. 2016, 59, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Koenig, W.; Habib, A.; Merval, R.; Lebret, M.; Torra, I.P.; Delerive, P.; Fadel, A.; Chinetti, G.; Fruchart, J.C.; et al. Activation of human aortic smooth-muscle cells is inhibited by PPARalpha but not by PPARgamma activators. Nature 1998, 393, 790–793. [Google Scholar] [CrossRef] [PubMed]
- Rival, Y.; Benéteau, N.; Taillandier, T.; Pezet, M.; Dupont-Passelaigue, E.; Patoiseau, J.F.; Junquéro, D.; Colpaert, F.C.; Delhon, A. PPARalpha and PPARdelta activators inhibit cytokine-induced nuclear translocation of NF-kappaB and expression of VCAM-1 in EAhy926 endothelial cells. Eur. J. Pharmacol. 2002, 435, 143–151. [Google Scholar] [CrossRef]
- Dubrac, S.; Stoitzner, P.; Pirkebner, D.; Elentner, A.; Schoonjans, K.; Auwerx, J.; Saeland, S.; Hengster, P.; Fritsch, P.; Romani, N.; et al. Peroxisome proliferator-activated receptor-alpha activation inhibits Langerhans cell function. J. Immunol. 2007, 178, 4362–4372. [Google Scholar] [CrossRef]
- Ramanan, S.; Kooshki, M.; Zhao, W.; Hsu, F.-C.; Robbins, M.E. PPARalpha ligands inhibit radiation-induced microglial inflammatory responses by negatively regulating NF-kappaB and AP-1 pathways. Free Radic. Biol. Med. 2008, 45, 1695–1704. [Google Scholar] [CrossRef]
- Zingarelli, B.; Piraino, G.; Hake, P.W.; O’Connor, M.; Denenberg, A.; Fan, H.; Cook, J.A. Peroxisome proliferator-activated receptor {delta} regulates inflammation via NF-{kappa}B signaling in polymicrobial sepsis. Am. J. Pathol. 2010, 177, 1834–1847. [Google Scholar] [CrossRef]
- Barroso, E.; Eyre, E.; Palomer, X.; Vázquez-Carrera, M. The peroxisome proliferator-activated receptor β/δ (PPARβ/δ) agonist GW501516 prevents TNF-α-induced NF-κB activation in human HaCaT cells by reducing p65 acetylation through AMPK and SIRT1. Biochem. Pharmacol. 2011, 81, 534–543. [Google Scholar] [CrossRef]
- Han, S.; Inoue, H.; Flowers, L.C.; Sidell, N. Control of COX-2 gene expression through peroxisome proliferator-activated receptor gamma in human cervical cancer cells. Clin. Cancer Res. 2003, 9, 4627–4635. [Google Scholar]
- He, X.; Liu, W.; Shi, M.; Yang, Z.; Zhang, X.; Gong, P. Docosahexaenoic acid attenuates LPS-stimulated inflammatory response by regulating the PPARγ/NF-κB pathways in primary bovine mammary epithelial cells. Res. Vet. Sci. 2017, 112, 7–12. [Google Scholar] [CrossRef]
- Korbecki, J.; Bobiński, R.; Dutka, M. Self-regulation of the inflammatory response by peroxisome proliferator-activated receptors. Inflamm. Res. 2019, 68, 443–458. [Google Scholar] [CrossRef]
- Delerive, P.; De Bosscher, K.; Besnard, S.; Vanden Berghe, W.; Peters, J.M.; Gonzalez, F.J.; Fruchart, J.C.; Tedgui, A.; Haegeman, G.; Staels, B. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J. Biol. Chem. 1999, 274, 32048–32054. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.-J.; Tseng, C.-P.; Yang, C.-M.; Ma, Y.-H. 20-Hydroxyeicosatetraenoic acid inhibits ATP-induced COX-2 expression via peroxisome proliferator activator receptor-α in vascular smooth muscle cells: 20-HETE inhibits COX-2 expression. Br. J. Pharmacol. 2011, 163, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Subbaramaiah, K.; Lin, D.T.; Hart, J.C.; Dannenberg, A.J. Peroxisome Proliferator-activated Receptor γ Ligands Suppress the Transcriptional Activation of Cyclooxygenase-2: EVIDENCE FOR INVOLVEMENT OF ACTIVATOR PROTEIN-1 AND CREB-BINDING PROTEIN/p300. J. Biol. Chem. 2001, 276, 12440–12448. [Google Scholar] [CrossRef] [PubMed]
- Khandoudi, N.; Delerive, P.; Berrebi-Bertrand, I.; Buckingham, R.E.; Staels, B.; Bril, A. Rosiglitazone, a Peroxisome Proliferator-Activated Receptor-, Inhibits the Jun NH2-Terminal Kinase/Activating Protein 1 Pathway and Protects the Heart From Ischemia/Reperfusion Injury. Diabetes 2002, 51, 1507–1514. [Google Scholar] [CrossRef]
- Ji, H.-G.; Piao, J.-Y.; Kim, S.-J.; Kim, D.-H.; Lee, H.-N.; Na, H.-K.; Surh, Y.-J. Docosahexaenoic acid inhibits Helicobacter pylori-induced STAT3 phosphorylation through activation of PPARγ. Mol. Nutr. Food Res. 2016, 60, 1448–1457. [Google Scholar] [CrossRef]
- Yu, J.H.; Kim, K.H.; Kim, H. SOCS 3 and PPAR-gamma ligands inhibit the expression of IL-6 and TGF-beta1 by regulating JAK2/STAT3 signaling in pancreas. Int. J. Biochem. Cell Biol. 2008, 40, 677–688. [Google Scholar] [CrossRef]
- Cipolletta, D.; Feuerer, M.; Li, A.; Kamei, N.; Lee, J.; Shoelson, S.E.; Benoist, C.; Mathis, D. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 2012, 486, 549–553. [Google Scholar] [CrossRef]
