WIN55,212-2-Induced Expression of Mir-29b1 Favours the Suppression of Osteosarcoma Cell Migration in a SPARC-Independent Manner



 ,
,  ,
,  ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
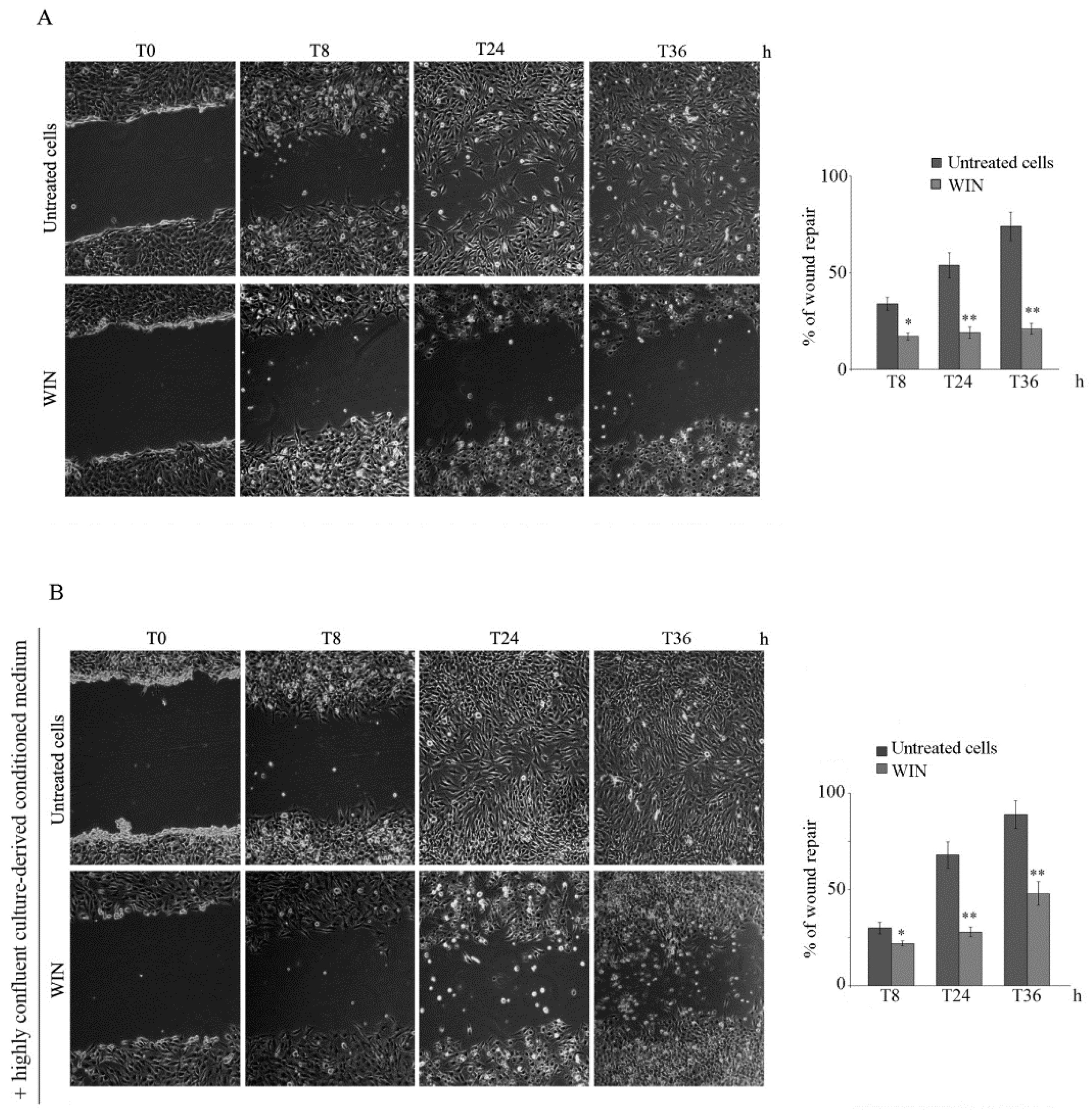
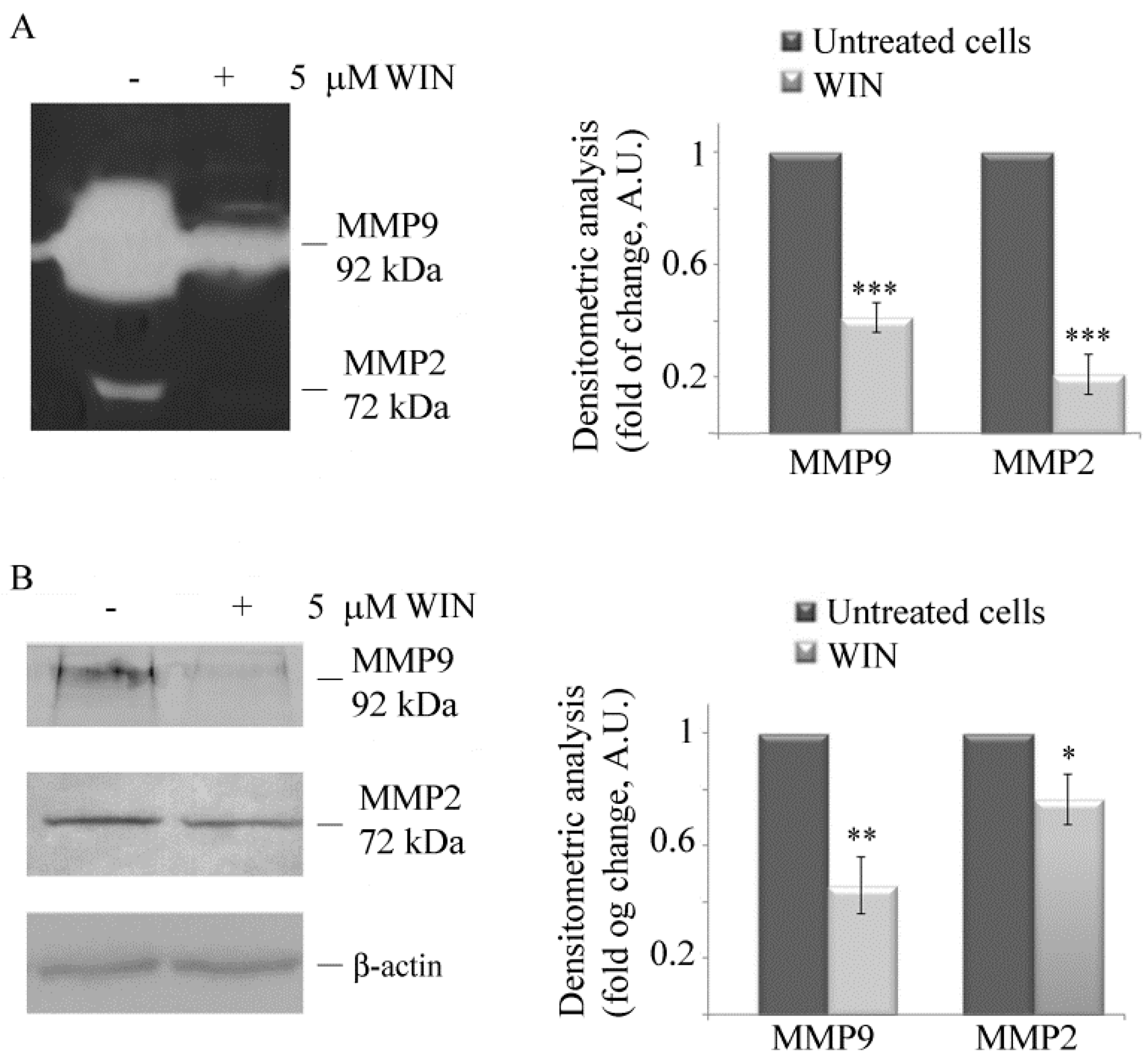
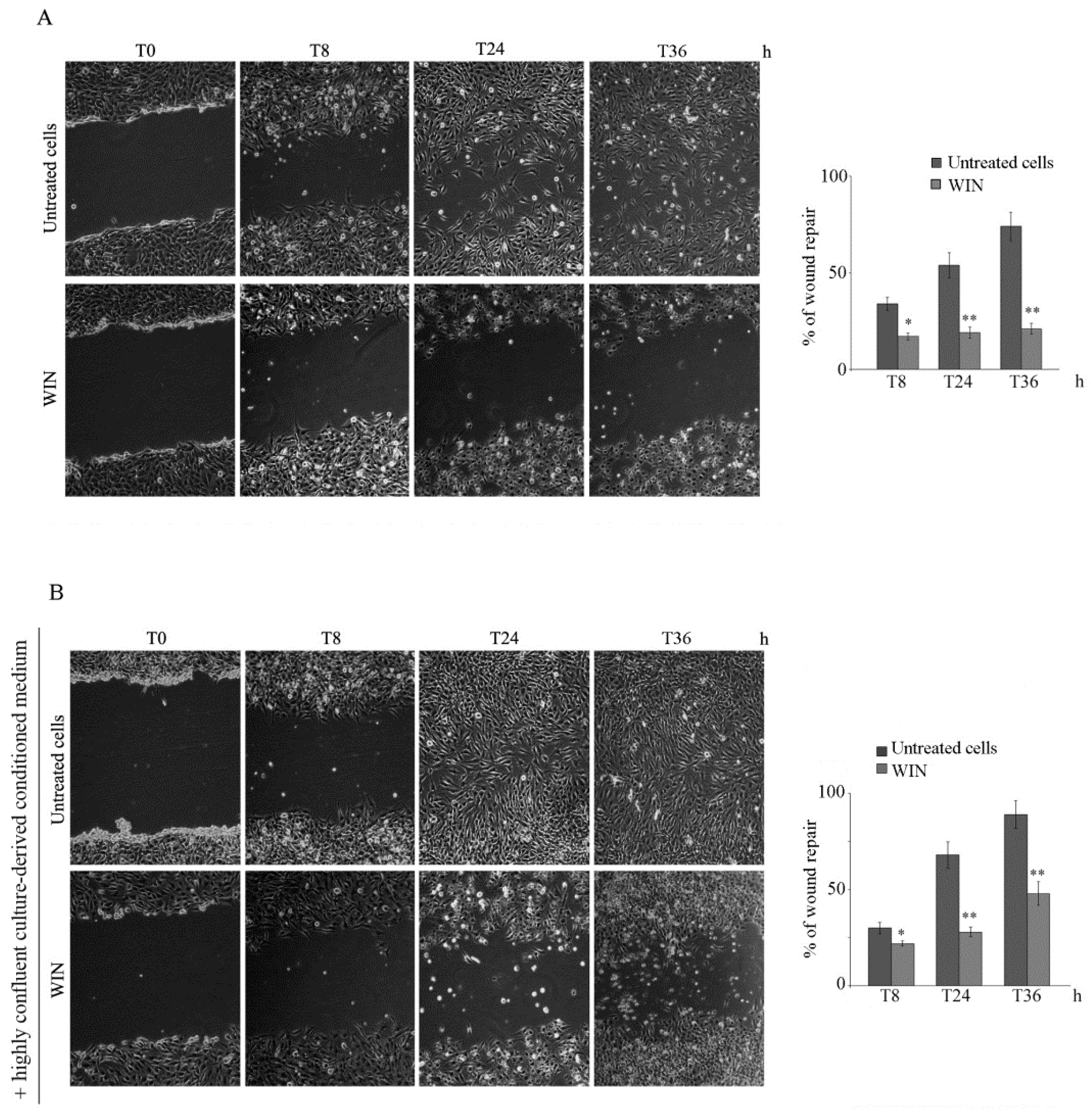
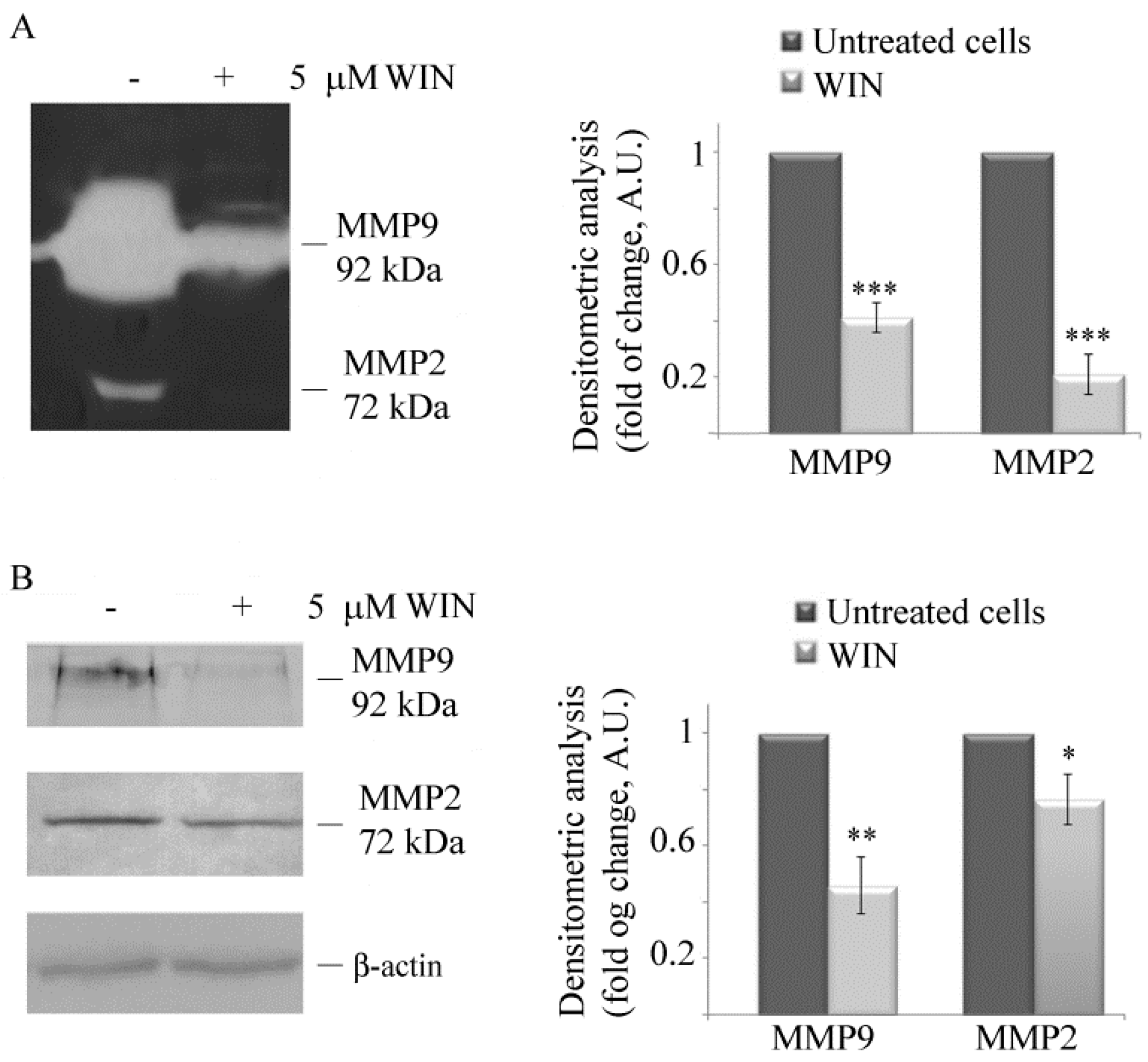
2.1. WIN Treatment Reduces the Migratory Ability of MG63 Cells and Affects MMP Activity
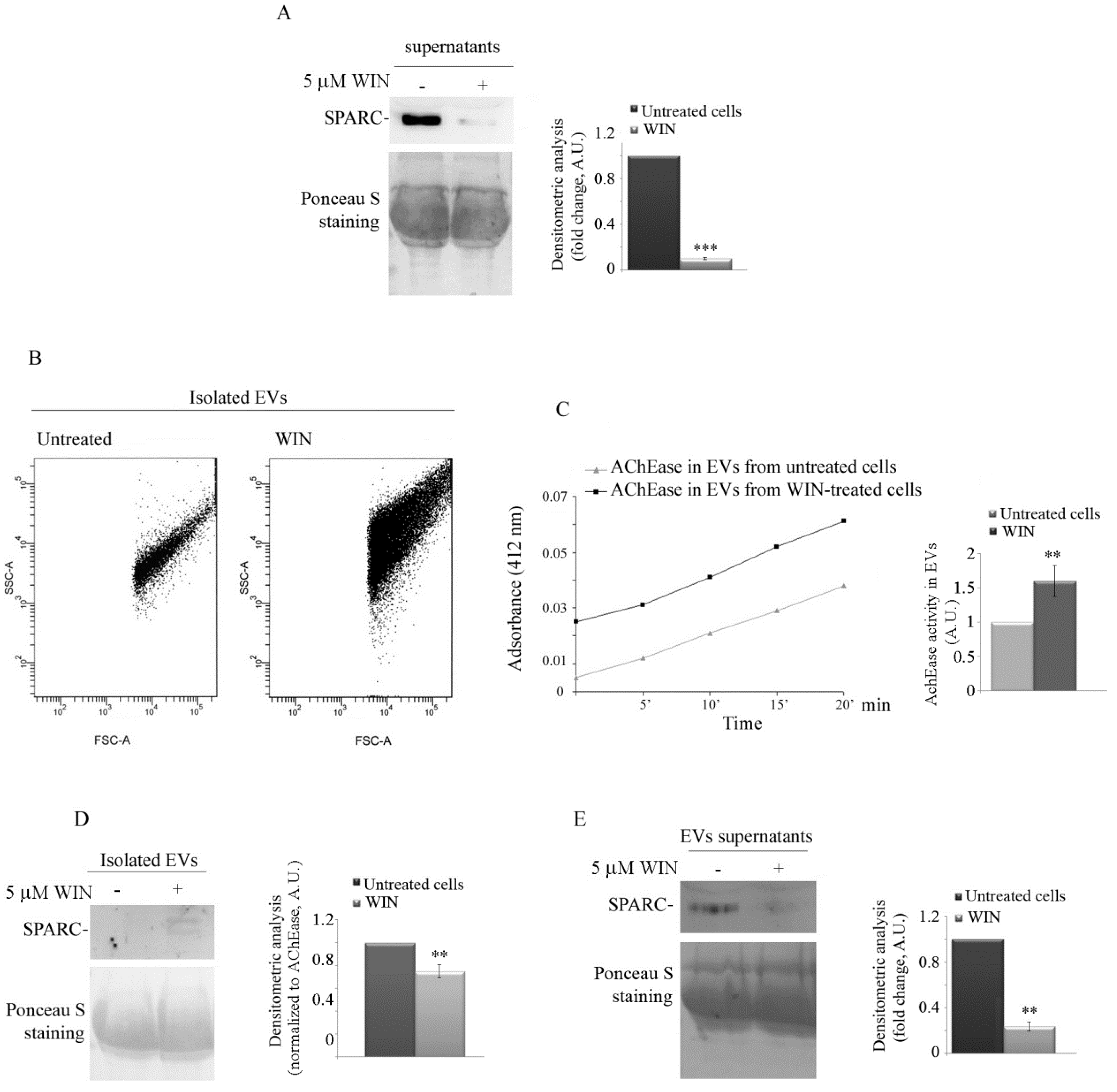
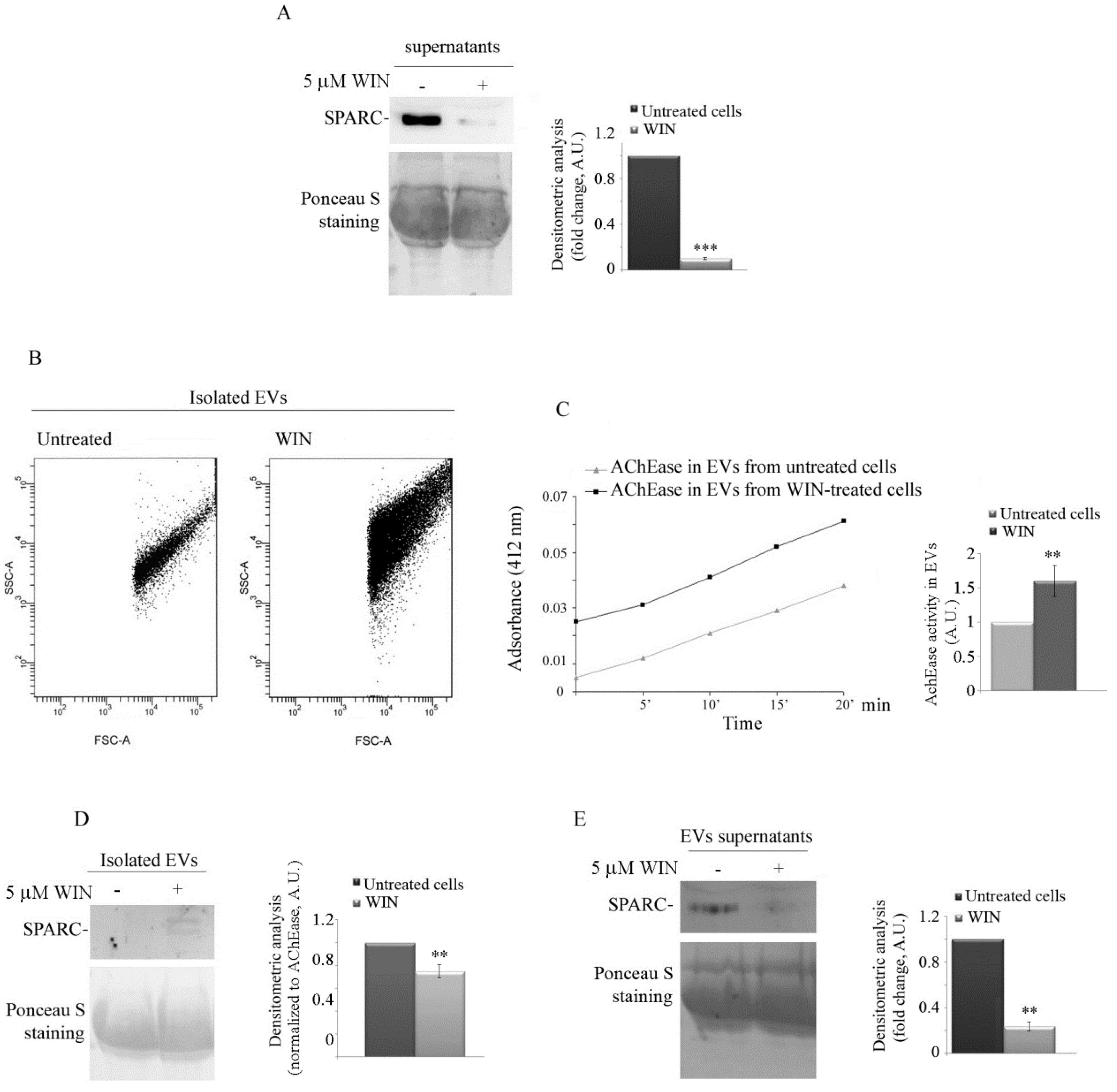
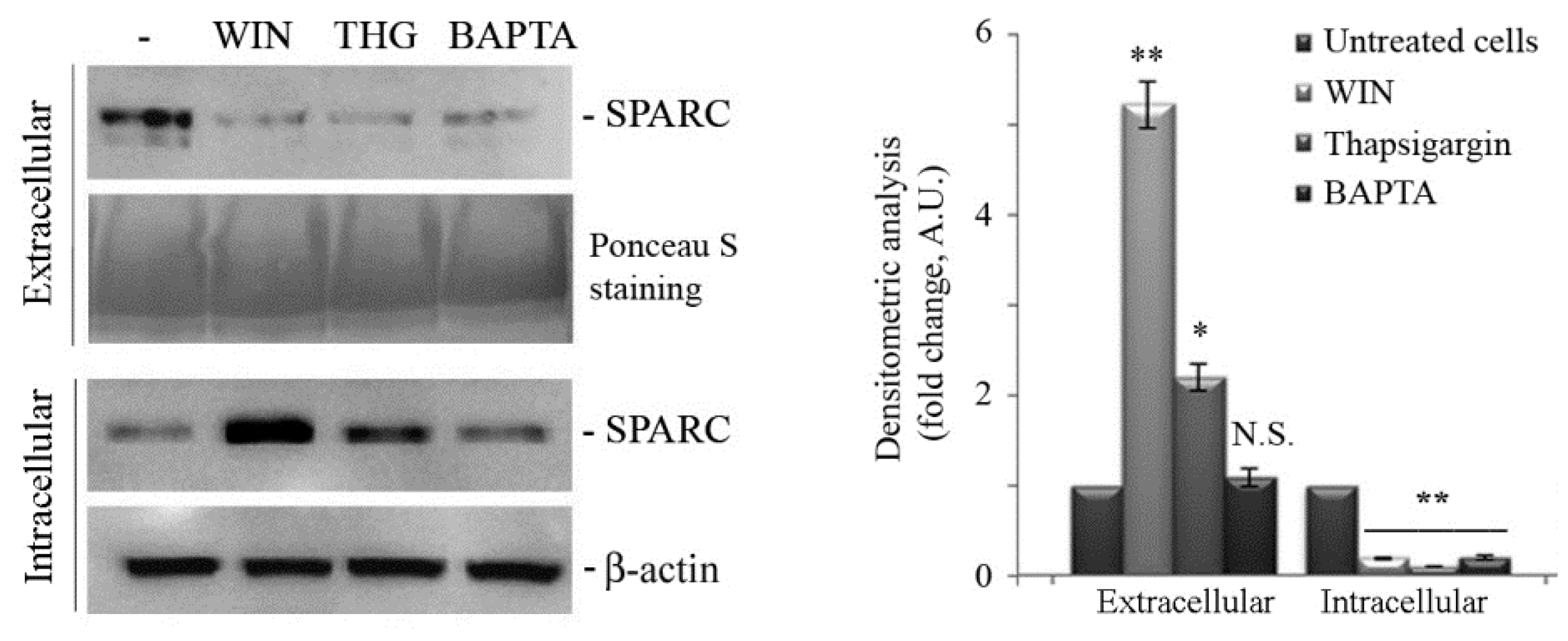
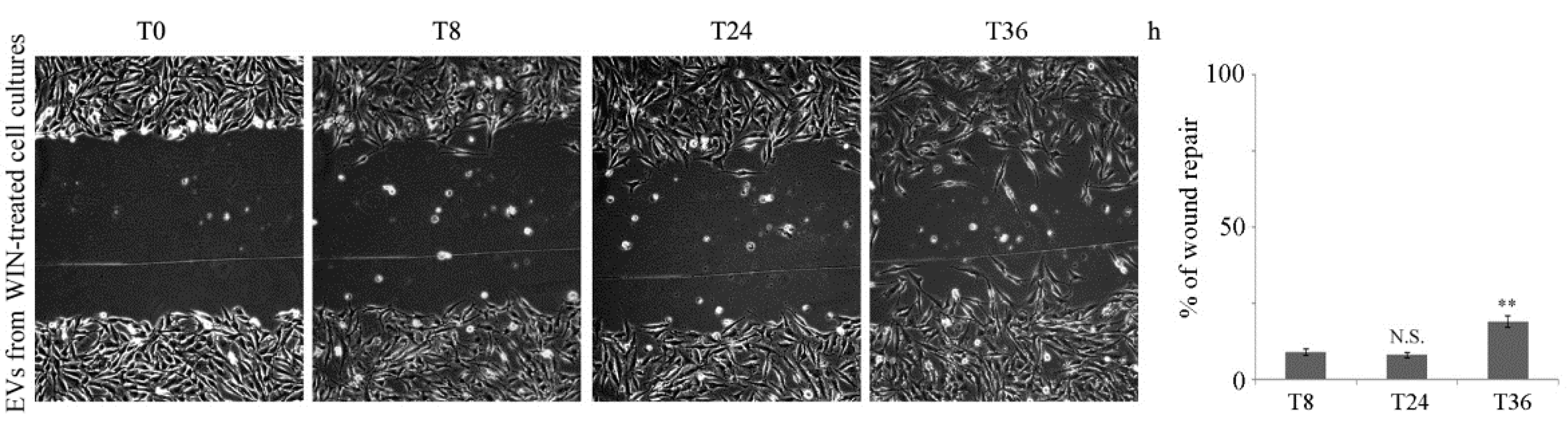
2.2. WIN Treatment Prevents SPARC Release in the Extracellular Environment and Increases the Release of Extracellular Vesicles
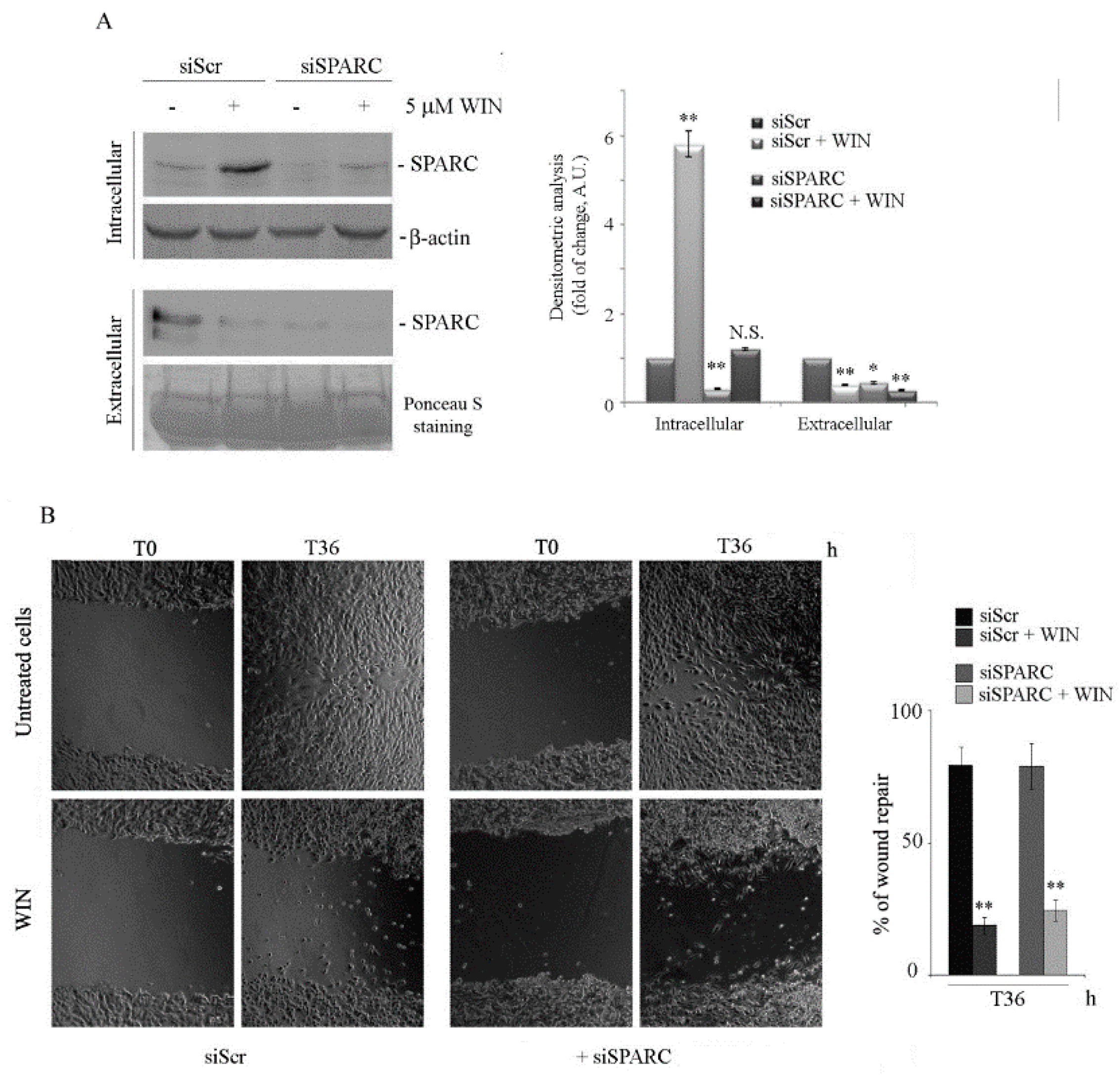
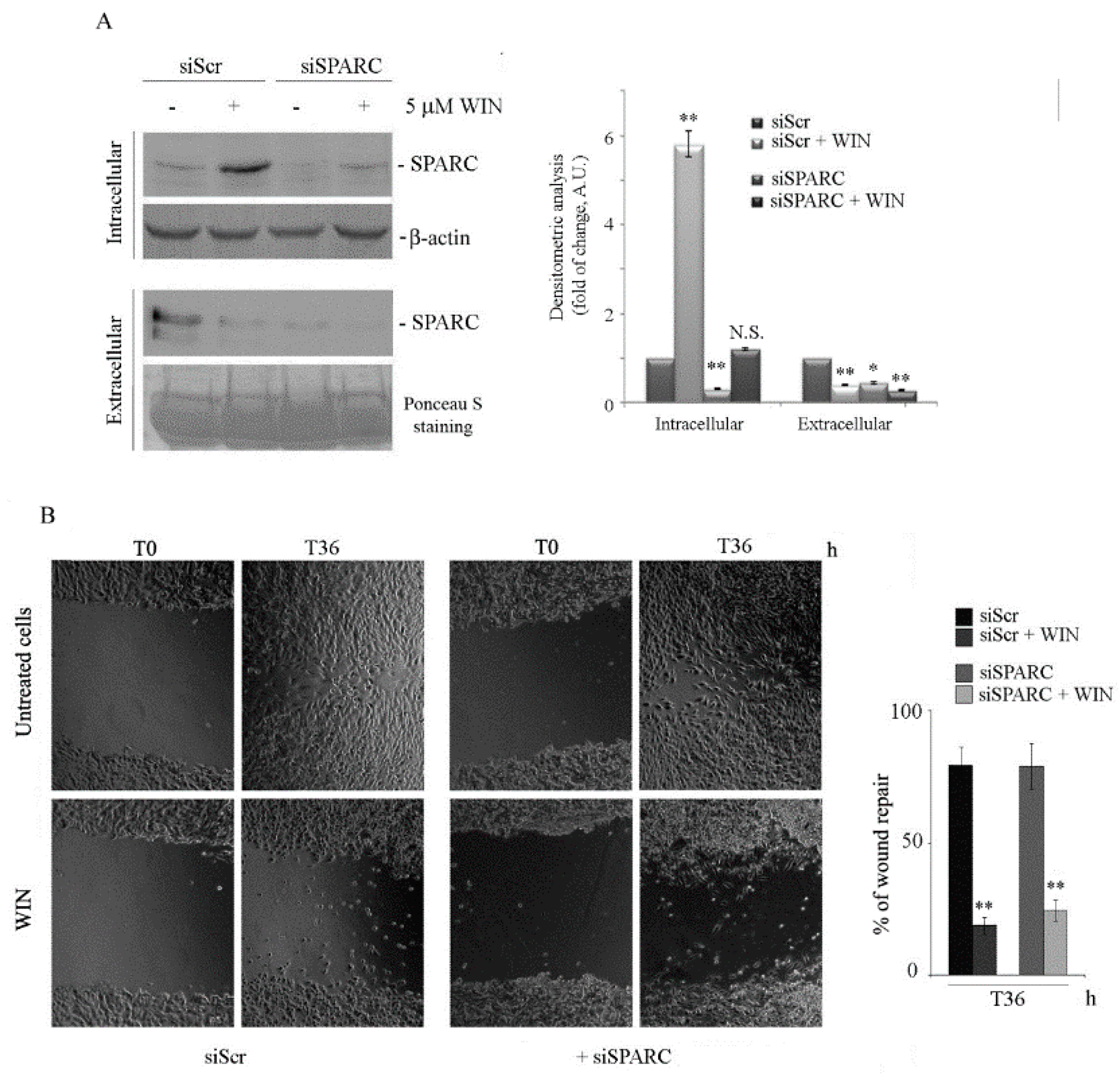
2.3. Silencing SPARC did not Affect the Anti-Migratory Effect of WIN
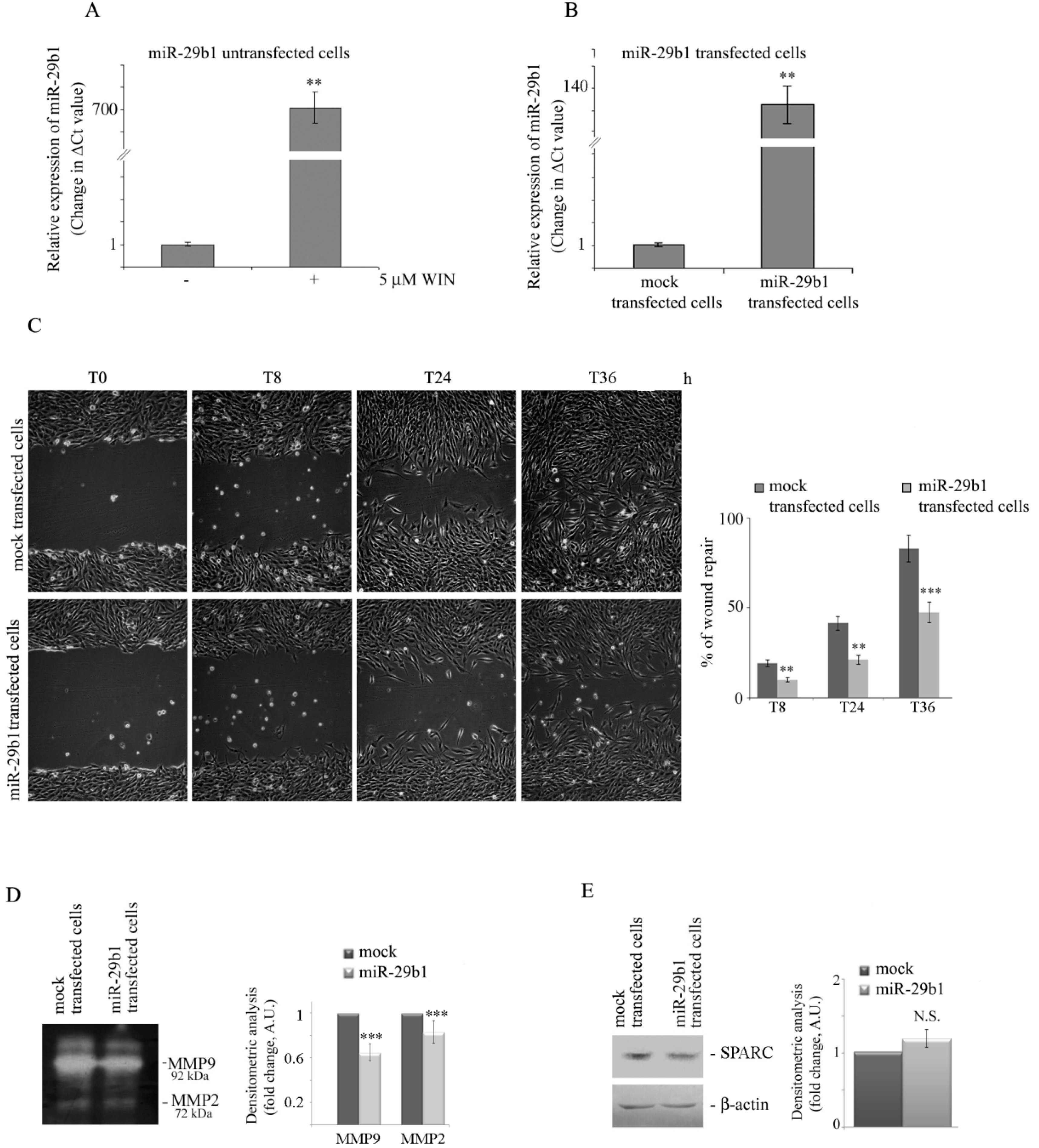
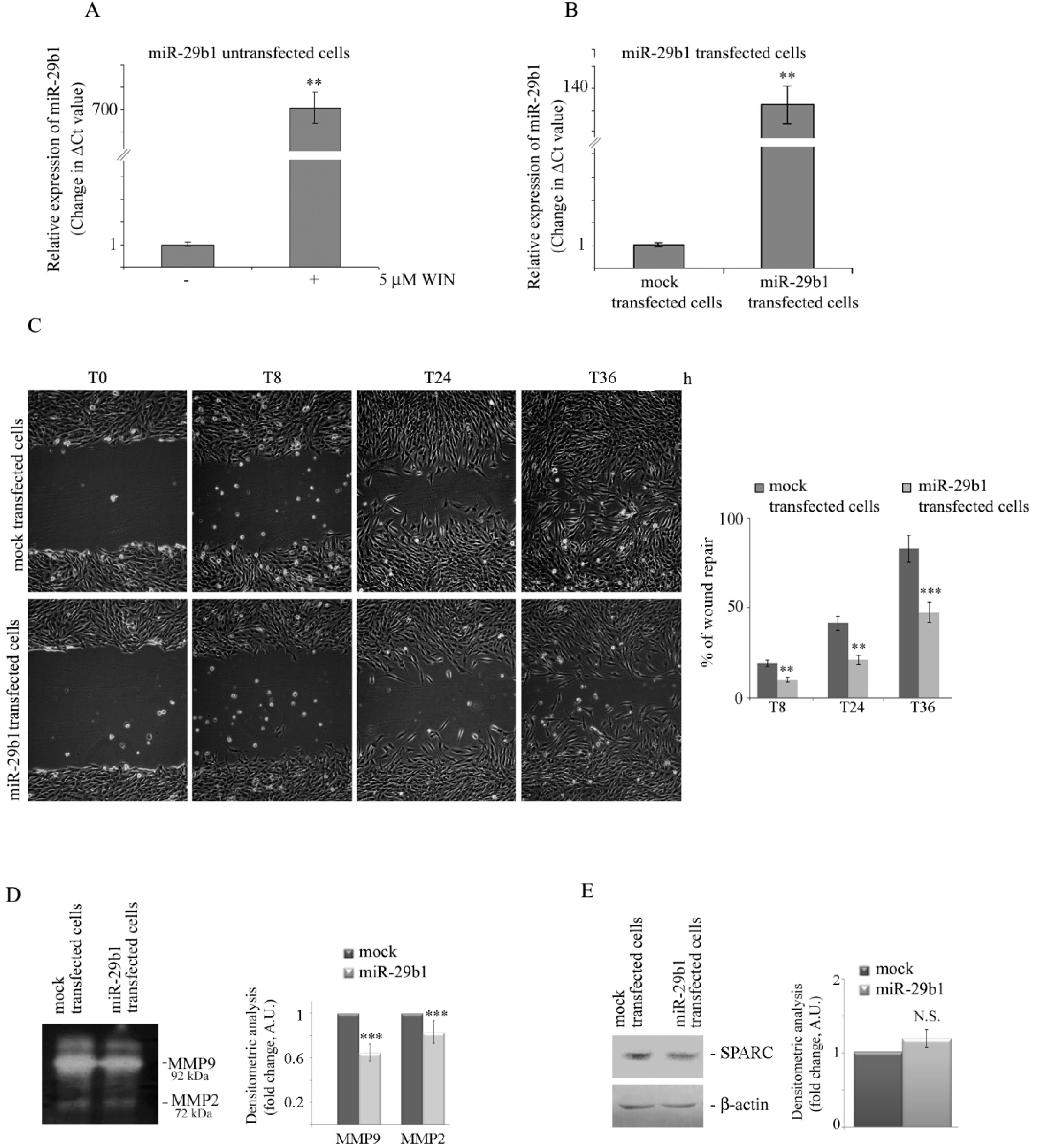
2.4. WIN Induces the Upregulation of miR-29b1 which Plays a Role in Cell Migration
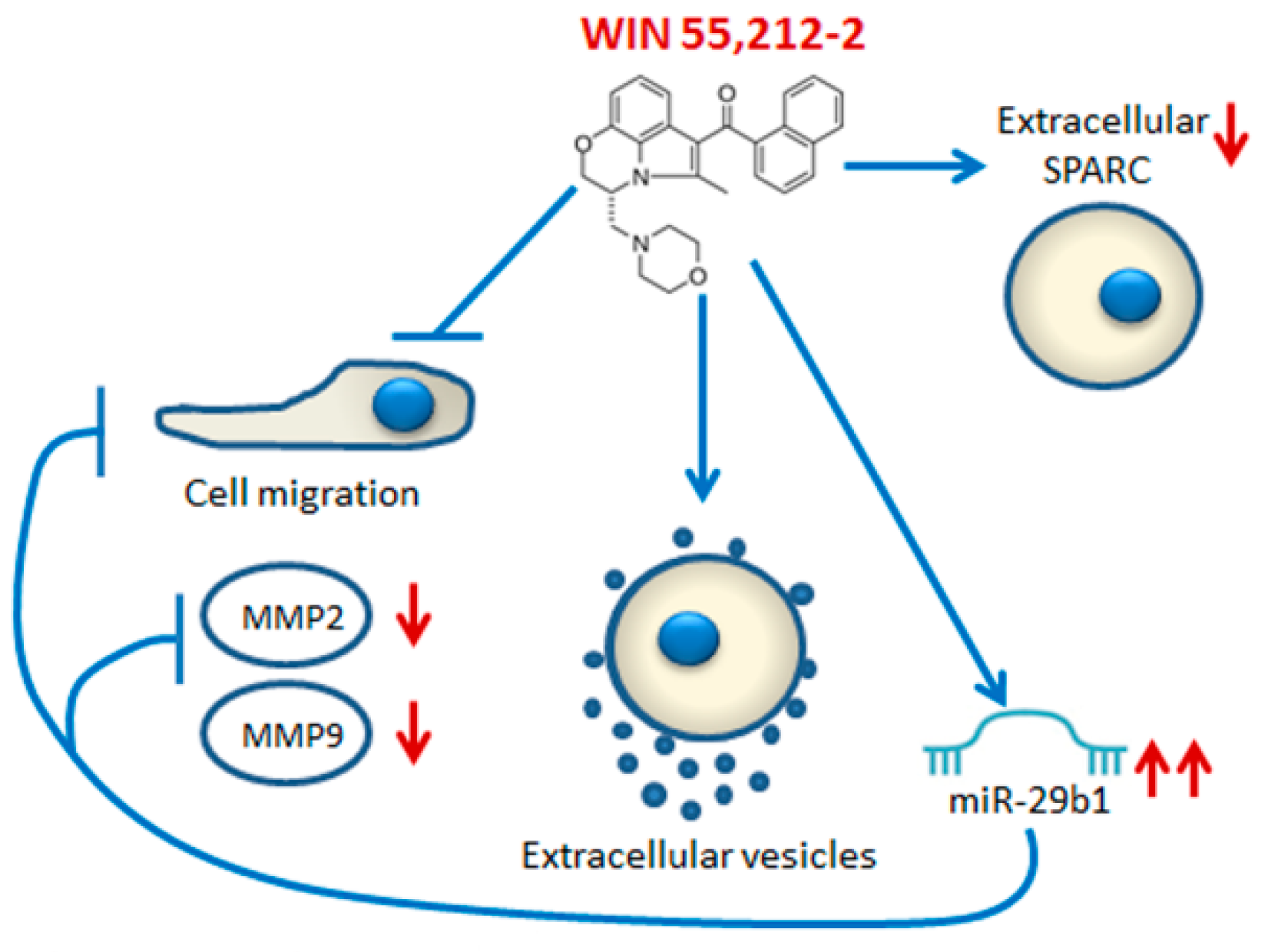
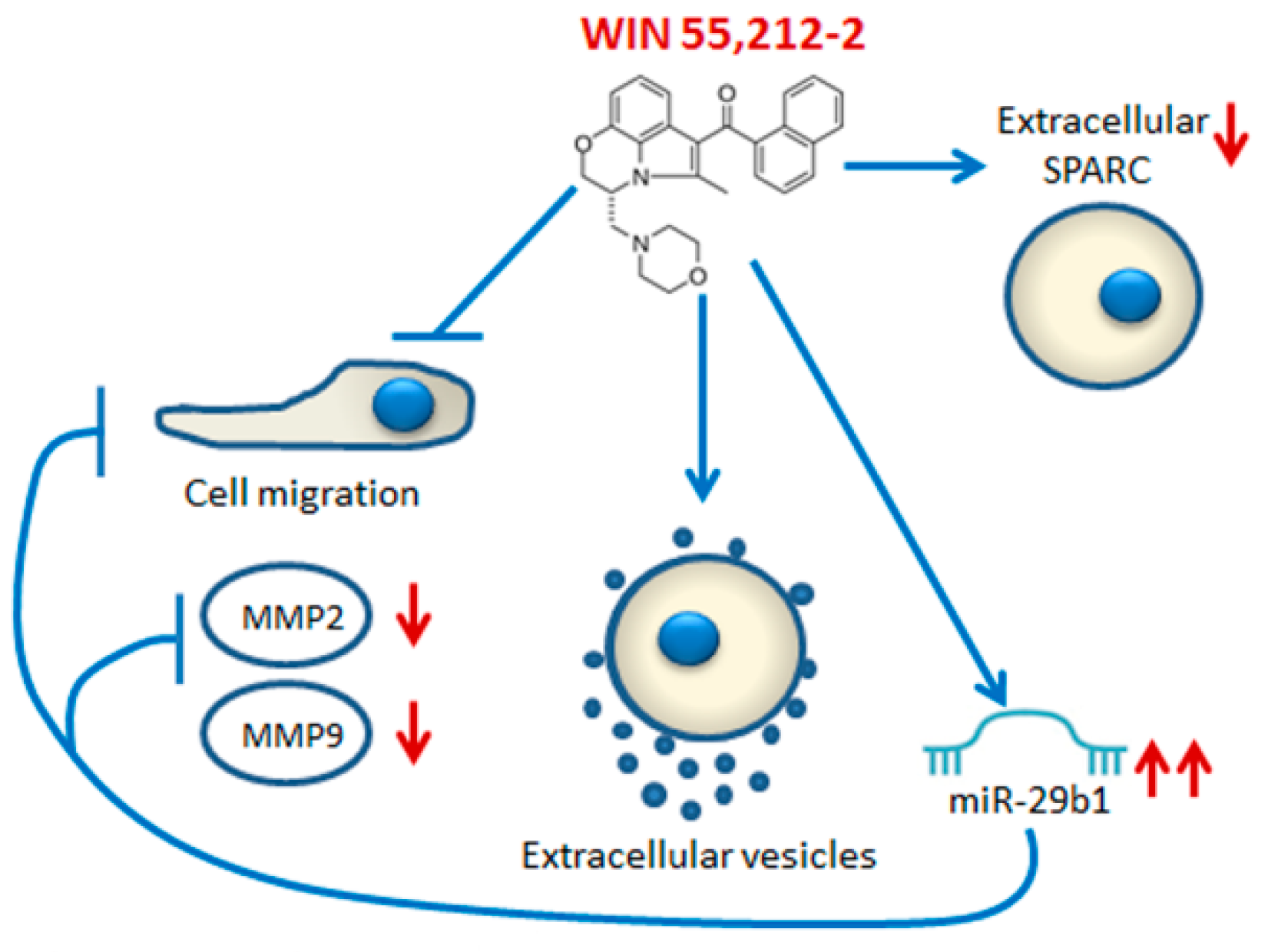
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Cultures
4.3. Wound Healing Assay
4.4. Gelatin Zymography
4.5. Western Blot Analysis
4.6. Isolation and Quantification of Extracellular Vesicles from Culture Media
4.7. Gene Silencing Using siRNA
4.8. Real-Time PCR for miR-29b1 Expression
4.9. Stable Transfection of MG63 Cells with miR-29b1 Plasmid Vector
4.10. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| SPARC | Secreted protein acidic and rich in cysteine |
| MMPs | Matrix metalloproteinases |
| EVs | Extracellular vesicles |
| AchEase | Acetylcholinesterase |
