Molecular Cloning and Exploration of the Biochemical and Functional Analysis of Recombinant Glucose-6-Phosphate Dehydrogenase from Gluconoacetobacter diazotrophicus PAL5

 , , , , ,
, , , , ,  , , , ,
, , , ,  , , ,
, , ,  , and
, and
Abstract
1. Introduction
2. Results and Discussion
2.1. zwf cDNA Isolation and Characterization
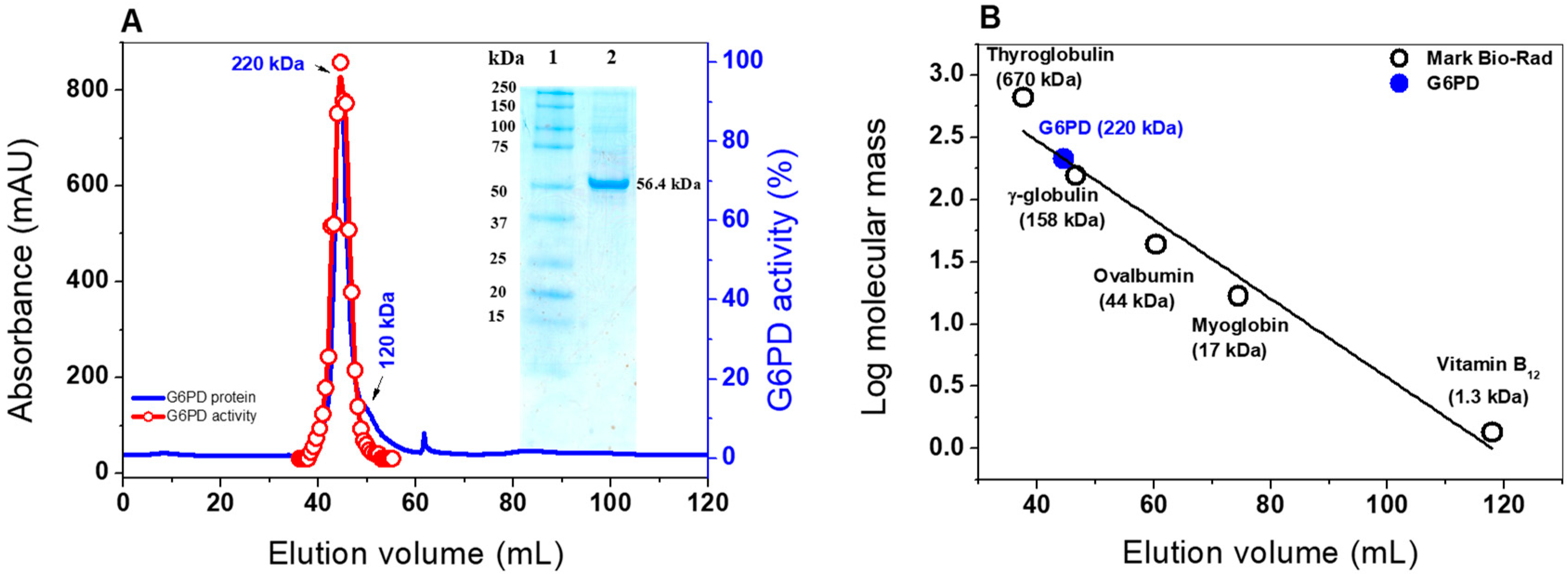
2.2. Heterologous Expression and Purification of G6PD
2.3. Biochemical Analysis of the G6PD Protein
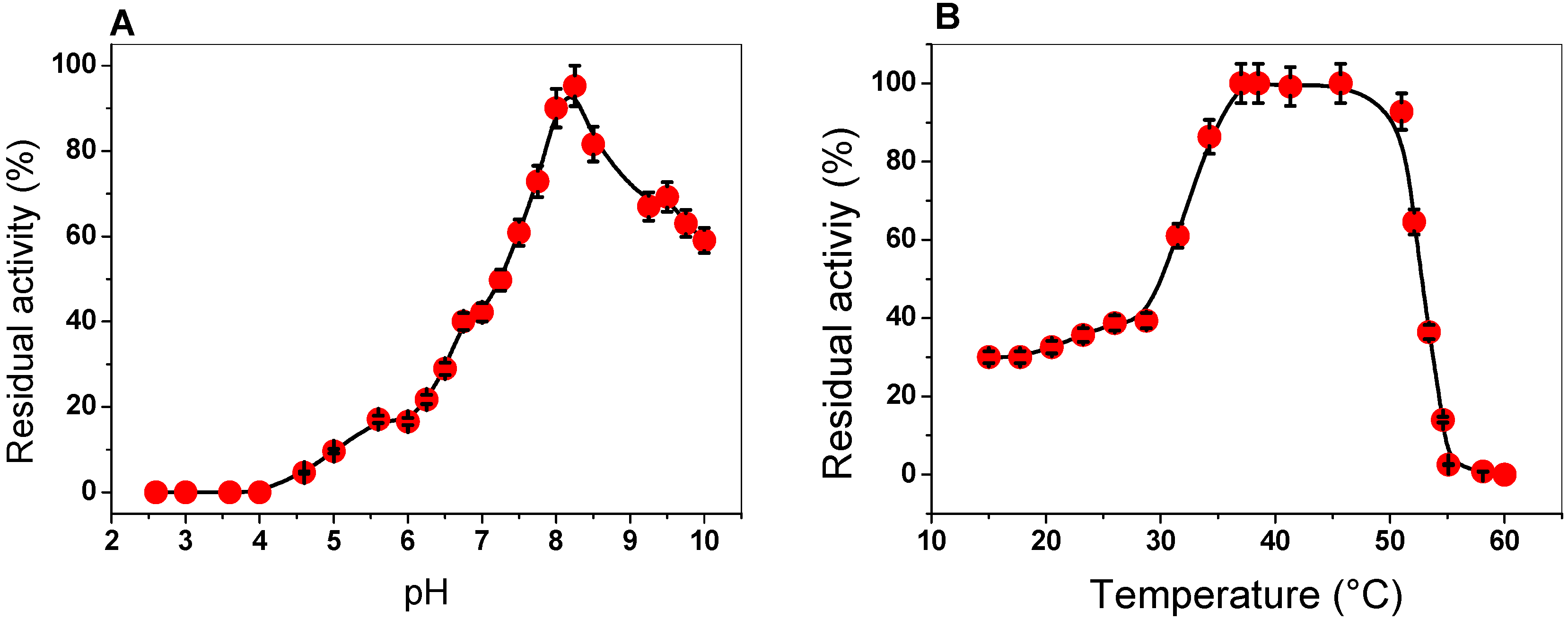
2.3.1. Effect of pH and Temperature on G6PD Activity
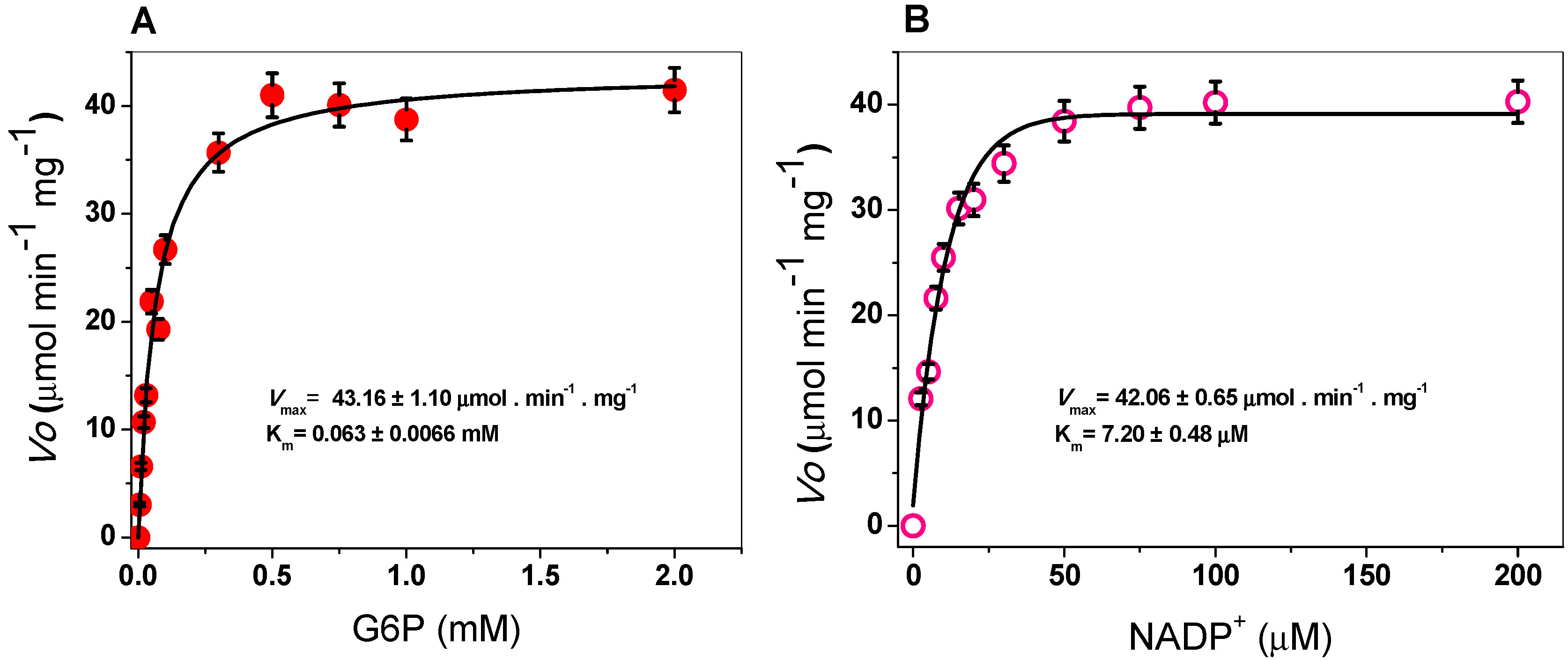
2.3.2. Kinetic Study of the G6PD Enzyme
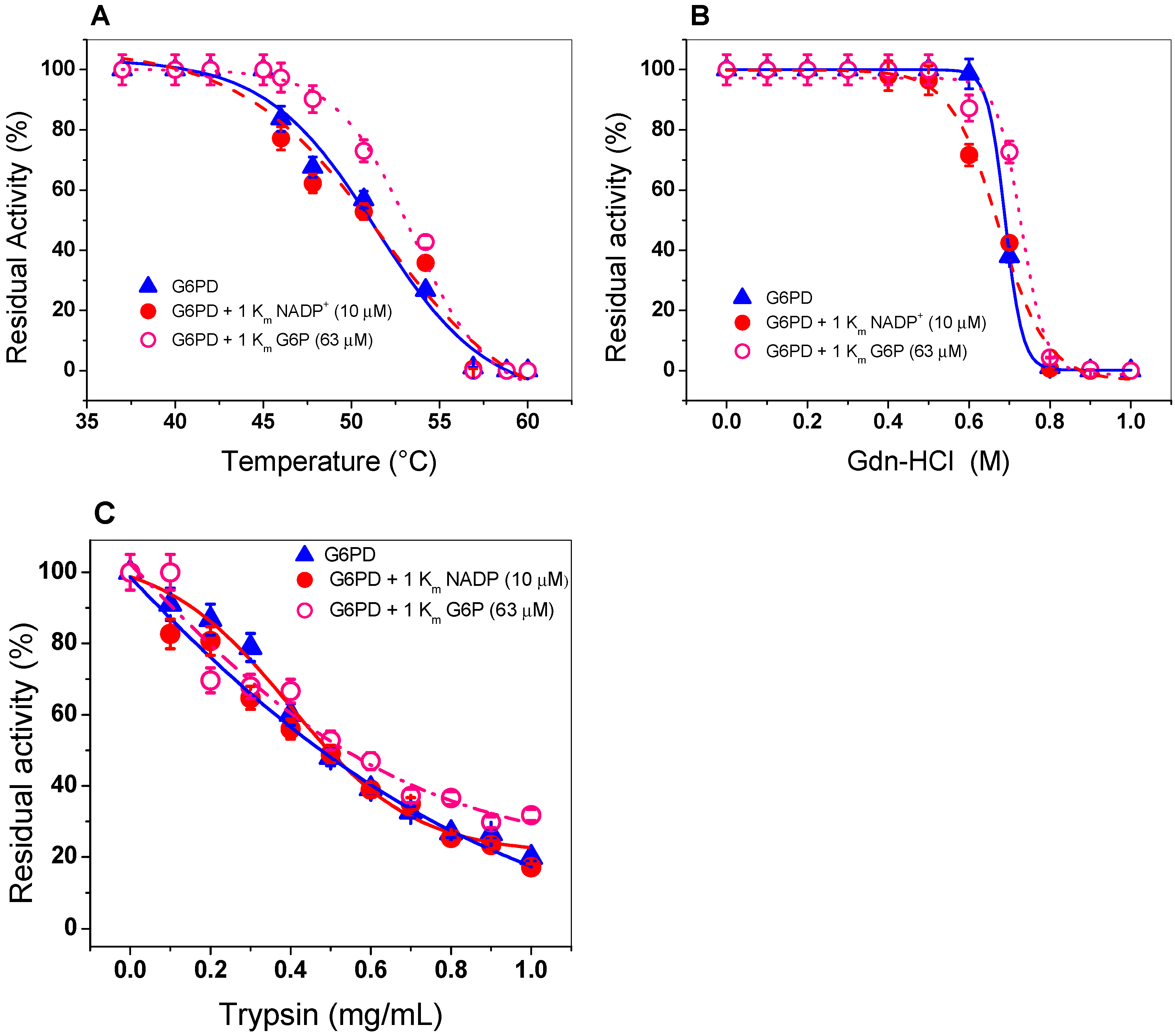
2.4. Evaluation of Protein Stability
2.4.1. Thermal Inactivation Analysis
2.4.2. Stability of G6PD in the Presence of Guanidine Hydrochloride (Gdn-HCl) or Protease Digestion
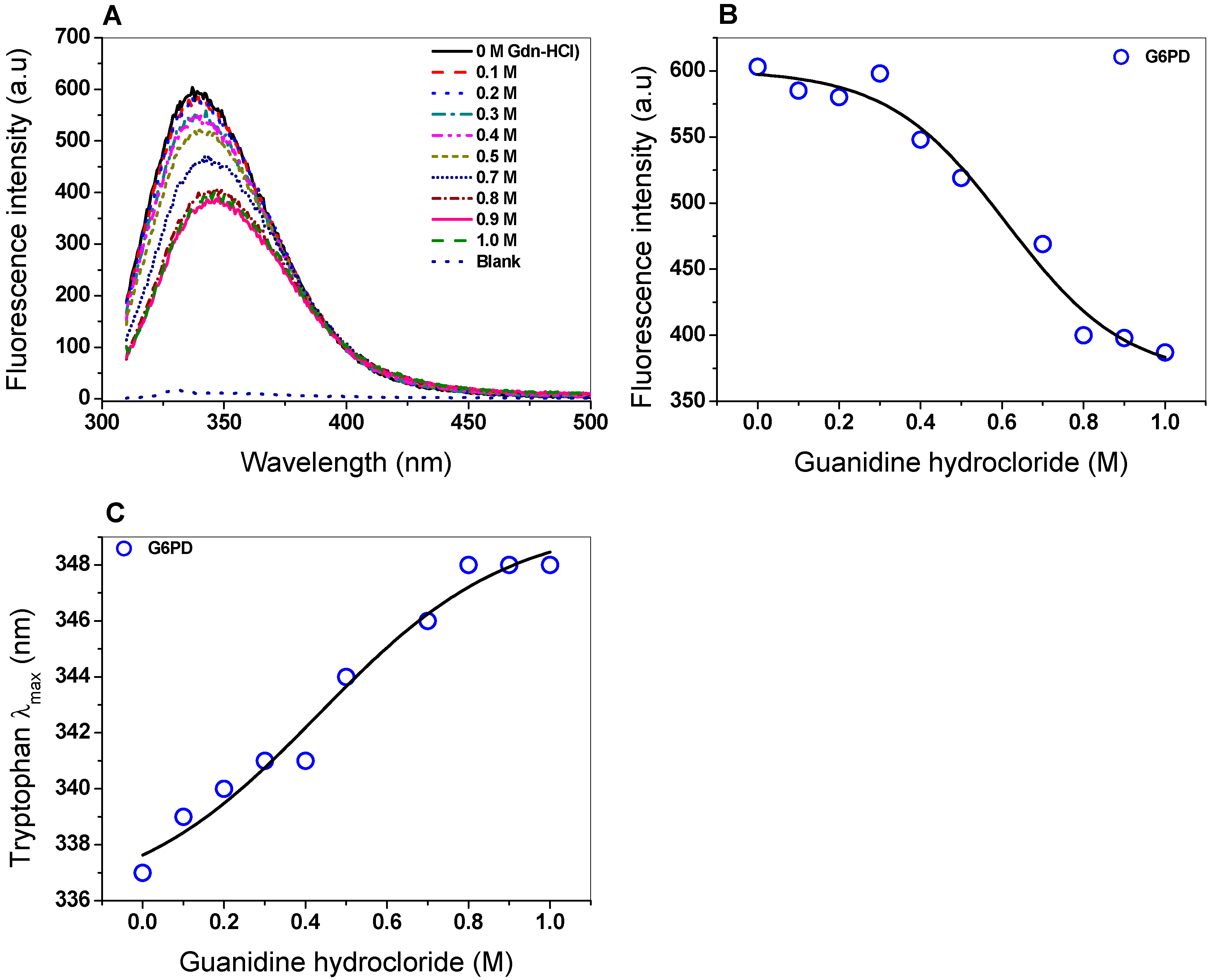
2.5. Spectroscopic Characterization
2.5.1. Circular Dichroism (CD) Analysis and Thermal Stability
2.5.2. Intrinsic Fluorescence
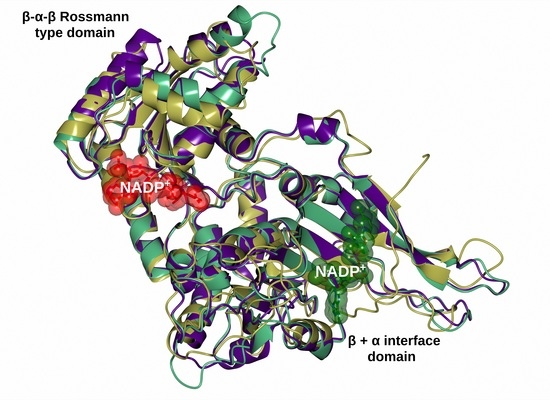
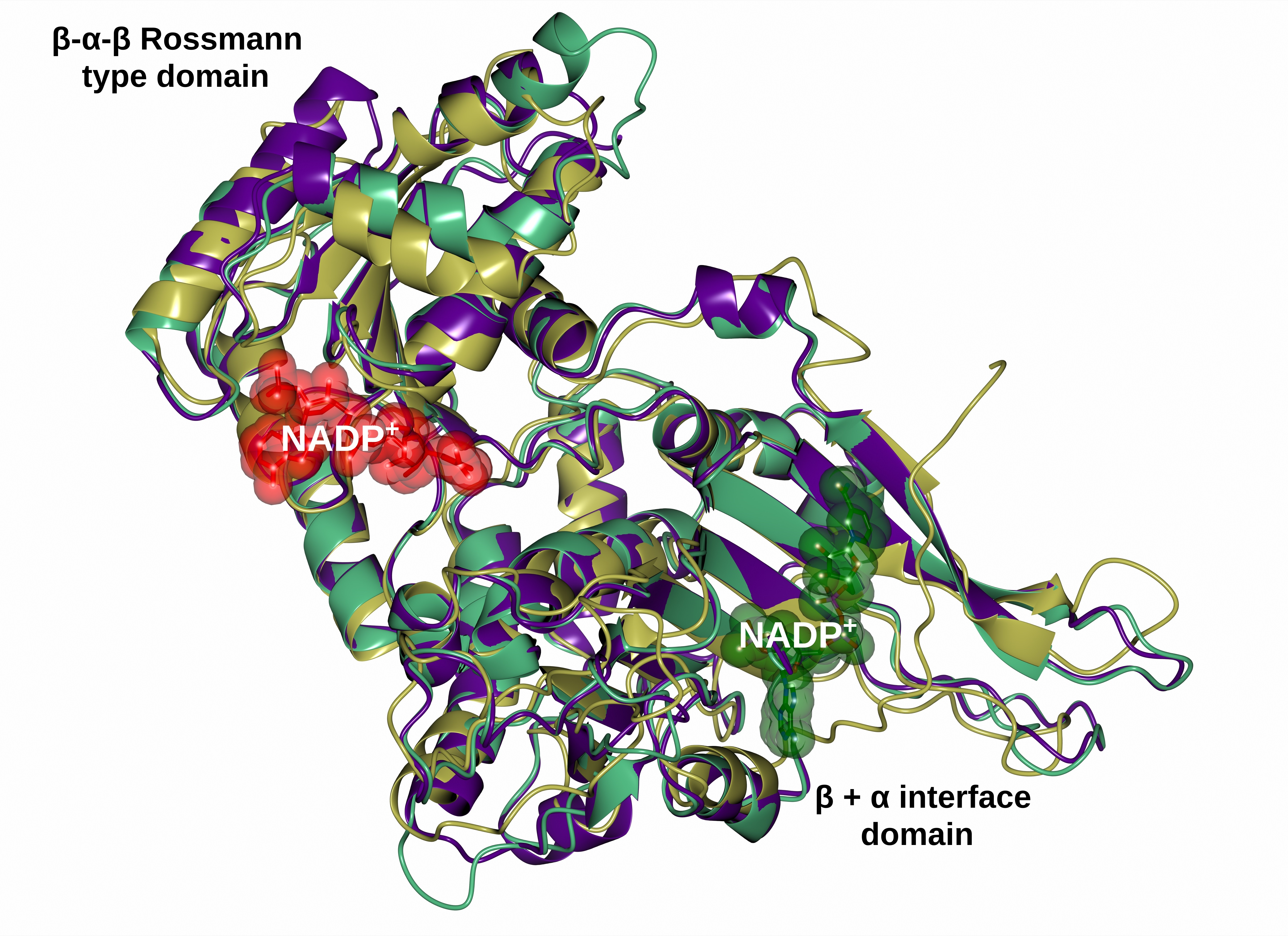
2.6. Homology Modeling of the Recombinant G6PD
3. Materials and Methods
3.1. Cloning of zwf from GDI
3.2. Alignment of the G6PD Protein of GDI
3.3. Expression and Purification of the G6PD Protein
3.4. Biochemical Analysis of G6PD Protein from GDI
3.4.1. Native Status of the G6PD protein
3.4.2. Effect of pH and Temperature on G6PD Activity
3.4.3. Kinetic Study of the G6PD Enzyme
3.5. Evaluation of Protein Stability
3.5.1. Thermal Inactivation Analysis
3.5.2. Stability of G6PD in the Presence of Gdn-HCl
3.5.3. Stability of G6PD in the Presence of Protease Digestion
3.6. Homology Modeling of the Structure of G6PD Protein
3.7. Spectroscopic Characterization
3.7.1. Circular Dichroism (CD) and Thermal Stability
3.7.2. Intrinsic Fluorescence of the G6PD Protein
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| G6PD | Glucose-6-phosphate dehydrogenase |
| PPP | Pentose Phosphate Pathway |
| GDI | Gluconacetobacter diazotrophicus |
| NADP | Nicotinamide adenine dinucleotide phosphate |
| DC | Dichroism Circular |
References
- Cavalcante, V.A.; Döbereiner, J. A new acid-tolerant nitrogen fixing bacterium associated with sugarcane. Plant Soil. 1988, 108, 23–31. [Google Scholar] [CrossRef]
