MicroRNA Mediate Visfatin and Resistin Induction of Oxidative Stress in Human Osteoarthritic Synovial Fibroblasts Via NF-κB Pathway


 , ,
, , 
Abstract
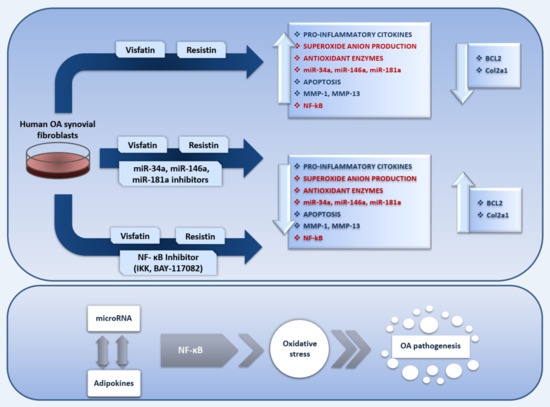
:
1. Introduction
2. Results
2.1. Cell viability Evaluation in Visfatin and Resistin Treated Cells
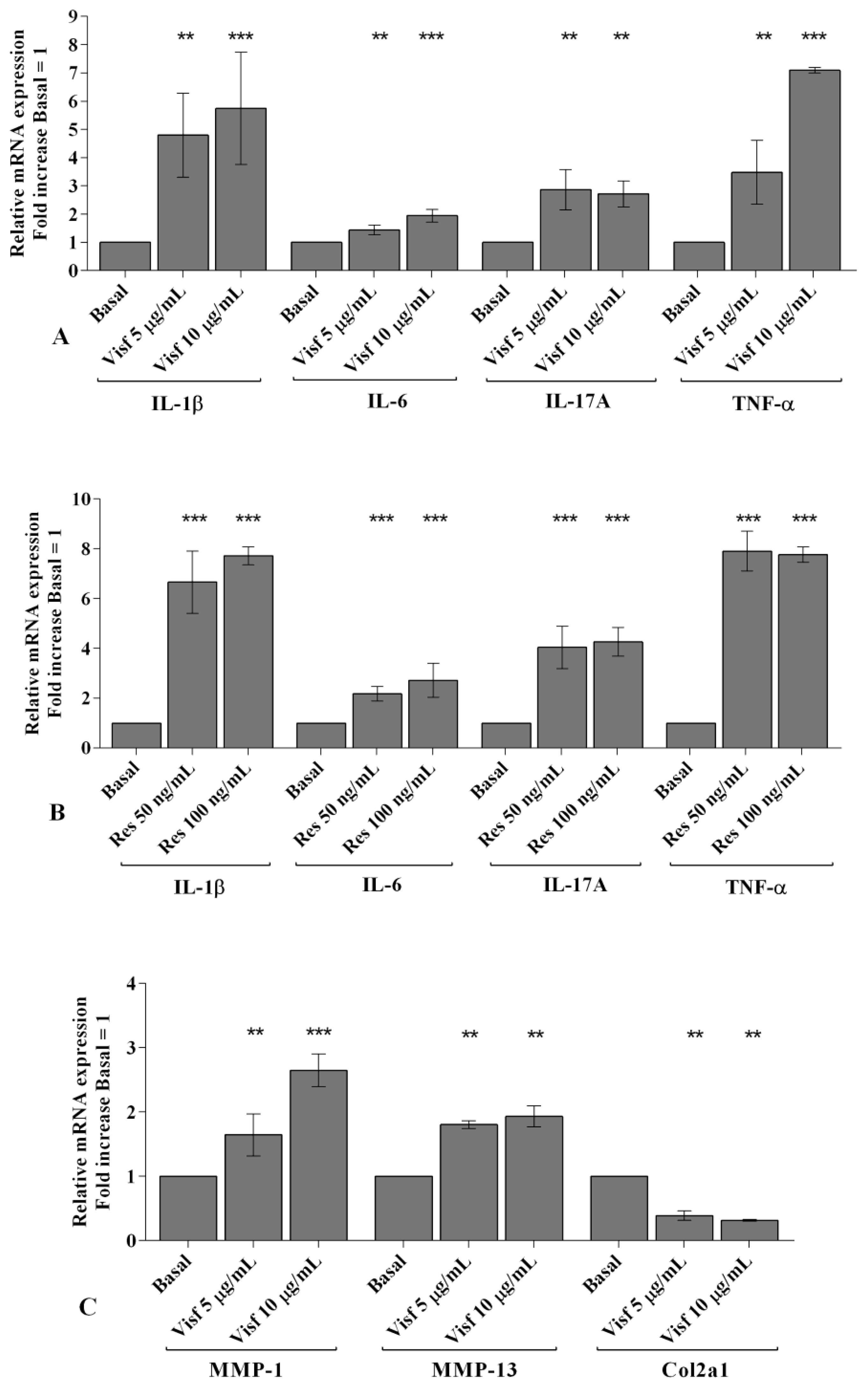
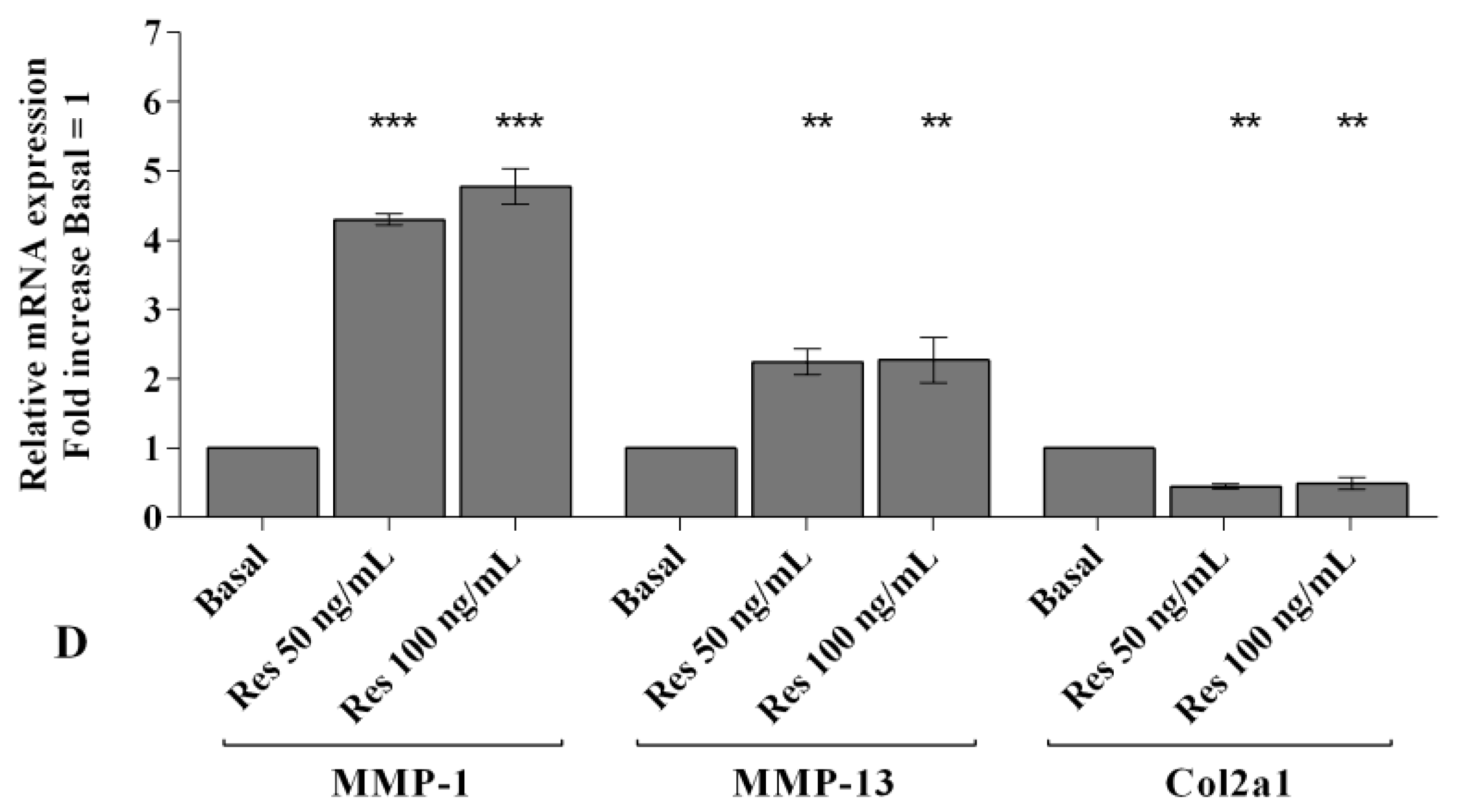
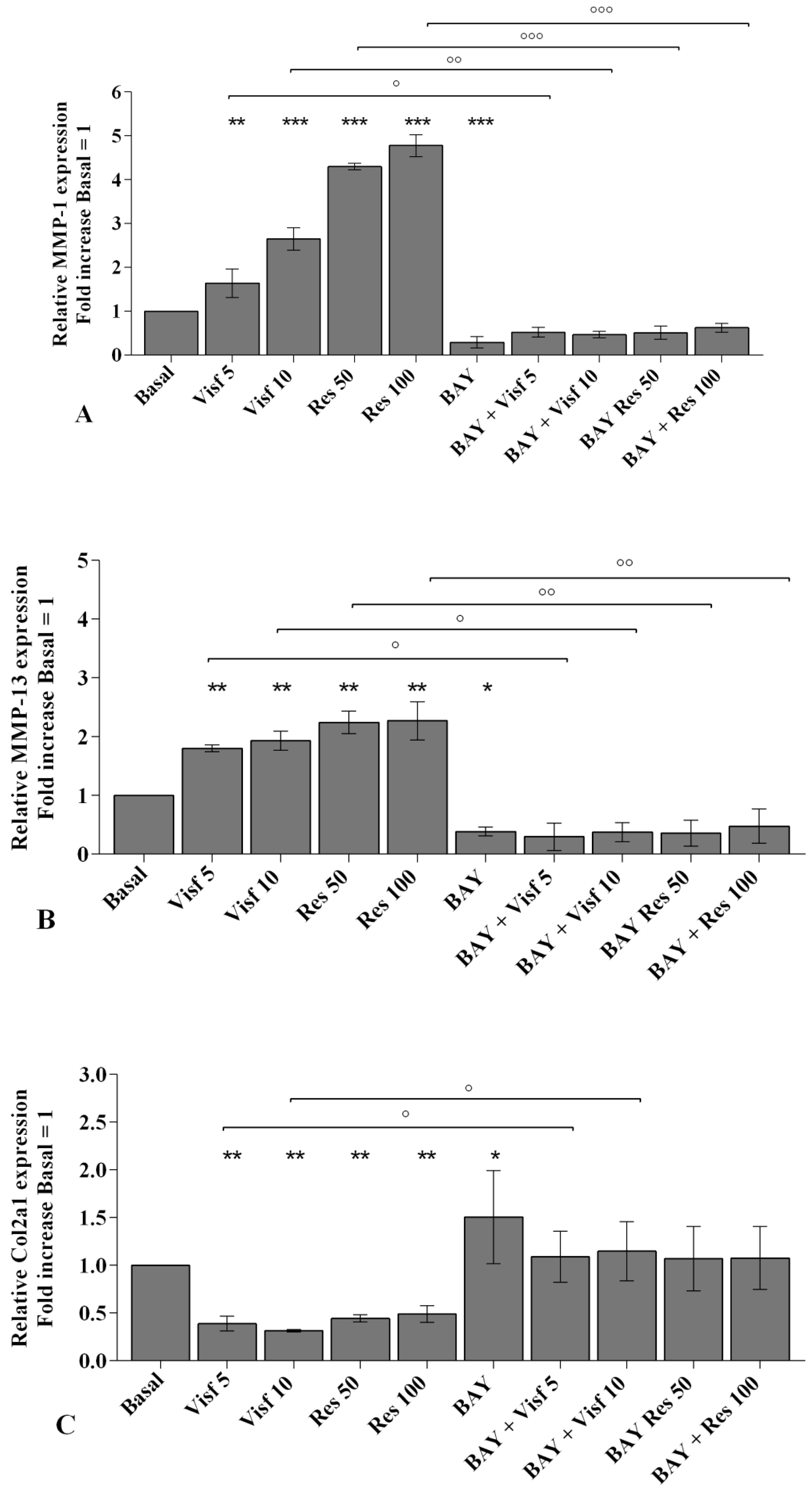
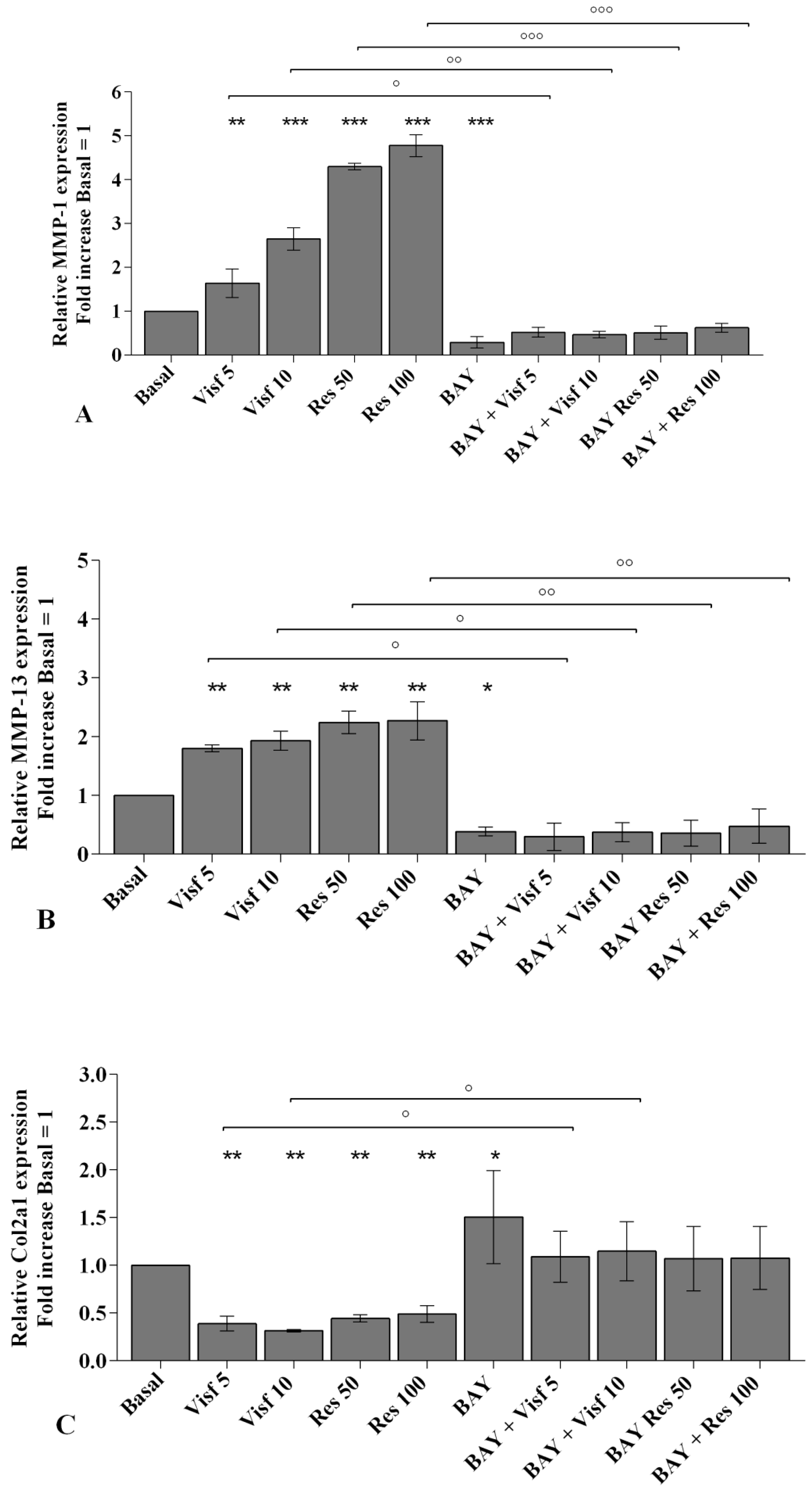