- Schipper, H.S.; Prakken, B.; Kalkhoven, E.; Boes, M. Adipose tissue-resident immune cells: Key players in immunometabolism. Trends Endocrinol. Metab. 2012, 23, 407–415. [Google Scholar] [CrossRef]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nature 2001, 409, 307–312. [Google Scholar] [CrossRef]
- Peraldi, P.; Xu, M.; Spiegelman, B.M. Thiazolidinediones block tumor necrosis factor-alpha-induced inhibition of insulin signaling. J. Clin. Investig. 1997, 100, 1863–1869. [Google Scholar] [CrossRef]
- Hammarstedt, A.; Andersson, C.X.; Rotter Sopasakis, V.; Smith, U. The effect of PPARγ ligands on the adipose tissue in insulin resistance. Prostaglandins Leukot. Essent. Fat. Acids 2005, 73, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Chawla, A. Control of Macrophage Activation and Function by PPARs. Circ. Res. 2010, 106, 1559–1569. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.T.; Welch, J.S.; Ricote, M.; Binder, C.J.; Willson, T.M.; Kelly, C.; Witztum, J.L.; Funk, C.D.; Conrad, D.; Glass, C.K. Interleukin-4-dependent production of PPAR-γ ligands in macrophages by 12/15-lipoxygenase. Nature 1999, 400, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Red Eagle, A.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Kratz, M.; Coats, B.R.; Hisert, K.B.; Hagman, D.; Mutskov, V.; Peris, E.; Schoenfelt, K.Q.; Kuzma, J.N.; Larson, I.; Billing, P.S.; et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014, 20, 614–625. [Google Scholar] [CrossRef]
- Hevener, A.L.; Olefsky, J.M.; Reichart, D.; Nguyen, M.T.A.; Bandyopadyhay, G.; Leung, H.-Y.; Watt, M.J.; Benner, C.; Febbraio, M.A.; Nguyen, A.-K.; et al. Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J. Clin. Investig. 2007, 117, 1658–1669. [Google Scholar] [CrossRef]
- Bertola, A.; Ciucci, T.; Rousseau, D.; Bourlier, V.; Duffaut, C.; Bonnafous, S.; Blin-Wakkach, C.; Anty, R.; Iannelli, A.; Gugenheim, J.; et al. Identification of adipose tissue dendritic cells correlated with obesity-associated insulin-resistance and inducing Th17 responses in mice and patients. Diabetes 2012, 61, 2238–2247. [Google Scholar] [CrossRef]
- Macdougall, C.E.; Wood, E.G.; Loschko, J.; Scagliotti, V.; Cassidy, F.C.; Robinson, M.E.; Feldhahn, N.; Castellano, L.; Voisin, M.-B.; Marelli-Berg, F.; et al. Visceral Adipose Tissue Immune Homeostasis Is Regulated by the Crosstalk between Adipocytes and Dendritic Cell Subsets. Cell Metab. 2018, 27, 588–601. [Google Scholar] [CrossRef]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory Mechanisms in Obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef]
- Sears, B.; Perry, M. The role of fatty acids in insulin resistance. Lipids Health Dis. 2015, 14, 121. [Google Scholar] [CrossRef] [PubMed]
- Vessby, B.; Uusitupa, M.; Hermansen, K.; Riccardi, G.; Rivellese, A.A.; Tapsell, L.C.; Nälsén, C.; Berglund, L.; Louheranta, A.; Rasmussen, B.M.; et al. Substituting dietary saturated for monounsaturated fat impairs insulin sensitivity in healthy men and women: The KANWU study. Diabetologia 2001, 44, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Mayer-Davis, E.J. Intensity and Amount of Physical Activity in Relation to Insulin Sensitivity: The Insulin Resistance Atherosclerosis Study. JAMA 1998, 279, 669. [Google Scholar] [CrossRef] [PubMed]
- Cefalu, W.T. Insulin Resistance: Cellular and Clinical Concepts. Exp. Biol. Med. 2001, 226, 13–26. [Google Scholar] [CrossRef]
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-α function. Nature 1997, 389, 610–614. [Google Scholar] [CrossRef]
- Yuan, M. Reversal of Obesity- and Diet-Induced Insulin Resistance with Salicylates or Targeted Disruption of Ikkbeta. Science 2001, 293, 1673–1677. [Google Scholar] [CrossRef]
- Kim, J.K.; Kim, Y.-J.; Fillmore, J.J.; Chen, Y.; Moore, I.; Lee, J.; Yuan, M.; Li, Z.W.; Karin, M.; Perret, P.; et al. Prevention of fat-induced insulin resistance by salicylate. J. Clin. Investig. 2001, 108, 437–446. [Google Scholar] [CrossRef]
- Aguirre, V.; Uchida, T.; Yenush, L.; Davis, R.; White, M.F. The c-Jun NH2-terminal Kinase Promotes Insulin Resistance during Association with Insulin Receptor Substrate-1 and Phosphorylation of Ser307*. J. Biol. Chem. 2000, 275, 9047–9054. [Google Scholar] [CrossRef]
- Thaler, J.P.; Schwartz, M.W. Minireview: Inflammation and Obesity Pathogenesis: The Hypothalamus Heats Up. Endocrinology 2010, 151, 4109–4115. [Google Scholar] [CrossRef]