- Chawla, N.; Phour, M.; Suneja, S.; Sangwaan, S.; Goyal, S. Gluconacetobacter diazotrophicus: An overview. Res. Environ. Life Sci. 2014, 7, 1–10. [Google Scholar]
- Cocking, E.C.; Stone, P.J.; Davey, M.R. Intracellular colonization of roots of Arabidopsis and crop plants by Gluconacetobacter diazotrophicus. Vitro Cell. Dev. Biol. Plant 2006, 42, 74–82. [Google Scholar] [CrossRef]
- Saravanan, V.S.; Madhaiyan, M.; Osborne, J.; Thangaraju, M.; Sa, T.M. Ecological occurrence of Gluconacetobacter diazotrophicus and nitrogen-fixing acetobacteraceae members: Their possible role in plant growth promotion. Microb. Ecol. 2008, 55, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Paula, M.A.; Reis, V.M.; Döbereiner, J. Interacction of Glomus clarum with Acetobacter diazotrophicus in infection of sweet potato (Ipomoea batatas), sugarcane (Saccharum spp.), and sweet sorghum (Sorghum vulgare). Biol. Fertil. Soils 1991, 11, 111–115. [Google Scholar] [CrossRef]
- Jiménez-Salgado, T.; Fuentes-Ramírez, L.E.; Tapia-Hernández, A.; Mascarúa-Esparza, M.A.; Martínez-Romero, E.; Caballero-Mellado, J. Coffea arabica L., a new host plant for Acetobacter diazotrophicus, and isolation of other nitrogen-fixing acetobacteria. Appl. Environ. Microbiol. 1997, 63, 3676–3683. [Google Scholar] [PubMed]
- Bertalan, M.; Alvano, R.; de Padua, V.; Rouws, L.; Rojas, C.; Hemerly, A.; Teixeira, K.; Schwab, S.; Araujo, J.; Oliveira, A.; et al. Complete genome sequence of the sugarcane nitrogen-fixing endophyte Gluconacetobacter diazotrophicus PAL5. BMC Genom. 2009, 10, 450. [Google Scholar] [CrossRef]
- Giongo, A.; Tyler, H.L.; Zipperer, U.N.; Triplett, E.W. Two genome sequences of the same bacterial strain, Gluconacetobacter diazotrophicus PAl 5, suggest a new standard in genome sequence submission. Stand. Genom. Sci. 2010, 2, 309–317. [Google Scholar] [CrossRef]
- Luna, M.F.; Bernardelli, C.E.; Galar, M.L.; Boiardi, J.L. Glucose metabolism in batch and continuous cultures of Gluconacetobacter diazotrophicus PAl 3. Cur. Microbiol. 2006, 52, 163–168. [Google Scholar] [CrossRef]
- Alvarez, B.; Martínez-Drets, G. Metabolic characterization of Acetobacter diazotrophicus. Can. J. Microbiol. 1995, 41, 918–924. [Google Scholar] [CrossRef]