2.2. Visfatin and Resistin Promote Inflammation and Regulate Cartilage Turnover
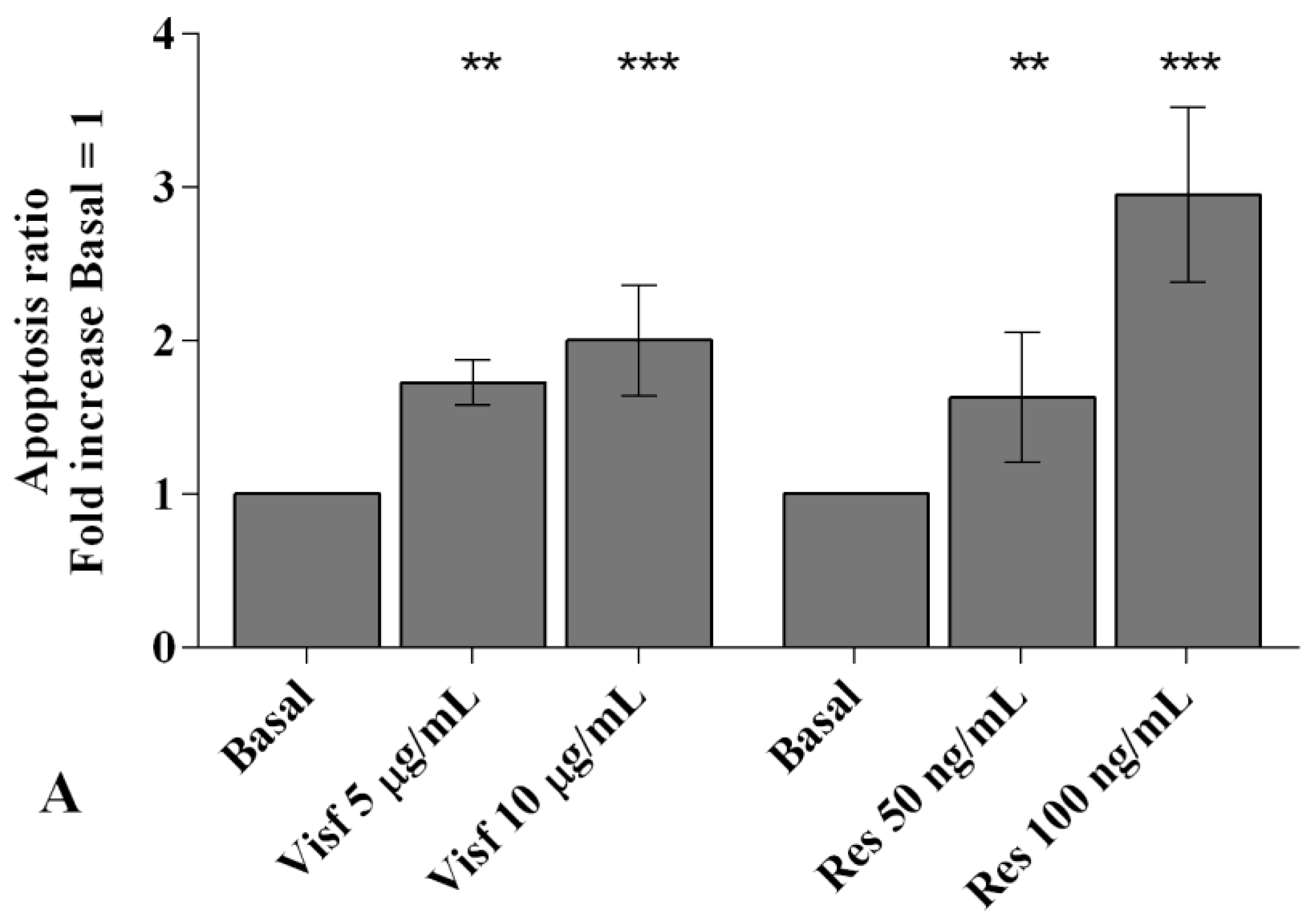
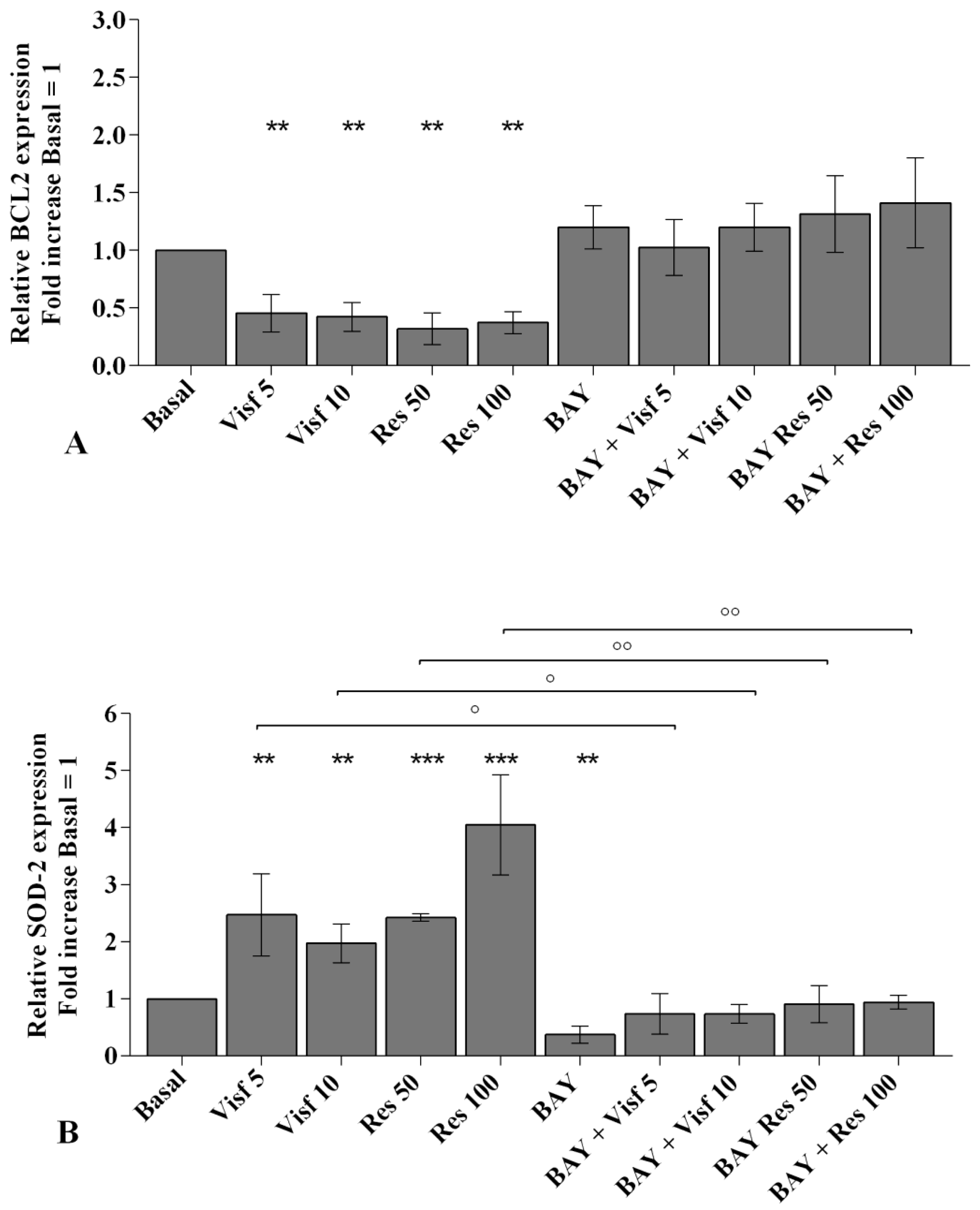