- Velloso, L.A.; Schwartz, M.W. Altered hypothalamic function in diet-induced obesity. Int. J. Obes. 2011, 35, 1455–1465. [Google Scholar] [CrossRef]
- Valdearcos, M.; Robblee, M.M.; Benjamin, D.I.; Nomura, D.K.; Xu, A.W.; Koliwad, S.K. Microglia Dictate the Impact of Saturated Fat Consumption on Hypothalamic Inflammation and Neuronal Function. Cell Rep. 2014, 9, 2124–2138. [Google Scholar] [CrossRef] [PubMed]
- Spencer, S.J.; Tilbrook, A. The glucocorticoid contribution to obesity. Stress 2011, 14, 233–246. [Google Scholar] [CrossRef] [PubMed]
- De Souza, C.T.; Araujo, E.P.; Bordin, S.; Ashimine, R.; Zollner, R.L.; Boschero, A.C.; Saad, M.J.A.; Velloso, L.A. Consumption of a Fat-Rich Diet Activates a Proinflammatory Response and Induces Insulin Resistance in the Hypothalamus. Endocrinology 2005, 146, 4192–4199. [Google Scholar] [CrossRef] [PubMed]
- Posey, K.A.; Clegg, D.J.; Printz, R.L.; Byun, J.; Morton, G.J.; Vivekanandan-Giri, A.; Pennathur, S.; Baskin, D.G.; Heinecke, J.W.; Woods, S.C.; et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am. J. Physiol.-Endocrinol. Metab. 2009, 296, E1003–E1012. [Google Scholar] [CrossRef]
- Jaworski, K.; Sarkadi-Nagy, E.; Duncan, R.E.; Ahmadian, M.; Sul, H.S. Regulation of Triglyceride Metabolism.IV. Hormonal regulation of lipolysis in adipose tissue. Am. J. Physiol.-Gastrointest. Liver Physiol. 2007, 293, G1–G4. [Google Scholar] [CrossRef]
- Wilcox, G. Insulin and insulin resistance. Clin. Biochem. Rev. 2005, 26, 19–39. [Google Scholar]
- Tokarz, V.L.; MacDonald, P.E.; Klip, A. The cell biology of systemic insulin function. J. Cell Biol. 2018, 217, 2273–2289. [Google Scholar] [CrossRef]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef]
- Olefsky, J.M.; Glass, C.K. Macrophages, Inflammation, and Insulin Resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef]
- Bachanek, M.; Abdalla, N.; Cendrowski, K.; Sawicki, W. Value of ultrasonography in the diagnosis of polycystic ovary syndrome—literature review. J. Ultrason. 2015, 15, 410–422. [Google Scholar] [CrossRef]
- Bachelot, A. Polycystic ovarian syndrome: Clinical and biological diagnosis. Ann. Biol. Clin. 2016, 74, 661–667. [Google Scholar] [CrossRef]
- Glintborg, D.; Andersen, M. MANAGEMENT OF ENDOCRINE DISEASE: Morbidity in polycystic ovary syndrome. Eur. J. Endocrinol. 2017, 176, R53–R65. [Google Scholar] [CrossRef]
- Pourghassem Gargari, B.; Houjeghani, S.; Farzadi, L.; Houjeghani, S.; Safaeiyan, A. Relationship between Serum Leptin, Ghrelin and Dietary Macronutrients in Women with Polycystic Ovary Syndrome. Int. J. Fertil. Steril. 2015, 9, 313–321. [Google Scholar] [PubMed]
- Jalilian, A.; Kiani, F.; Sayehmiri, F.; Sayehmiri, K.; Khodaee, Z.; Akbari, M. Prevalence of polycystic ovary syndrome and its associated complications in Iranian women: A meta-analysis. Iran. J. Reprod. Med. 2015, 13, 591–604. [Google Scholar] [PubMed]
- Carvalho, L.M.L.; Dos Reis, F.M.; Candido, A.L.; Nunes, F.F.C.; Ferreira, C.N.; Gomes, K.B. Polycystic Ovary Syndrome as a systemic disease with multiple molecular pathways: A narrative review. Endocr. Regul. 2018, 52, 208–221. [Google Scholar] [CrossRef]
- Jacobs, H.S.; Conway, G.S. Leptin, polycystic ovaries and polycystic ovary syndrome. Hum. Reprod. Update 1999, 5, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Moran, L.; Brinkworth, G.; Norman, R. Dietary Therapy in Polycystic Ovary Syndrome. Semin. Reprod. Med. 2008, 26, 085–092. [Google Scholar] [CrossRef]
- Faghfoori, Z.; Fazelian, S.; Shadnoush, M.; Goodarzi, R. Nutritional management in women with polycystic ovary syndrome: A review study. Diabetes Metab. Syndr. Clin. Res. Rev. 2017, 11, S429–S432. [Google Scholar] [CrossRef]
- Jena, D.; Choudhury, A.K.; Mangaraj, S.; Singh, M.; Mohanty, B.K.; Baliarsinha, A.K. Study of Visceral and Subcutaneous Abdominal Fat Thickness and Its Correlation with Cardiometabolic Risk Factors and Hormonal Parameters in Polycystic Ovary Syndrome. Indian J. Endocrinol. Metab. 2018, 22, 321–327. [Google Scholar]
- Frøssing, S.; Nylander, M.C.; Chabanova, E.; Kistorp, C.; Skouby, S.O.; Faber, J. Quantification of visceral adipose tissue in polycystic ovary syndrome: Dual-energy X-ray absorptiometry versus magnetic resonance imaging. Acta Radiol. 2018, 59, 13–17. [Google Scholar] [CrossRef]
- Fain, J.N.; Bahouth, S.W.; Madan, A.K. TNFα release by the nonfat cells of human adipose tissue. Int. J. Obes. 2004, 28, 616–622. [Google Scholar] [CrossRef]