- Attwood, M.M.; van Dijken, J.P.; Pronk, J.T. Glucose metabolism and gluconic acid production by Acetobacter diazotrophiucs. J. Ferment. Bioeng. 1991, 72, 101–105. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2012, 9, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Kotaka, M.; Gover, S.; Vandeputte-Rutten, L.; Au, S.W.N.; Lam, V.M.S.; Adams, M.J. Structural studies of glucose-6-phosphate and NADP+ binding to human glucose-6-phosphate dehydrogenase. Acta Crystallogr. D Biol. Crystallogr. 2005, 61, 495–504. [Google Scholar] [CrossRef]
- Bautista, J.M.; Mason, P.J.; Luzzatto, L. Human glucose-6-phosphate dehydrogenase Lysine 205 is dispensable for substrate binding but essential for catalysis. FEBS Lett. 1995, 366, 61–64. [Google Scholar] [CrossRef]
- Rowland, P.; Basak, A.K.; Gover, S.; Levy, H.R.; Adams, M.J. The three-dimensional structure of glucose 6-phosphate dehydrogenase from Leuconostoc mesenteroides refined at 2.0 A resolution. Structure 1994, 2, 1073–1087. [Google Scholar] [CrossRef]
- Hansen, T.; Schlichting, B.; Schonheit, P. Glucose-6-phosphate dehydrogenase from the hyperthermophilic bacterium Thermotoga maritima: Expression of the g6pd gene and characterization of an extremely thermophilic enzyme. FEMS Microbiol. Lett. 2002, 216, 249–253. [Google Scholar] [CrossRef]
- Acero-Navarro, K.E.; Jiménez-Ramírez, M.; Villalobos, M.A.; Vargas-Martínez, R.; Perales-Vela, H.V.; Velasco-García, R. Cloning, overexpression, and purification of glucose-6-phosphate dehydrogenase of Pseudomonas aeruginosa. Protein Expr. Purif. 2018, 142, 53–61. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; Ghazy, A.M.; Salem, A.M.H.; Ghazy, M.A.; Abdel-Monsef, M.M. Purification and characterization of glucose-6-phosphate dehydrogenase from camel liver. Enzym. Res. 2014, 2014, 714054. [Google Scholar] [CrossRef] [PubMed]
- Adem, S.; Ciftci, M. Purification of rat kidney glucose 6-phosphate dehydrogenase, 6-phosphogluconate dehydrogenase, and glutathione reductase enzymes using 2’,5’-ADP Sepharose 4B affinity in a single chromatography step. Protein Expr. Purif. 