| THG | Thapsigargin |
References
- Lindsey, B.A.; Markel, J.E.; Kleinerman, E.S. Osteosarcoma Overview. Rheumatol. Ther. 2017, 4, 25–43. [Google Scholar] [CrossRef] [PubMed]
- Ottaviani, G.; Jaffe, N. The etiology of osteosarcoma. Cancer Treat Res. 2009, 152, 15–32. [Google Scholar] [PubMed]
- Taran, S.; Taran, R.; Malipatil, N. Pediatric osteosarcoma: An updated review. Indian J. Med Paediatr. Oncol. 2017, 38, 33. [Google Scholar] [CrossRef] [PubMed]
- Aljubran, A.H.; Griffin, A.; Pintilie, M.; Blackstein, M. Osteosarcoma in adolescents and adults: Survival analysis with and without lung metastases. Ann. Oncol. 2009, 20, 1136–1141. [Google Scholar] [CrossRef] [PubMed]
- Calvaruso, G.; Pellerito, O.; Notaro, A.; Giuliano, M. Cannabinoid-associated cell death mechanisms in tumor models (review). Int. J. Oncol. 2012, 41, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Ramer, R. Anti-tumour actions of cannabinoids. Br. J. Pharmacol. 2019, 176, 1384–1394. [Google Scholar] [CrossRef]
- Bifulco, M.; Laezza, C.; Pisanti, S.; Gazzerro, P. Cannabinoids and cancer: Pros and cons of an antitumour strategy. Br. J. Pharmacol. 2006, 148, 123–135. [Google Scholar] [CrossRef]
- Velasco, G.; Sánchez, C.; Guzmán, M. Anticancer mechanisms of cannabinoids. Curr. Oncol. 2016, 23, S23–S32. [Google Scholar] [CrossRef]
- Lu, Y.; Anderson, H.D. Cannabinoid signaling in health and disease. Can. J. Physiol. Pharmacol. 2017, 95, 311–327. [Google Scholar] [CrossRef]
- Velasco, G.; Galve-Roperh, I.; Sánchez, C.; Blázquez, C.; Haro, A.; Guzmán, M. Cannabinoids and ceramide: Two lipids acting hand-by-hand. Life Sci. 2005, 77, 1723–1731. [Google Scholar] [CrossRef]