- Duleba, A.J.; Dokras, A. Is PCOS an inflammatory process? Fertil. Steril. 2012, 97, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Mortada, R.; Kallail, K.J.; Dong, F.; Karakas, S. HbA1c in Patients with Polycystic Ovary Syndrome: A Potential Marker of Inflammation. J. Reprod. Infertil. 2015, 16, 203–206. [Google Scholar]
- Nehir Aytan, A.; Bastu, E.; Demiral, I.; Bulut, H.; Dogan, M.; Buyru, F. Relationship between hyperandrogenism, obesity, inflammation and polycystic ovary syndrome. Gynecol. Endocrinol. 2016, 32, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Faubert, J.; Battista, M.-C.; Baillargeon, J.-P. PHYSIOLOGY AND ENDOCRINOLOGY SYMPOSIUM: Insulin action and lipotoxicity in the development of polycystic ovary syndrome: A review1. J. Anim. Sci. 2016, 94, 1803–1811. [Google Scholar] [CrossRef]
- Dunaif, A. Perspectives in Polycystic Ovary Syndrome: From Hair to Eternity. J. Clin. Endocrinol. Metab. 2016, 101, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Kahsar-Miller, M.; Azziz, R. The development of the polycystic ovary syndrome: Family history as a risk factor. Trends Endocrinol. Metab. 1998, 9, 55–58. [Google Scholar] [CrossRef]
- Vink, J.M.; Sadrzadeh, S.; Lambalk, C.B.; Boomsma, D.I. Heritability of Polycystic Ovary Syndrome in a Dutch Twin-Family Study. J. Clin. Endocrinol. Metab. 2006, 91, 2100–2104. [Google Scholar] [CrossRef]
- Jia, H.; Yu, L.; Guo, X.; Gao, W.; Jiang, Z. Associations of adiponectin gene polymorphisms with polycystic ovary syndrome: A meta-analysis. Endocrine 2012, 42, 299–306. [Google Scholar] [CrossRef]
- Ruan, Y.; Ma, J.; Xie, X. Association of IRS-1 and IRS-2 genes polymorphisms with polycystic ovary syndrome: A meta-analysis. Endocr. J. 2012, 59, 601–609. [Google Scholar] [CrossRef]
- Reddy, T.V.; Govatati, S.; Deenadayal, M.; Shivaji, S.; Bhanoori, M. Polymorphisms in the TFAM and PGC1-α genes and their association with polycystic ovary syndrome among South Indian women. Gene 2018, 641, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Sales, M.; Sóter, M.; Candido, A.; Fernandes, A.; Oliveira, F.; Ferreira, A.; Sousa, M.; Ferreira, C.; Gomes, K. Correlation between plasminogen activator inhibitor-1 (PAI-1) promoter 4G/5G polymorphism and metabolic/proinflammatory factors in polycystic ovary syndrome. Gynecol. Endocrinol. 2013, 29, 936–939. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Lv, Y.; Li, L.; Chen, Z.-J. Genetic Studies on Polycystic Ovary Syndrome. Best Pract. Res. Clin. Obstet. Gynaecol. 2016, 37, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, A.H.; Kohan, L.; Aledavood, A.; Rostami, S. Association of miR-146a rs2910164 and miR-222 rs2858060 polymorphisms with the risk of polycystic ovary syndrome in Iranian women: A case–control study. Taiwan. J. Obstet. Gynecol. 2017, 56, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, S. Polymorphism in the peroxisome proliferator-activated receptor-gamma gene in women with polycystic ovary syndrome. Hum. Reprod. 2003, 18, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Ergün, M.A.; Karakoç, A.; Yurtçu, E.; Yetkin, İ.; Ayvaz, G.; Çakir, N.; Arslan, M. Pro12Ala polymorphism of the peroxisome proliferator-activated receptor-γ gene in first-degree relatives of subjects with polycystic ovary syndrome. Gynecol. Endocrinol. 2005, 21, 206–210. [Google Scholar] [CrossRef]
- Yilmaz, M.; Ali Ergün, M.; Karakoç, A.; Yurtçu, E.; Çakir, N.; Arslan, M. Pro12Ala polymorphism of the peroxisome proliferator-activated receptor-γ gene in women with polycystic ovary syndrome. Gynecol. Endocrinol. 2006, 22, 336–342. [Google Scholar] [CrossRef]
- Orio, F.; Matarese, G.; Di Biase, S.; Palomba, S.; Labella, D.; Sanna, V.; Savastano, S.; Zullo, F.; Colao, A.; Lombardi, G. Exon 6 and 2 Peroxisome Proliferator-Activated Receptor-γ Polymorphisms in Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2003, 88, 5887–5892. [Google Scholar] [CrossRef][Green Version]
- Orio, F.; Palomba, S.; Cascella, T.; Di Biase, S.; Labella, D.; Russo, T.; Savastano, S.; Zullo, F.; Colao, A.; Vettor, R.; et al. Lack of an Association between Peroxisome Proliferator-Activated Receptor-γ Gene Pro12Ala Polymorphism and Adiponectin Levels in the Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2004, 89, 5110–5115. [Google Scholar] [CrossRef][Green Version]
- Hahn, S.; Fingerhut, A.; Khomtsiv, U.; Khomtsiv, L.; Tan, S.; Quadbeck, B.; Herrmann, B.L.; Knebel, B.; Muller-Wieland, D.; Mann, K.; et al. The peroxisome proliferator activated receptor gamma Pro12Ala polymorphism is associated with a lower hirsutism score and increased insulin sensitivity in women with polycystic ovary syndrome. Clin. Endocrinol. 2005, 62, 573–579. [Google Scholar] [CrossRef]