2012, 81, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.; Ghazy, A.H.; Salem, A.M.; Ghazy, M.A.; Abdel-Monsef, M.M. Biochemical characterization of buffalo liver glucose-6-phosphate dehydrogenase isoforms. Protein J. 2015, 34, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Suthar, M.K.; Doharey, P.K.; Gupta, S.; Yadav, S.; Chauhan, P.M.; Saxena, J.K. Molecular cloning and characterization of glucose-6-phosphate dehydrogenase from Brugia malayi. Parasitology 2013, 140, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Pickl, A.; Schönheit, P. The oxidative pentose phosphate pathway in the haloarchaeon Haloferax volcanii involves a novel type of glucose-6-phosphate dehydrogenase—The archaeal Zwischenferment. FEBS Lett. 2015, 589, 1105–1111. [Google Scholar] [CrossRef]
- Naylor, C.E.; Gover, S.; Basak, A.K.; Cosgrove, M.S.; Levy, H.R.; Adams, M.J. NADP and NAD binding to the dual coenzyme specific enzyme Leuconostoc mesenteroides glucose 6-phosphate dehydrogenase: Different interdomain hinge angles are seen in different binary and ternary complexes. Acta crystallogr. D Biol. Crystallogr. 2001, 57, 635–648. [Google Scholar] [CrossRef]
- Wang, X.T.; Lam, V.M.; Engel, P.C. Marked decrease in specific activity contributes to disease phenotype in two human glucose-6-phosphate dehydrogenase mutants, G6PD Union and G6PD Andalus. Hum. Mutat. 2005, 26, 284–293. [Google Scholar] [CrossRef]
- Özer, N.; Bilgi, C.; Ögüsa, I.H. Dog liver glucose-6-phosphate dehydrogenase: Purification and kinetic properties. Int. J. Biochem. Cell. Biol. 2002, 34, 253–262. [Google Scholar] [CrossRef]
- Rendón, J.L.; del Arenal, I.P.; Guevara-Flores, A.; Mendoza-Hernández, G.; Pardo, J.P. Glucose 6-phosphate dehydrogenase from larval Taenia crassiceps (cysticerci): Purification and properties. Parasitol. Res. 2008, 102, 1351–1357. [Google Scholar] [CrossRef]
- Ortiz, C.; Moraca, F.; Medeiros, A.; Botta, M.; Hamilton, N.; Comini, M.A. Binding mode and selectivity of steroids towards glucose-6-phosphate dehydrogenase from the pathogen Trypanosoma cruzi. Molecules 2016, 21, 368. [Google Scholar] [CrossRef]