- Chakravarti, B.; Ravi, J.; Ganju, R.K. Cannabinoids as therapeutic agents in cancer: Current status and future implications. Oncotarget 2014, 5, 5852. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, K.J.; Kim, J.S.; Rho, J.G.; Shin, J.J.; Song, W.K.; Lee, E.K.; Egan, J.M.; Kim, W. Cannabinoids Regulate Bcl-2 and Cyclin D2 Expression in Pancreatic β Cells. PLoS ONE 2016, 11, e0150981. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-W.; Hah, J.H.; Oh, S.-M.; Jeong, W.-J.; Sung, M.-W. 5-lipoxygenase mediates docosahexaenoyl ethanolamide and N-arachidonoyl-L-alanine-induced reactive oxygen species production and inhibition of proliferation of head and neck squamous cell carcinoma cells. BMC Cancer 2016, 16, 458. [Google Scholar] [CrossRef] [PubMed]
- Chiarlone, A.; Börner, C.; Martín-Gómez, L.; Jiménez-González, A.; García-Concejo, A.; García-Bermejo, M.L.; Lorente, M.; Blázquez, C.; García-Taboada, E.; de Haro, A.; et al. MicroRNA let-7d is a target of cannabinoid CB1 receptor and controls cannabinoid signaling. Neuropharmacology 2016, 108, 345–352. [Google Scholar] [CrossRef]
- Sreevalsan, S.; Safe, S. The cannabinoid WIN 55,212-2 decreases specificity protein transcription factors and the oncogenic cap protein eIF4E in colon cancer cells. Mol. Cancer Ther. 2013, 12, 2483–2493. [Google Scholar] [CrossRef]
- Giuliano, M.; Pellerito, O.; Portanova, P.; Calvaruso, G.; Santulli, A.; De Blasio, A.; Vento, R.; Tesoriere, G. Apoptosis induced in HepG2 cells by the synthetic cannabinoid WIN: Involvement of the transcription factor PPARgamma. Biochimie 2009, 91, 457–465. [Google Scholar] [CrossRef]