- Skov, V.; Glintborg, D.; Knudsen, S.; Tan, Q.; Jensen, T.; Kruse, T.A.; Beck-Nielsen, H.; Højlund, K. Pioglitazone enhances mitochondrial biogenesis and ribosomal protein biosynthesis in skeletal muscle in polycystic ovary syndrome. PLoS ONE 2008, 3, e2466. [Google Scholar] [CrossRef] [PubMed]
- Buse, J.B.; Tan, M.H.; Prince, M.J.; Erickson, P.P. The effects of oral anti-hyperglycaemic medications on serum lipid profiles in patients with type 2 diabetes. Diabetes Obes. Metab. 2004, 6, 133–156. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.F.; Carpentier, A.; Adeli, K.; Giacca, A. Disordered Fat Storage and Mobilization in the Pathogenesis of Insulin Resistance and Type 2 Diabetes. Endocr. Rev. 2002, 23, 201–229. [Google Scholar] [CrossRef] [PubMed]
- Norman, R.J.; Dewailly, D.; Legro, R.S.; Hickey, T.E. Polycystic ovary syndrome. Lancet 2007, 370, 685–697. [Google Scholar] [CrossRef]
- Al-Eisa, E.; Gabr, S.A.; Alghadir, A.H. Effects of supervised aerobic training on the levels of anti-Mullerian hormone and adiposity measures in women with normo-ovulatory and polycystic ovary syndrome. J. Pak. Med. Assoc. 2017, 67, 499–507. [Google Scholar]
- Palioura, E.; Diamanti-Kandarakis, E. Industrial endocrine disruptors and polycystic ovary syndrome. J. Endocrinol. Investig. 2013, 36, 1105–1111. [Google Scholar] [CrossRef]
- Varlamov, O.; Bishop, C.V.; Handu, M.; Takahashi, D.; Srinivasan, S.; White, A.; Roberts, C.T. Combined androgen excess and Western-style diet accelerates adipose tissue dysfunction in young adult, female nonhuman primates. Hum. Reprod. 2017, 32, 1892–1902. [Google Scholar] [CrossRef]
- Rizkalla, S.W.; Taghrid, L.; Laromiguiere, M.; Huet, D.; Boillot, J.; Rigoir, A.; Elgrably, F.; Slama, G. Improved plasma glucose control, whole-body glucose utilization, and lipid profile on a low-glycemic index diet in type 2 diabetic men: A randomized controlled trial. Diabetes Care 2004, 27, 1866–1872. [Google Scholar] [CrossRef]
- Ebbeling, C.B.; Leidig, M.M.; Sinclair, K.B.; Seger-Shippee, L.G.; Feldman, H.A.; Ludwig, D.S. Effects of an ad libitum low-glycemic load diet on cardiovascular disease risk factors in obese young adults. Am. J. Clin. Nutr. 2005, 81, 976–982. [Google Scholar] [CrossRef]
- Haqq, L.; McFarlane, J.; Dieberg, G.; Smart, N. Effect of lifestyle intervention on the reproductive endocrine profile in women with polycystic ovarian syndrome: A systematic review and meta-analysis. Endocr. Connect. 2014, 3, 36–46. [Google Scholar] [CrossRef]
- Teede, H.J.; Misso, M.L.; Costello, M.F.; Dokras, A.; Laven, J.; Moran, L.; Piltonen, T.; Norman, R.J.; International PCOS Network. Recommendations from the international evidence-based guideline for the assessment and management of polycystic ovary syndrome†‡. Hum. Reprod. 2018, 33, 1602–1618. [Google Scholar] [CrossRef] [PubMed]
- Valsamakis, G.; Lois, K.; Kumar, S.; Mastorakos, G. Metabolic and other effects of pioglitazone as an add-on therapy to metformin in the treatment of polycystic ovary syndrome (PCOS). Hormones 2013, 12, 363–378. [Google Scholar] [CrossRef] [PubMed]
- Froment, P.; Touraine, P. Thiazolidinediones and Fertility in Polycystic Ovary Syndrome (PCOS). PPAR Res. 2006, 2006, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Baillargeon, J.-P.; Jakubowicz, D.J.; Iuorno, M.J.; Jakubowicz, S.; Nestler, J.E. Effects of metformin and rosiglitazone, alone and in combination, in nonobese women with polycystic ovary syndrome and normal indices of insulin sensitivity. Fertil. Steril. 2004, 82, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Veldhuis, J.D.; Zhang, G.; Garmey, J.C. Troglitazone, an insulin-sensitizing thiazolidinedione, represses combined stimulation by LH and insulin of de novo androgen biosynthesis by thecal cells in vitro. J. Clin. Endocrinol. Metab. 2002, 87, 1129–1133. [Google Scholar] [CrossRef]
- Kempná, P.; Hofer, G.; Mullis, P.E.; Flück, C.E. Pioglitazone inhibits androgen production in NCI-H295R cells by regulating gene expression of CYP17 and HSD3B2. Mol. Pharm. 2007, 71, 787–798. [Google Scholar] [CrossRef]
- Li, M.; Pan, L.C.; Simmons, H.A.; Li, Y.; Healy, D.R.; Robinson, B.S.; Ke, H.Z.; Brown, T.A. Surface-specific effects of a PPARgamma agonist, darglitazone, on bone in mice. Bone 2006, 39, 796–806. [Google Scholar] [CrossRef]