- Schuurmann, J.; Quehl, P.; Lindhorst, F.; Lang, K.; Jose, J. Autodisplay of glucose-6-phosphate dehydrogenase for redox cofactor regeneration at the cell surface. Biotechnol. Bioeng. 2017, 114, 1658–1669. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.H.; Han, J.; Wu, J.; Chen, H. Heteroexpression and functional characterization of glucose 6-phosphate dehydrogenase from industrial Aspergillus oryzae. J. Microbiol. Biotechnol. 2019, 29, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Wennekes, L.M.; Goosen, T.; van den Broek, P.J.; van den Broek, H.W. Purification and characterization of glucose-6-phosphate dehydrogenase from Aspergillus niger and Aspergillus nidulans. J. Gen. Microbiol. 1993, 139, 2793–2800. [Google Scholar] [CrossRef]
- Gómez-Manzo, S.; Terrón-Hernández, J.; de la Mora-de la Mora, I.; García Torres, I.; López-Velázquez, G.; Reyes-Vivas, H.; Oria-Hernández, J. Cloning, expression, purification and characterization of His-tagged human glucose-6-phosphate dehydrogenase: A simplified method for protein yield. Protein J. 2013, 32, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Manzo, S.; Terrón-Hernández, J.; De la Mora-De la Mora, I.; González-Valdez, A.; Marcial-Quino, J.; García-Torres, I.; Vanoye-Carlo, A.; López-Velázquez, G.; Hernández-Alcantara, G.; Oria-Hernández, J.; et al. The stability of G6PD is affected by mutations with different clinical phenotypes. Int. J. Mol. Sci. 2014, 15, 21179–21201. [Google Scholar] [CrossRef] [PubMed]
- Bian, M.; Li, S.; Wei, H.; Huang, S.; Zhou, F.; Zhu, Y.; Zhu, G. Heteroexpression and biochemical characterization of a glucose-6-phosphate dehydrogenase from oleaginous yeast Yarrowia lipolytica. Protein Expr. Purif. 2018, 148, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Morales-Luna, L.; Serrano-Posada, H.; González-Valdez, A.; Ortega-Cuellar, D.; Vanoye-Carlo, A.; Hernández-Ochoa, B.; Sierra-Palacios, E.; Rufino-González, Y.; Castillo-Rodríguez, R.A.; Pérez de la Cruz, V.; et al. Biochemical characterization and structural modeling of fused glucose-6-phosphate dehydrogenase-phosphogluconolactonase from Giardia lamblia. Int. J. Mol. Sci. 2018, 25, 19. [Google Scholar] [CrossRef] [PubMed]
- Jortzik, E.; Mailu, B.M.; Preuss, J.; Fischer, M.; Bode, L.; Rahlfs, S.; Becker, K. Glucose-6-phosphate dehydrogenase–6-phosphogluconolactonase: A unique bifunctional enzyme from Plasmodium falciparum. Biochem. J. 2011, 436, 641–650. [Google Scholar] [CrossRef]