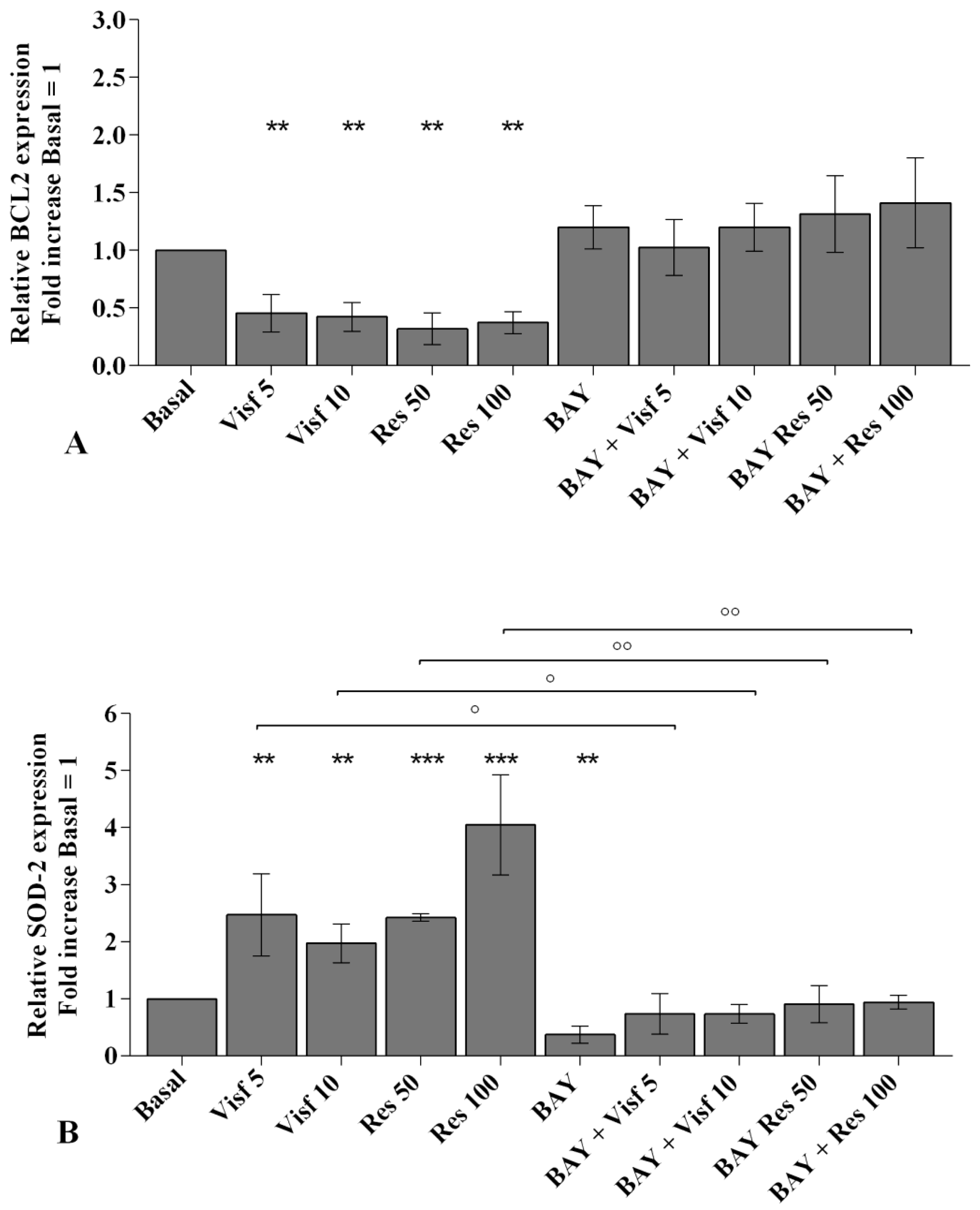
2.3. Adipokines Induce Apoptosis and Regulate BCL2 Expression
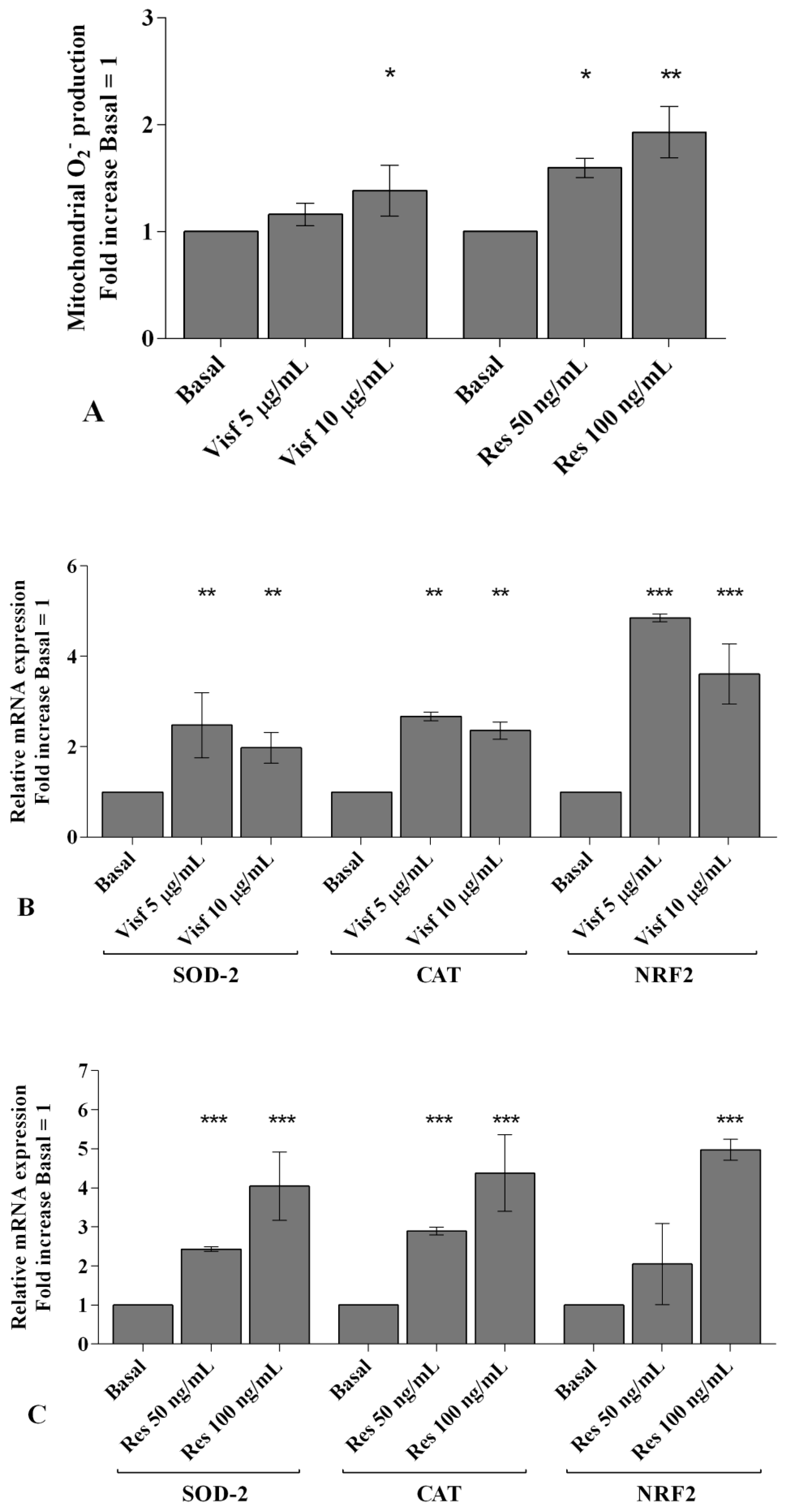
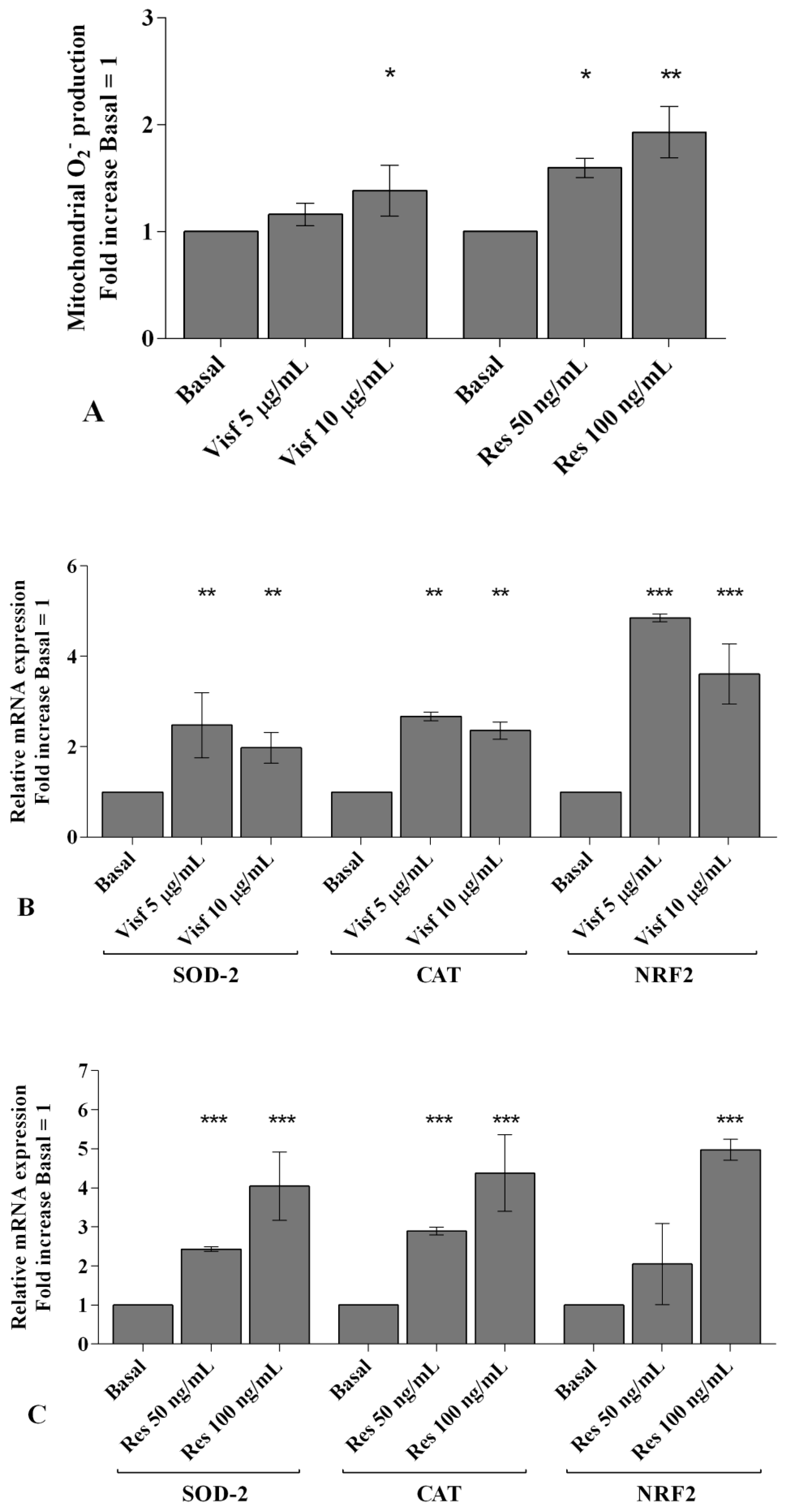