- Abbas, A.; Blandon, J.; Rude, J.; Elfar, A.; Mukherjee, D. PPAR-γ agonist in treatment of diabetes: Cardiovascular safety considerations. Cardiovasc. Hematol. Agents Med. Chem. 2012, 10, 124–134. [Google Scholar] [CrossRef]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults: Systematic review: Epidemiology of NAFLD and NASH. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef]
- Thoma, C.; Day, C.P.; Trenell, M.I. Lifestyle interventions for the treatment of non-alcoholic fatty liver disease in adults: A systematic review. J. Hepatol. 2012, 56, 255–266. [Google Scholar] [CrossRef]
- Sayiner, M.; Koenig, A.; Henry, L.; Younossi, Z.M. Epidemiology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis in the United States and the Rest of the World. Clin. Liver Dis. 2016, 20, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, P.; Kowdley, K.V. The Metabolic Syndrome and Its Influence on Nonalcoholic Steatohepatitis. Clin. Liver Dis. 2016, 20, 225–243. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M.; Tiniakos, D.G. Histopathology of nonalcoholic fatty liver disease. World J. Gastroenterol. 2010, 16, 5286–5296. [Google Scholar] [CrossRef] [PubMed]
- Bugianesi, E.; McCullough, A.J.; Marchesini, G. Insulin resistance: A metabolic pathway to chronic liver disease. Hepatology 2005, 42, 987–1000. [Google Scholar] [CrossRef]
- Chao, H.-W.; Chao, S.-W.; Lin, H.; Ku, H.-C.; Cheng, C.-F. Homeostasis of Glucose and Lipid in Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2019, 20, 298. [Google Scholar] [CrossRef]
- Dowman, J.K.; Tomlinson, J.W.; Newsome, P.N. Pathogenesis of non-alcoholic fatty liver disease. QJM 2010, 103, 71–83. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F.W. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2018, 2018, 9547613. [Google Scholar] [CrossRef]
- Ucar, F.; Sezer, S.; Erdogan, S.; Akyol, S.; Armutcu, F.; Akyol, O. The relationship between oxidative stress and nonalcoholic fatty liver disease: Its effects on the development of nonalcoholic steatohepatitis. Redox Rep. 2013, 18, 127–133. [Google Scholar] [CrossRef]
- Souza-Mello, V.; Gregório, B.M.; Cardoso-de-Lemos, F.S.; de Carvalho, L.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Comparative effects of telmisartan, sitagliptin and metformin alone or in combination on obesity, insulin resistance, and liver and pancreas remodelling in C57BL/6 mice fed on a very high-fat diet. Clin. Sci. 2010, 119, 239–250. [Google Scholar] [CrossRef]
- Doege, H.; Baillie, R.A.; Ortegon, A.M.; Tsang, B.; Wu, Q.; Punreddy, S.; Hirsch, D.; Watson, N.; Gimeno, R.E.; Stahl, A. Targeted Deletion of FATP5 Reveals Multiple Functions in Liver Metabolism: Alterations in Hepatic Lipid Homeostasis. Gastroenterology 2006, 130, 1245–1258. [Google Scholar] [CrossRef] [PubMed]
- Falcon, A.; Doege, H.; Fluitt, A.; Tsang, B.; Watson, N.; Kay, M.A.; Stahl, A. FATP2 is a hepatic fatty acid transporter and peroxisomal very long-chain acyl-CoA synthetase. Am. J. Physiol.-Endocrinol. Metab. 2010, 299, E384–E393. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Pirola, C.J. Genetic predisposition in nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2017, 23, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Petersen, K.F.; Dufour, S.; Savage, D.B.; Bilz, S.; Solomon, G.; Yonemitsu, S.; Cline, G.W.; Befroy, D.; Zemany, L.; Kahn, B.B.; et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 12587–12594. [Google Scholar] [CrossRef]
- Motomura, W.; Inoue, M.; Ohtake, T.; Takahashi, N.; Nagamine, M.; Tanno, S.; Kohgo, Y.; Okumura, T. Up-regulation of ADRP in fatty liver in human and liver steatosis in mice fed with high fat diet. Biochem. Biophys. Res. Commun. 2006, 340, 1111–1118. [Google Scholar] [CrossRef]
- Schadinger, S.E.; Bucher, N.L.R.; Schreiber, B.M.; Farmer, S.R. PPARγ2 regulates lipogenesis and lipid accumulation in steatotic hepatocytes. Am. J. Physiol.-Endocrinol. Metab. 2005, 288, E1195–E1205. [Google Scholar] [CrossRef]
- Wijarnpreecha, K.; Thongprayoon, C.; Panjawatanan, P.; Ungprasert, P. Short sleep duration and risk of nonalcoholic fatty liver disease: A systematic review and meta-analysis: Coffee and nonalcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2016, 31, 1802–1807. [Google Scholar] [CrossRef]