- Gómez-Manzo, S.; Marcial-Quino, J.; Vanoye-Carlo, A.; Serrano-Posada, H.; González-Valdez, A.; Martínez-Rosas, V.; Hernández-Ochoa, B.; Sierra-Palacios, E.; Castillo-Rodríguez, R.A.; Cuevas-Cruz, M.; et al. Functional and biochemical characterization of three recombinant human glucose-6-phosphate dehydrogenase mutants: Zacatecas, vanua-lava and viangchan. Int. J. Mol. Sci. 2016, 17, 787. [Google Scholar] [CrossRef]
- Ramírez-Nava, E.J.; Ortega-Cuellar, D.; Serrano-Posada, H.; González-Valdez, A.; Vanoye-Carlo, A.; Hernández-Ochoa, B.; Sierra-Palacios, E.; Hernández-Pineda, J.; Rodríguez-Bustamante, E.; Arreguin-Espinosa, R.; et al. Biochemical analysis of two single mutants that give rise to a polymorphic g6pd a-double mutant. Int. J. Mol. Sci. 2017, 26, 18. [Google Scholar] [CrossRef]
- Cortés-Morales, Y.Y.; Vanoye-Carlo, A.; Castillo-Rodríguez, R.A.; Serrano-Posada, H.; González-Valdez, A.; Ortega-Cuellar, D.; Hernández-Ochoa, B.; Moreno-Vargas, L.M.; Prada-Gracia, D.; Sierra-Palacios, E.; et al. Cloning and biochemical characterization of three glucose-6-phosphate dehydrogenase mutants presents in the Mexican population. Int. J. Biol. Macromol. 2018, 119, 926–936. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Manzo, S.; Marcial-Quino, J.; Vanoye-Carlo, A.; Enríquez-Flores, S.; De la Mora-De la Mora, I.; González-Valdez, A.; García-Torres, I.; Martínez-Rosas, V.; Sierra-Palacios, E.; Lazcano-Pérez, F.; et al. Mutations of glucose-6-phosphate dehydrogenase Durham, Santa-Maria and A+ variants are associated with loss functional and structural stability of the protein. Int. J. Mol. Sci. 2015, 16, 28657–28668. [Google Scholar] [CrossRef] [PubMed]
- Boonyuen, U.; Chamchoy, K.; Swangsri, T.; Saralamba, T.; Day, N.P.J.; Imwong, M. Detailed functional analysis of two clinical glucose-6-phosphate dehydrogenase (G6PD) variants, G6PD Viangchan and G6PD Viangchan + Mahidol: Decreased stability and catalytic efficiency contribute to the clinical phenotype. Mol. Genet. Metab. 2016, 2, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Boonyuen, U.; Chamchoy, K.; Swangsri, T.; Junkree, T.; Day, M.; White, N.; Imwong, M. A trade-off between catalytic activity and protein stability determines the clinical manifestations of glucose-6-phosphate dehydrogenase (G6PD) deficiency. Int. J. Biol. Macromol. 2017, 104, 145–156. [Google Scholar] [CrossRef]