- Pellerito, O.; Calvaruso, G.; Portanova, P.; De Blasio, A.; Santulli, A.; Vento, R.; Tesoriere, G.; Giuliano, M. The synthetic cannabinoid WIN 55,212-2 sensitizes hepatocellular carcinoma cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis by activating p8/CCAAT/enhancer binding protein homologous protein (CHOP)/death receptor 5 (DR5) axis. Mol. Pharmacol. 2010, 77, 854–863. [Google Scholar]
- Pellerito, O.; Notaro, A.; Sabella, S.; De Blasio, A.; Vento, R.; Calvaruso, G.; Giuliano, M. WIN induces apoptotic cell death in human colon cancer cells through a block of autophagic flux dependent on PPARγ down-regulation. Apoptosis 2014, 19, 1029–1042. [Google Scholar] [CrossRef]
- Notaro, A.; Sabella, S.; Pellerito, O.; Di Fiore, R.; De Blasio, A.; Vento, R.; Calvaruso, G.; Giuliano, M. Involvement of PAR-4 in cannabinoid-dependent sensitization of osteosarcoma cells to TRAIL-induced apoptosis. Int. J. Biol. Sci. 2014, 10, 466–478. [Google Scholar] [CrossRef]
- Notaro, A.; Sabella, S.; Pellerito, O.; Vento, R.; Calvaruso, G.; Giuliano, M. The secreted protein acidic and rich in cysteine is a critical mediator of cell death program induced by WIN/TRAIL combined treatment in osteosarcoma cells. Int. J. Oncol. 2016, 48, 1039–1044. [Google Scholar] [CrossRef]
- Arnold, S.A.; Brekken, R.A. SPARC: A matricellular regulator of tumorigenesis. J. Cell Commun. Signal. 2009, 3, 255–273. [Google Scholar] [CrossRef] [PubMed]
- Freimuth, N.; Ramer, R.; Hinz, B. Antitumorigenic effects of cannabinoids beyond apoptosis. J. Pharmacol. Exp. Ther. 2010, 332, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Brekken, R.A.; Sage, E.H. SPARC, a matricellular protein: At the crossroads of cell-matrix communication. Matrix Biol. 2001, 19, 816–827. [Google Scholar] [CrossRef]