- Santaliestra-Pasías, A.M.; Mouratidou, T.; Huybrechts, I.; Beghin, L.; Cuenca-García, M.; Castillo, M.J.; Galfo, M.; Hallstrom, L.; Kafatos, A.; Manios, Y.; et al. Increased sedentary behaviour is associated with unhealthy dietary patterns in European adolescents participating in the HELENA study. Eur. J. Clin. Nutr. 2014, 68, 300–308. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Farrell, G.C.; Wong, V.W.-S.; Chitturi, S. NAFLD in Asia—as common and important as in the West. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Perumpail, B.J.; Cholankeril, R.; Yoo, E.R.; Kim, D.; Ahmed, A. An Overview of Dietary Interventions and Strategies to Optimize the Management of Non-Alcoholic Fatty Liver Disease. Diseases 2017, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Mosca, A.; Nobili, V.; De Vito, R.; Crudele, A.; Scorletti, E.; Villani, A.; Alisi, A.; Byrne, C.D. Serum uric acid concentrations and fructose consumption are independently associated with NASH in children and adolescents. J. Hepatol. 2017, 66, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Bawden, S.; Stephenson, M.; Falcone, Y.; Lingaya, M.; Ciampi, E.; Hunter, K.; Bligh, F.; Schirra, J.; Taylor, M.; Morris, P.; et al. Increased liver fat and glycogen stores after consumption of high versus low glycaemic index food: A randomized crossover study: BAWDEN et al. Diabetes Obes. Metab. 2017, 19, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Mager, D.R.; Iñiguez, I.R.; Gilmour, S.; Yap, J. The Effect of a Low Fructose and Low Glycemic Index/Load (FRAGILE) Dietary Intervention on Indices of Liver Function, Cardiometabolic Risk Factors, and Body Composition in Children and Adolescents With Nonalcoholic Fatty Liver Disease (NAFLD). J. Parenter. Enter. Nutr. 2015, 39, 73–84. [Google Scholar] [CrossRef]
- Misciagna, G.; del Pilar Díaz, M.; Caramia, D.V.; Bonfiglio, C.; Franco, I.; Noviello, M.R.; Chiloiro, M.; Abbrescia, D.I.; Mirizzi, A.; Tanzi, M.; et al. Effect of a low glycemic index Mediterranean diet on non-alcoholic fatty liver disease. A randomized controlled clinici trial. J. Nutr. Health Aging 2017, 21, 404–412. [Google Scholar] [CrossRef]
- Hashida, R.; Kawaguchi, T.; Bekki, M.; Omoto, M.; Matsuse, H.; Nago, T.; Takano, Y.; Ueno, T.; Koga, H.; George, J.; et al. Aerobic vs. resistance exercise in non-alcoholic fatty liver disease: A systematic review. J. Hepatol. 2017, 66, 142–152. [Google Scholar] [CrossRef]
- Koltyn, K.F.; Brellenthin, A.G.; Cook, D.B.; Sehgal, N.; Hillard, C. Mechanisms of Exercise-Induced Hypoalgesia. J. Pain 2014, 15, 1294–1304. [Google Scholar] [CrossRef]
- Tutunchi, H.; Ostadrahimi, A.; Saghafi-Asl, M.; Maleki, V. The effects of oleoylethanolamide, an endogenous PPAR-α agonist, on risk factors for NAFLD: A systematic review. Obes. Rev. 2019, 20, 1057–1069. [Google Scholar] [CrossRef]
- Lu, W.; Li, S.; Li, J.; Wang, J.; Zhang, R.; Zhou, Y.; Yin, Q.; Zheng, Y.; Wang, F.; Xia, Y.; et al. Effects of Omega-3 Fatty Acid in Nonalcoholic Fatty Liver Disease: A Meta-Analysis. Gastroenterol. Res. Pract. 2016, 2016, 1–11. [Google Scholar] [CrossRef]
- Bellentani, S.; Dalle Grave, R.; Suppini, A.; Marchesini, G. Behavior therapy for nonalcoholic fatty liver disease: The need for a multidisciplinary approach. Hepatology 2007, 47, 746–754. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, E.M.; Rinella, M.E. The Role of Diet and Nutrient Composition in Nonalcoholic Fatty Liver Disease. J. Acad. Nutr. Diet. 2012, 112, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Mouzaki, M.; Allard, J.P. The Role of Nutrients in the Development, Progression, and Treatment of Nonalcoholic Fatty Liver Disease. J. Clin. Gastroenterol. 2012, 46, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol. Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Desvergne, B.; Wahli, W. Roles of PPARs in health and disease. Nature 2000, 405, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.D.I. Molecular mechanism for polyunsaturated fatty acid regulation of gene transcription. Am. J. Physiol.-Gastrointest. Liver Physiol. 2001, 281, G865–G869. [Google Scholar] [CrossRef]
- Jump, D.B.; Botolin, D.; Wang, Y.; Xu, J.; Christian, B.; Demeure, O. Fatty Acid Regulation of Hepatic Gene Transcription. J. Nutr. 2005, 135, 2503–2506. [Google Scholar] [CrossRef]
- Lombardo, Y.B.; Chicco, A.G. Effects of dietary polyunsaturated n-3 fatty acids on dyslipidemia and insulin resistance in rodents and humans. A review. J. Nutr. Biochem. 2006, 17, 1–13. [Google Scholar] [CrossRef]