2.4. Visfatin and Resistin Regulate Oxidant/Antioxidant Balance
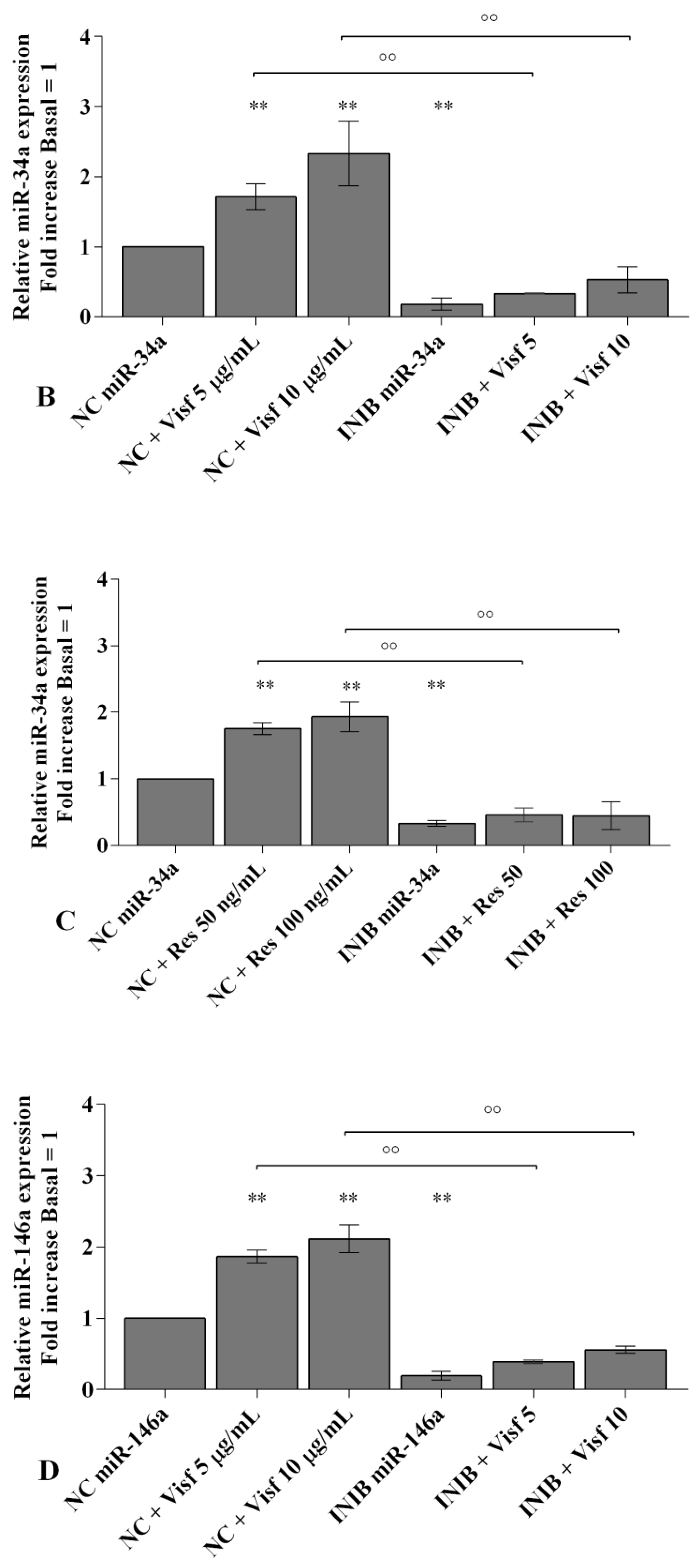
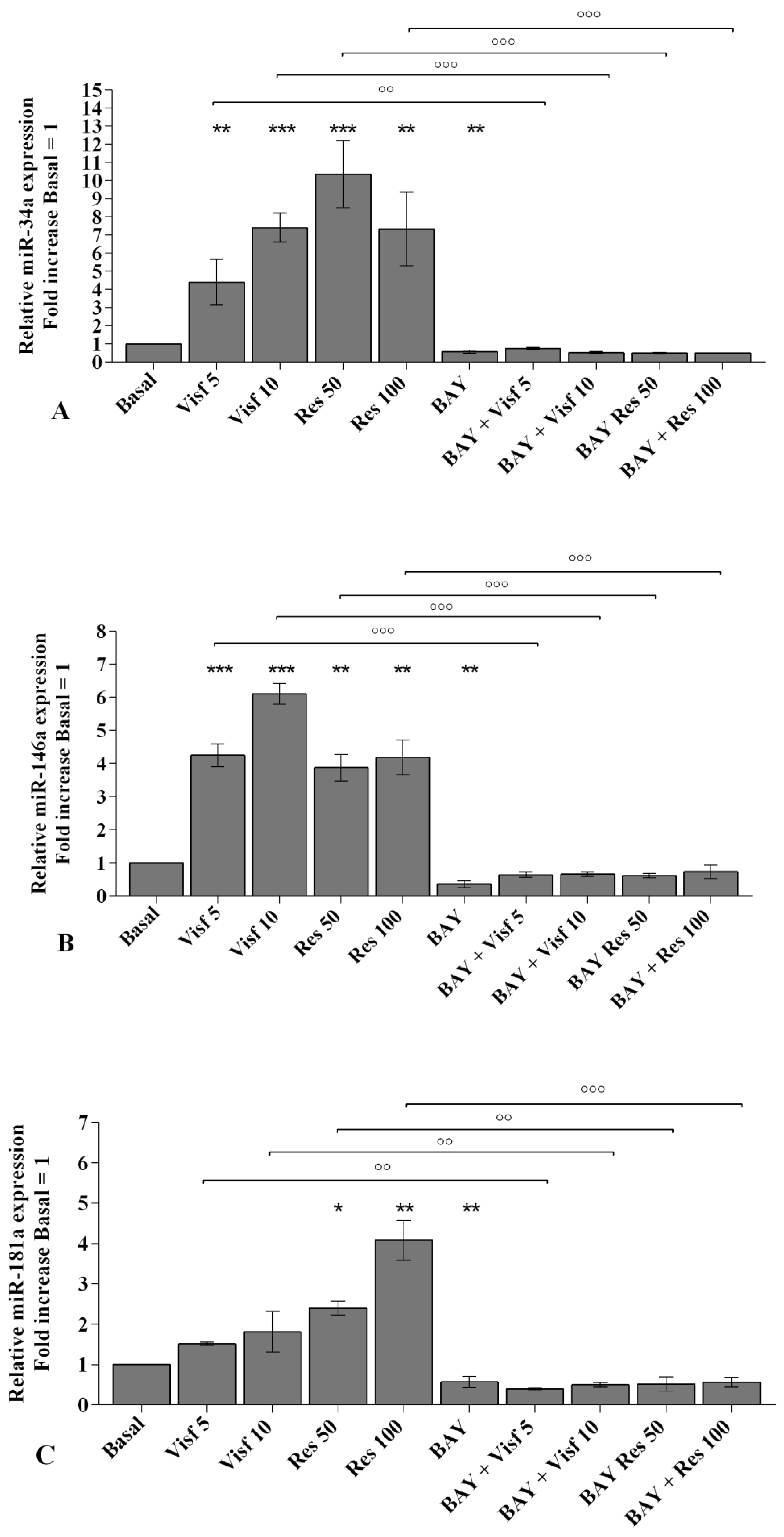