- Nagaraju, G.P.; Dontula, R.; El-Rayes, B.F.; Lakka, S.S. Molecular mechanisms underlying the divergent roles of SPARC in human carcinogenesis. Carcinogenesis 2014, 35, 967–973. [Google Scholar] [CrossRef]
- Yan, B.; Guo, Q.; Fu, F.-J.; Wang, Z.; Yin, Z.; Wei, Y.-B.; Yang, J.-R. The role of miR-29b in cancer: Regulation, function, and signaling. Onco. Targets 2015, 8, 539–548. [Google Scholar]
- Jones, K.B.; Salah, Z.; Del Mare, S.; Galasso, M.; Gaudio, E.; Nuovo, G.J.; Lovat, F.; LeBlanc, K.; Palatini, J.; Randall, R.L.; et al. miRNA signatures associate with pathogenesis and progression of osteosarcoma. Cancer Res. 2012, 72, 1865–1877. [Google Scholar] [CrossRef]
- Schmitt, M.J.; Margue, C.; Behrmann, I.; Kreis, S. MiRNA-29: A microRNA family with tumor-suppressing and immune-modulating properties. Curr. Mol. Med. 2013, 13, 572–585. [Google Scholar] [CrossRef]
- Kapinas, K.; Kessler, C.B.; Delany, A.M. miR-29 suppression of osteonectin in osteoblasts: Regulation during differentiation and by canonical Wnt signaling. J. Cell. Biochem. 2009, 108, 216–224. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, A.D.; Sage, E.H. SPARC, a matricellular protein that functions in cellular differentiation and tissue response to injury. J. Clin. Investig. 2001, 107, 1049–1054. [Google Scholar] [CrossRef] [Green Version]
- Qiu, F.; Sun, R.; Deng, N.; Guo, T.; Cao, Y.; Yu, Y.; Wang, X.; Zou, B.; Zhang, S.; Jing, T.; et al. miR-29a/b enhances cell migration and invasion in nasopharyngeal carcinoma progression by regulating SPARC and COL3A1 gene expression. PLoS ONE 2015, 10, e0120969. [Google Scholar] [CrossRef]