- Pachikian, B.D.; Essaghir, A.; Demoulin, J.-B.; Neyrinck, A.M.; Catry, E.; De Backer, F.C.; Dejeans, N.; Dewulf, E.M.; Sohet, F.M.; Portois, L.; et al. Hepatic n-3 Polyunsaturated Fatty Acid Depletion Promotes Steatosis and Insulin Resistance in Mice: Genomic Analysis of Cellular Targets. PLoS ONE 2011, 6, e23365. [Google Scholar] [CrossRef]
- George, J.; Liddle, C. Nonalcoholic Fatty Liver Disease: Pathogenesis and Potential for Nuclear Receptors as Therapeutic Targets. Mol. Pharm. 2008, 5, 49–59. [Google Scholar] [CrossRef]
- Fraulob, J.C.; Souza-Mello, V.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Beneficial effects of rosuvastatin on insulin resistance, adiposity, inflammatory markers and non-alcoholic fatty liver disease in mice fed on a high-fat diet. Clin. Sci. 2012, 123, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, H.; Tehilla, M.; Attal-Singer, J.; Bruck, R.; Luzzatti, R.; Singer, P. The therapeutic potential of long-chain omega-3 fatty acids in nonalcoholic fatty liver disease. Clin. Nutr. 2011, 30, 6–19. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Núñez, B.; Pruimboom, L.; Dijck-Brouwer, D.A.J.; Muskiet, F.A.J. Lifestyle and nutritional imbalances associated with Western diseases: Causes and consequences of chronic systemic low-grade inflammation in an evolutionary context. J. Nutr. Biochem. 2013, 24, 1183–1201. [Google Scholar] [CrossRef] [PubMed]
- Giampietro, L.; Laghezza, A.; Cerchia, C.; Florio, R.; Recinella, L.; Capone, F.; Ammazzalorso, A.; Bruno, I.; De Filippis, B.; Fantacuzzi, M.; et al. Novel Phenyldiazenyl Fibrate Analogues as PPAR α/γ/δ Pan-Agonists for the Amelioration of Metabolic Syndrome. ACS Med. Chem. Lett. 2019, 10, 545–551. [Google Scholar] [CrossRef]

{kind=link}
| Isoforms | Tissues Distribution | Target Genes | Functions | Synthetic Ligands | Natural Ligands |
|---|---|---|---|---|---|
| PPARγ | White and brown adipose tissue, the large intestine, skeletal muscle, spleen, pancreas, and brain. | aP2, FATP, FAT/CD36. | Regulation of adipogenesis, energy balance, lipogenesis, gluconeogenesis, lipid storage, glucose uptake, metabolism uptake and differentiation. | Rosiglitazone, Pioglitazone, Troglitazone, T3D-959, DBZ. | 9-HODE, 13-HODE, 15d-PGJ2, EPA. |
| PPARα | Liver, heart, skeletal muscle, intestinal mucosa, white and brown adipose tissue, pancreas, and brain. | Acyl-CoA oxidase, Thiolase, Apolipoprotein A-I, Apolipoprotein A-II, CYP8B1, FATP, FAT/CD36, and Lipoprotein lipase. | Fatty acid metabolism, inflammation, thermogenesis, ketogenesis, glucose uptake, fatty acid oxidation and lipid storage. | Wy-14643, GW-2331, GW-9578, K-877 Fibrates. | Palmitic acid, Oleic acid, Linoleic acid, Arachidonic acid, DHA, oleoylethanolamide. |
| PPARβ/δ | Liver, intestine, kidney, abdominal white and brown adipose tissue, skeletal muscle, heart, pancreas, and brain. | Genes involved in lipid uptake, metabolism, and efflux, Lpin2, St3gal5. | Fatty acid oxidation, fatty acid metabolism, regulates blood cholesterol, glucose uptake, glucose utilization, insulin secretion, ketogenesis and inflammation. | L-796449, L-783483, GW-2433, MBX-8025, T3D-959, GW501516, GW610742. | Dihomo-γ-linolenic acid, Arachidonic acid, Methyl palmitate, 2-bromopalmitic acid, prostacyclin I2, 4-HNE. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
d’Angelo, M.; Castelli, V.; Tupone, M.G.; Catanesi, M.; Antonosante, A.; Dominguez-Benot, R.; Ippoliti, R.; Cimini, A.M.; Benedetti, E. Lifestyle and Food Habits Impact on Chronic Diseases: Roles of PPARs. Int. J. Mol. Sci. 2019, 20, 5422. https://doi.org/10.3390/ijms20215422
d’Angelo M, Castelli V, Tupone MG, Catanesi M, Antonosante A, Dominguez-Benot R, Ippoliti R, Cimini AM, Benedetti E. Lifestyle and Food Habits Impact on Chronic Diseases: Roles of PPARs. International Journal of Molecular Sciences. 2019; 20(21):5422. https://doi.org/10.3390/ijms20215422
Chicago/Turabian Styled’Angelo, Michele, Vanessa Castelli, Maria Grazia Tupone, Mariano Catanesi, Andrea Antonosante, Reyes Dominguez-Benot, Rodolfo Ippoliti, Anna Maria Cimini, and Elisabetta Benedetti. 2019. "Lifestyle and Food Habits Impact on Chronic Diseases: Roles of PPARs" International Journal of Molecular Sciences 20, no. 21: 5422. https://doi.org/10.3390/ijms20215422
APA Styled’Angelo, M., Castelli, V., Tupone, M. G., Catanesi, M., Antonosante, A., Dominguez-Benot, R., Ippoliti, R., Cimini, A. M., & Benedetti, E. (2019). Lifestyle and Food Habits Impact on Chronic Diseases: Roles of PPARs. International Journal of Molecular Sciences, 20(21), 5422. https://doi.org/10.3390/ijms20215422