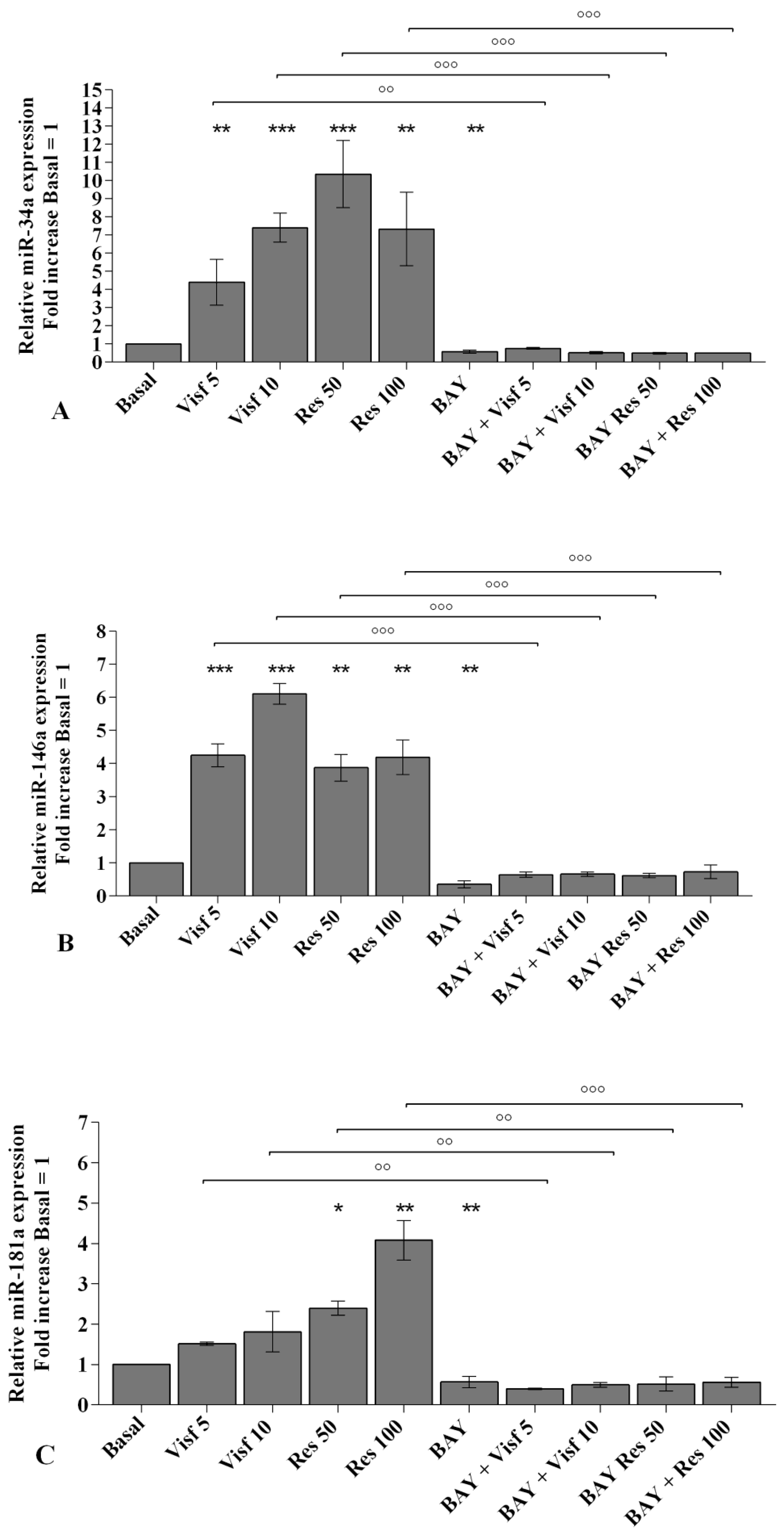
2.5. Visfatin and Resistin Modulate miRNA Gene Expression
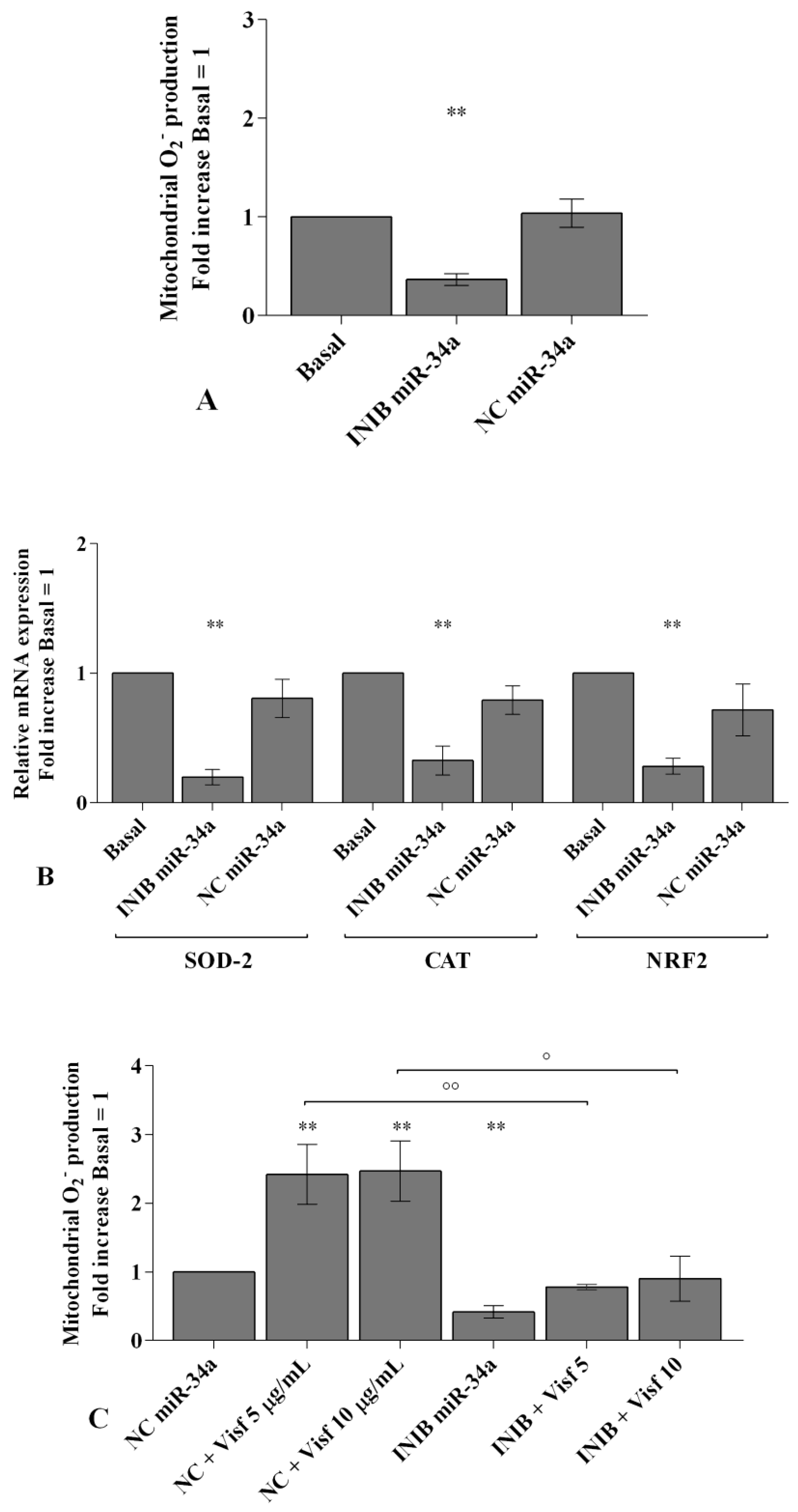