- Perissinotto, E.; Cavalloni, G.; Leone, F.; Fonsato, V.; Mitola, S.; Grignani, G.; Surrenti, N.; Sangiolo, D.; Bussolino, F.; Piacibello, W.; et al. Involvement of chemokine receptor 4/stromal cell-derived factor 1 system during osteosarcoma tumor progression. Clin. Cancer Res. 2005, 11, 490–497. [Google Scholar] [PubMed]
- Chu, Y.; Fang, Y.; Chi, J.; Li, J.; Zhang, D.; Zou, Y.; Wang, Z. Astragalus polysaccharides decrease proliferation, migration, and invasion but increase apoptosis of human osteosarcoma cells by up-regulation of microRNA-133a. Braz. J. Med Biol. Res. 2018, 51, 12. [Google Scholar] [CrossRef] [PubMed]
- Xian, X.; Huang, L.; Zhang, B.; Wu, C.; Cui, J.; Wang, Z. WIN 55,212-2 Inhibits the Epithelial Mesenchymal Transition of Gastric Cancer Cells via COX-2 Signals. Cell Physiol. Biochem. 2016, 39, 2149–2157. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Wang, J.; Zhou, Z.; He, Z.; Zhao, Q. Cannabinoid WIN55, 212–2 induces cell cycle arrest and inhibits the proliferation and migration of human BEL7402 hepatocellular carcinoma cells. Mol. Med. Rep. 2015, 12, 7963–7970. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Li, Z.; Zhu, X.; Xu, R.; Xu, Y. miR-29 Family Inhibits Resistance to Methotrexate and Promotes Cell Apoptosis by Targeting COL3A1 and MCL1 in Osteosarcoma. Med. Sci. Monit. 2018, 24, 8812–8821. [Google Scholar] [CrossRef]
- Zhu, K.; Liu, L.; Zhang, J.; Wang, Y.; Liang, H.; Fan, G.; Jiang, Z.; Zhang, C.-Y.; Chen, X.; Zhou, G. MiR-29b suppresses the proliferation and migration of osteosarcoma cells by targeting CDK6. Protein Cell 2016, 7, 434–444. [Google Scholar] [CrossRef] [Green Version]
- Kisková, T.; Mungenast, F.; Suváková, M.; Jäger, W.; Thalhammer, T. Future Aspects for Cannabinoids in Breast Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 1673. [Google Scholar] [CrossRef]