- Gómez-Manzo, S.; Marcial-Quino, J.; Ortega-Cuellar, D.; Serrano-Posada, H.; González-Valdez, H.; Vanoye-Carlo, A.; Hernández-Ochoa, B.; Sierra-Palacios, E.; Castillo-Villanueva, A.; Reyes-Vivas, H. Functional and biochemical analysis of glucose-6-phosphate dehydrogenase (G6PD) variants: Elucidating the molecular basis of G6PD deficiency. Catalysts 2017, 7, 135. [Google Scholar] [CrossRef]
- Wang, X.T.; Chan, T.F.; Lam, V.; Engel, P. What is the role of the second “structural” NADP+-binding site in human glucose-6-phosphate dehydrogenase? Protein Sci. 2008, 17, 1403–1411. [Google Scholar] [CrossRef]
- Huang, Y.; Choi, M.Y.; Au, S.W.; Au, D.M.; Lam, V.M.S.; Engel, P.C. Purification and detailed study of two clinically different human glucose 6-phosphate dehydrogenase variants, G6PD (Plymouth) and G6PD (Mahidol): Evidence for defective protein folding as the basis of disease. Mol. Genet. Metab. 2008, 93, 44–53. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Au, S.W.N.; Gover, S.; Lam, V.; Adams, M. Human glucose-6-phosphate dehydrogenase: The crystal structure reveals a structural NADP+ molecule and provides insights into enzyme deficiency. Structure 2000, 8, 293–303. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- McNicholas, S.; Potterton, E.; Wilson, K.S.; Noble, M.E.M. Presenting your structures: The CCP4mg molecular-graphics software. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 386–394. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | kcat (s−1) | Km G6P (µM) | Km NADP+ (µM) | Reference |
|---|---|---|---|---|
| Gluconacetobacter diazotrophichus | 293,181 | 63 | 7 | This study |
| Escherichia coli DH5α | 32 | 224 | 127 | [32] |
| Pseudomonas aeruginosa | 540 | 498 | 56 | [21] |
| Termotoga maritima | 35,000 | 200 | 40 | [20] |
| Haloferax volcanii | 11 | 370 | 520 | [26] |
| Giardia lamblia | 31 | 18 | 14 | [38] |
| Plasmodium falciparum | 8 | 19 | 6 | [39] |
| Trypanosoma cruzy | 62 | 77 | 16 | [31] |
| Aspergillus niger | NR | 153 | 26 | [34] |
| Aspergillus oryzae | 1000 | 109 | 6 | [33] |
| Brugia malayi | 40 | 245 | 14 | [25] |
| Dog liver | NR | 122 | 10 | [29] |
| Buffalo liver | NR | NR | 59 | [24] |