- Sherman, A.B.; Gilger, B.C.; Berglund, A.K.; Schnabel, L.V. Effect of bone marrow-derived mesenchymal stem cells and stem cell supernatant on equine corneal wound healing in vitro. Stem Cell Res. 2017, 8, 120. [Google Scholar] [CrossRef]
- Lauricella, M.; Carlisi, D.; Giuliano, M.; Calvaruso, G.; Cernigliaro, C.; Vento, R.; D’Anneo, A. The analysis of estrogen receptor-α positive breast cancer stem-like cells unveils a high expression of the serpin proteinase inhibitor PI-9: Possible regulatory mechanisms. Int. J. Oncol. 2016, 49, 352–360. [Google Scholar] [CrossRef]
- Cernigliaro, C.; D’Anneo, A.; Carlisi, D.; Giuliano, M.; Marino Gammazza, A.; Barone, R.; Longhitano, L.; Cappello, F.; Emanuele, S.; Distefano, A.; et al. Ethanol-Mediated Stress Promotes Autophagic Survival and Aggressiveness of Colon Cancer Cells via Activation of Nrf2/HO-1 Pathway. Cancers 2019, 11, 505. [Google Scholar] [CrossRef]
- Barreca, M.M.; Spinello, W.; Cavalieri, V.; Turturici, G.; Sconzo, G.; Kaur, P.; Tinnirello, R.; Asea, A.A.A.; Geraci, F. Extracellular Hsp70 Enhances Mesoangioblast Migration via an Autocrine Signaling Pathway. J. Cell. Physiol. 2017, 232, 1845–1861. [Google Scholar] [CrossRef] [PubMed]
- Geraci, F.; Ragonese, P.; Barreca, M.M.; Aliotta, E.; Mazzola, M.A.; Realmuto, S.; Vazzoler, G.; Savettieri, G.; Sconzo, G.; Salemi, G. Differences in Intercellular Communication During Clinical Relapse and Gadolinium-Enhanced MRI in Patients With Relapsing Remitting Multiple Sclerosis: A Study of the Composition of Extracellular Vesicles in Cerebrospinal Fluid. Front. Cell. Neurosci. 2018, 12, 418. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Feather-Stone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Di Fiore, R.; Drago-Ferrante, R.; Pentimalli, F.; Di Marzo, D.; Forte, I.M.; D’Anneo, A.; Carlisi, D.; De Blasio, A.; Giuliano, M.; Tesoriere, G.; et al. MicroRNA-29b-1 impairs in vitro cell proliferation, self-renewal and chemoresistance of human osteosarcoma 3AB-OS cancer stem cells. Int. J. Oncol. 2014, 45, 2013–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Notaro, A.; Emanuele, S.; Geraci, F.; D’Anneo, A.; Lauricella, M.; Calvaruso, G.; Giuliano, M. WIN55,212-2-Induced Expression of Mir-29b1 Favours the Suppression of Osteosarcoma Cell Migration in a SPARC-Independent Manner. Int. J. Mol. Sci. 2019, 20, 5235. https://doi.org/10.3390/ijms20205235
Notaro A, Emanuele S, Geraci F, D’Anneo A, Lauricella M, Calvaruso G, Giuliano M. WIN55,212-2-Induced Expression of Mir-29b1 Favours the Suppression of Osteosarcoma Cell Migration in a SPARC-Independent Manner. International Journal of Molecular Sciences. 2019; 20(20):5235. https://doi.org/10.3390/ijms20205235
Chicago/Turabian StyleNotaro, Antonietta, Sonia Emanuele, Fabiana Geraci, Antonella D’Anneo, Marianna Lauricella, Giuseppe Calvaruso, and Michela Giuliano. 2019. "WIN55,212-2-Induced Expression of Mir-29b1 Favours the Suppression of Osteosarcoma Cell Migration in a SPARC-Independent Manner" International Journal of Molecular Sciences 20, no. 20: 5235. https://doi.org/10.3390/ijms20205235
APA StyleNotaro, A., Emanuele, S., Geraci, F., D’Anneo, A., Lauricella, M., Calvaruso, G., & Giuliano, M. (2019). WIN55,212-2-Induced Expression of Mir-29b1 Favours the Suppression of Osteosarcoma Cell Migration in a SPARC-Independent Manner. International Journal of Molecular Sciences, 20(20), 5235. https://doi.org/10.3390/ijms20205235