| Camel liver | NR | 81 | 81 | [22] |
| Homo sapiens | 230 | 38 | 7 | [36] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramírez-Nava, E.J.; Ortega-Cuellar, D.; González-Valdez, A.; Castillo-Rodríguez, R.A.; Ponce-Soto, G.Y.; Hernández-Ochoa, B.; Cárdenas-Rodríguez, N.; Martínez-Rosas, V.; Morales-Luna, L.; Serrano-Posada, H.; et al. Molecular Cloning and Exploration of the Biochemical and Functional Analysis of Recombinant Glucose-6-Phosphate Dehydrogenase from Gluconoacetobacter diazotrophicus PAL5. Int. J. Mol. Sci. 2019, 20, 5279. https://doi.org/10.3390/ijms20215279
Ramírez-Nava EJ, Ortega-Cuellar D, González-Valdez A, Castillo-Rodríguez RA, Ponce-Soto GY, Hernández-Ochoa B, Cárdenas-Rodríguez N, Martínez-Rosas V, Morales-Luna L, Serrano-Posada H, et al. Molecular Cloning and Exploration of the Biochemical and Functional Analysis of Recombinant Glucose-6-Phosphate Dehydrogenase from Gluconoacetobacter diazotrophicus PAL5. International Journal of Molecular Sciences. 2019; 20(21):5279. https://doi.org/10.3390/ijms20215279
Chicago/Turabian StyleRamírez-Nava, Edson Jiovany, Daniel Ortega-Cuellar, Abigail González-Valdez, Rosa Angélica Castillo-Rodríguez, Gabriel Yaxal Ponce-Soto, Beatriz Hernández-Ochoa, Noemí Cárdenas-Rodríguez, Víctor Martínez-Rosas, Laura Morales-Luna, Hugo Serrano-Posada, and et al. 2019. "Molecular Cloning and Exploration of the Biochemical and Functional Analysis of Recombinant Glucose-6-Phosphate Dehydrogenase from Gluconoacetobacter diazotrophicus PAL5" International Journal of Molecular Sciences 20, no. 21: 5279. https://doi.org/10.3390/ijms20215279
APA StyleRamírez-Nava, E. J., Ortega-Cuellar, D., González-Valdez, A., Castillo-Rodríguez, R. A., Ponce-Soto, G. Y., Hernández-Ochoa, B., Cárdenas-Rodríguez, N., Martínez-Rosas, V., Morales-Luna, L., Serrano-Posada, H., Sierra-Palacios, E., Arreguin-Espinosa, R., Cuevas-Cruz, M., Rocha-Ramírez, L. M., Pérez de la Cruz, V., Marcial-Quino, J., & Gómez-Manzo, S. (2019). Molecular Cloning and Exploration of the Biochemical and Functional Analysis of Recombinant Glucose-6-Phosphate Dehydrogenase from Gluconoacetobacter diazotrophicus PAL5. International Journal of Molecular Sciences, 20(21), 5279. https://doi.org/10.3390/ijms20215279