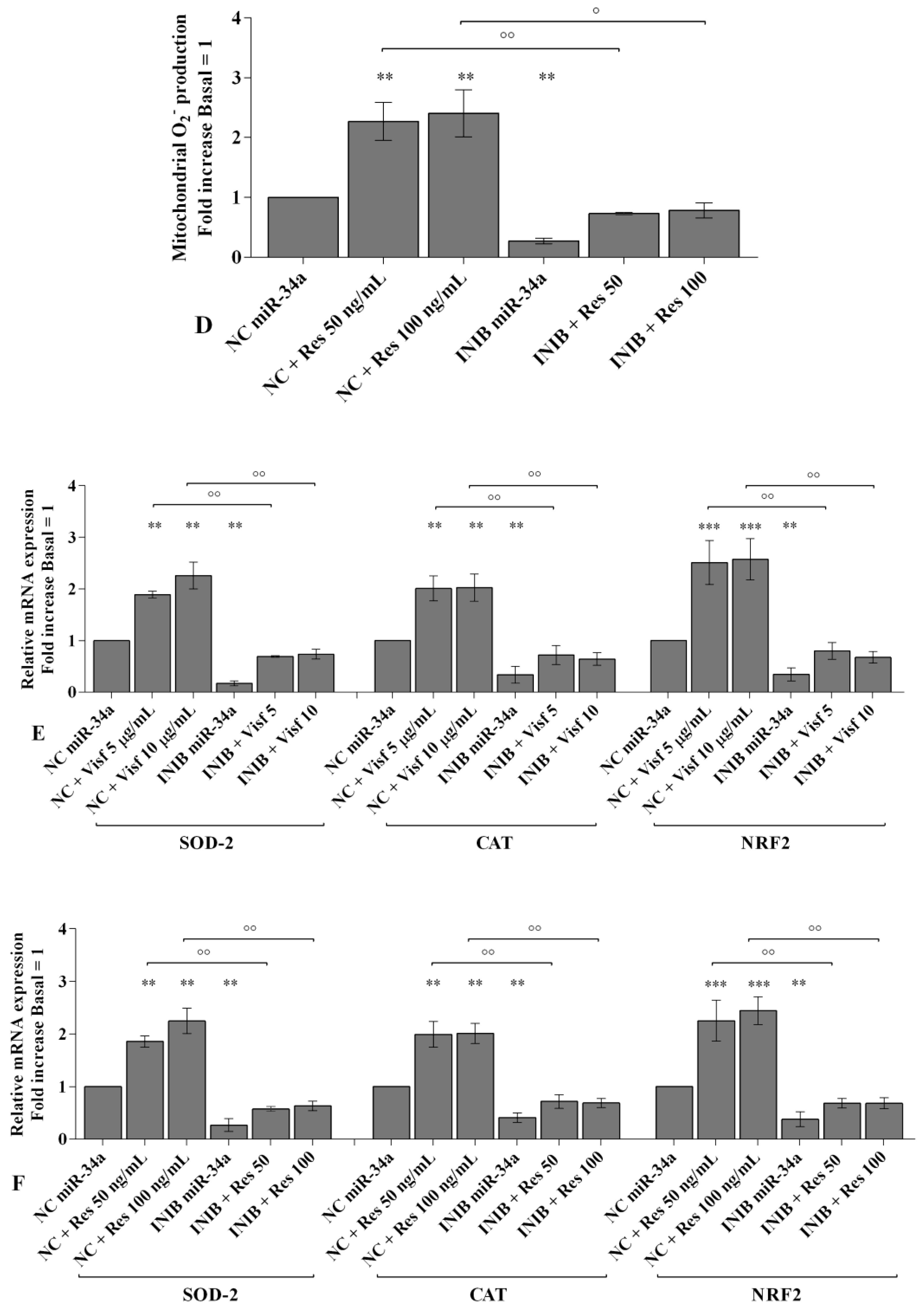
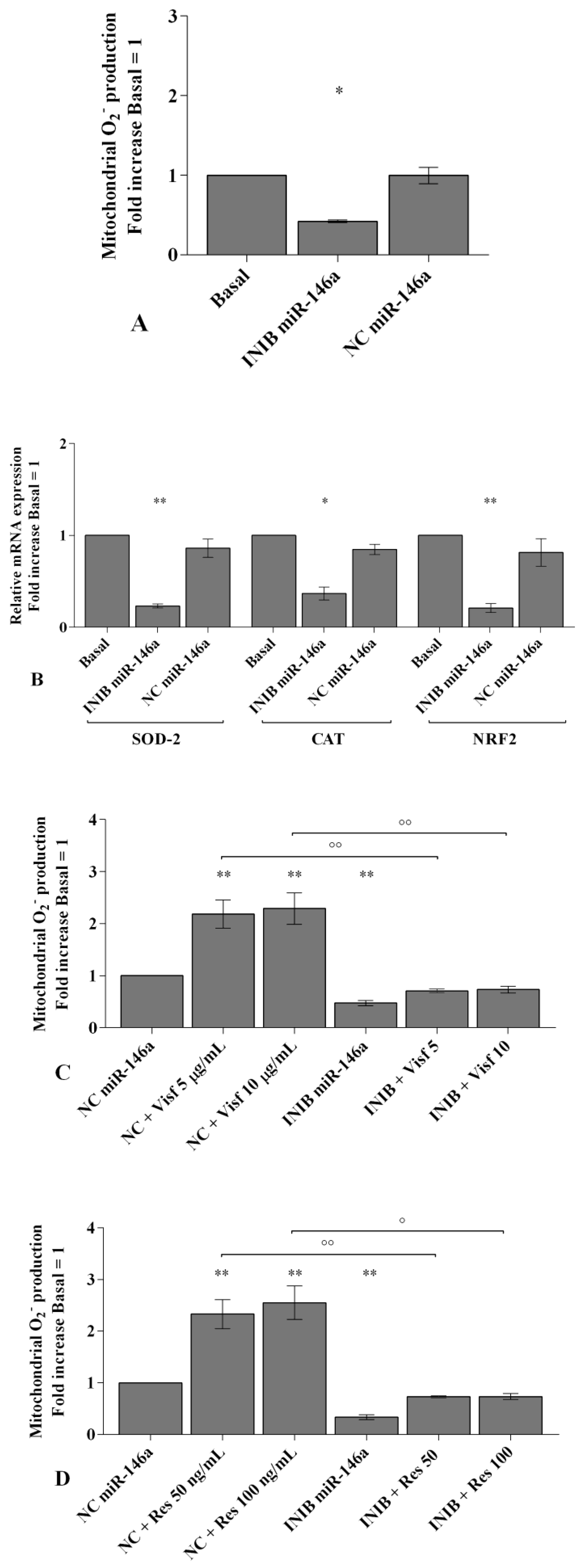
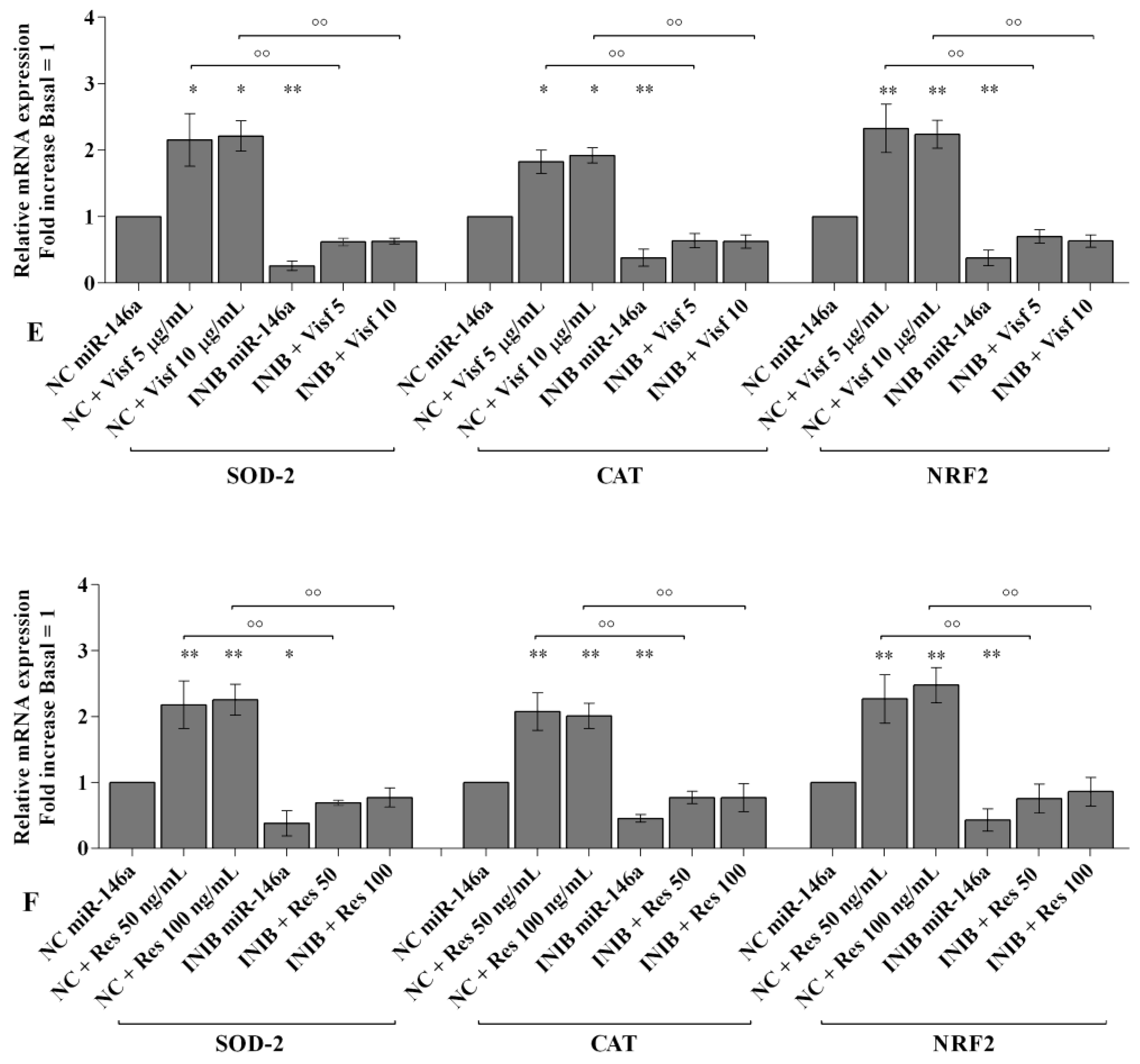
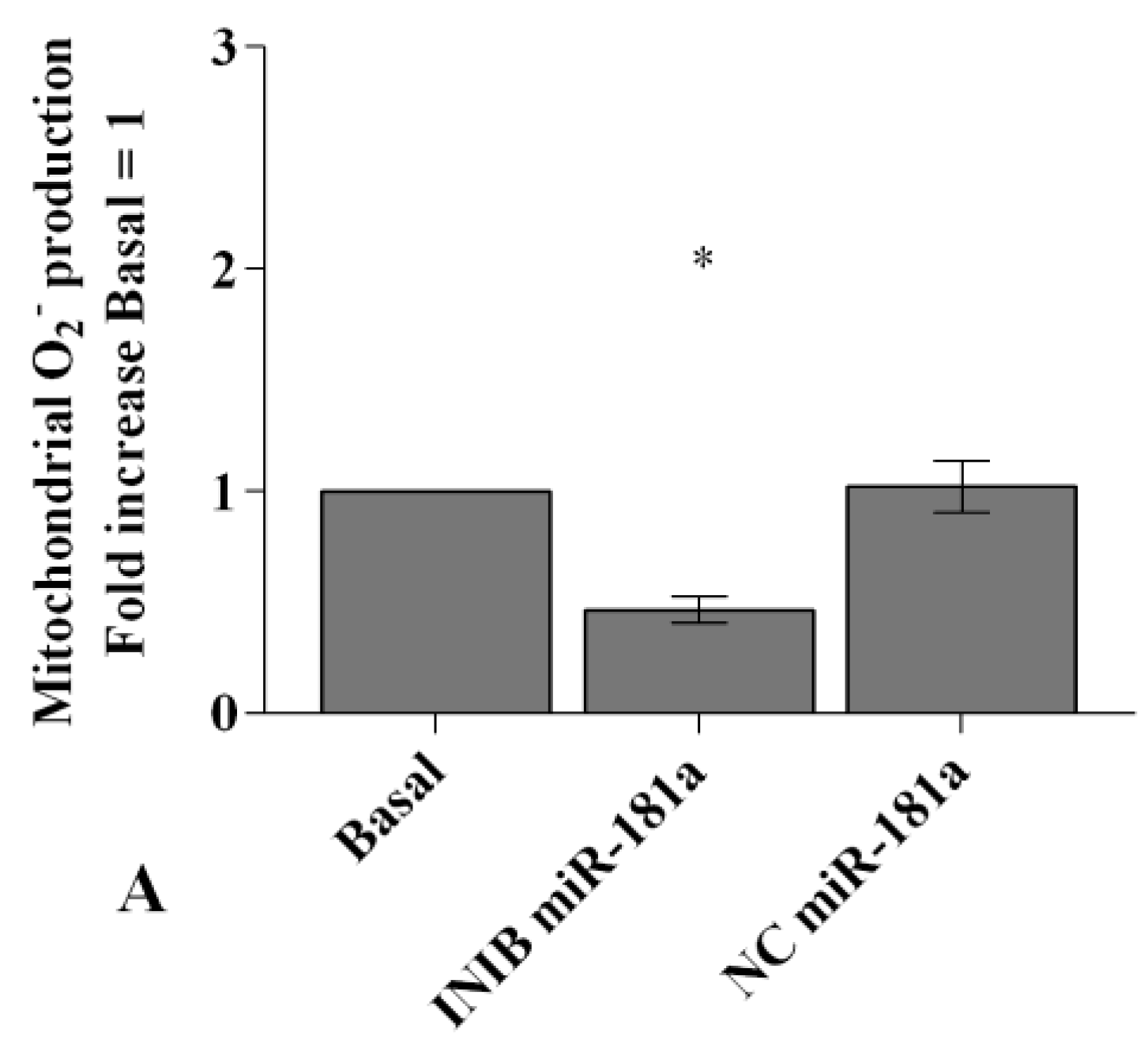
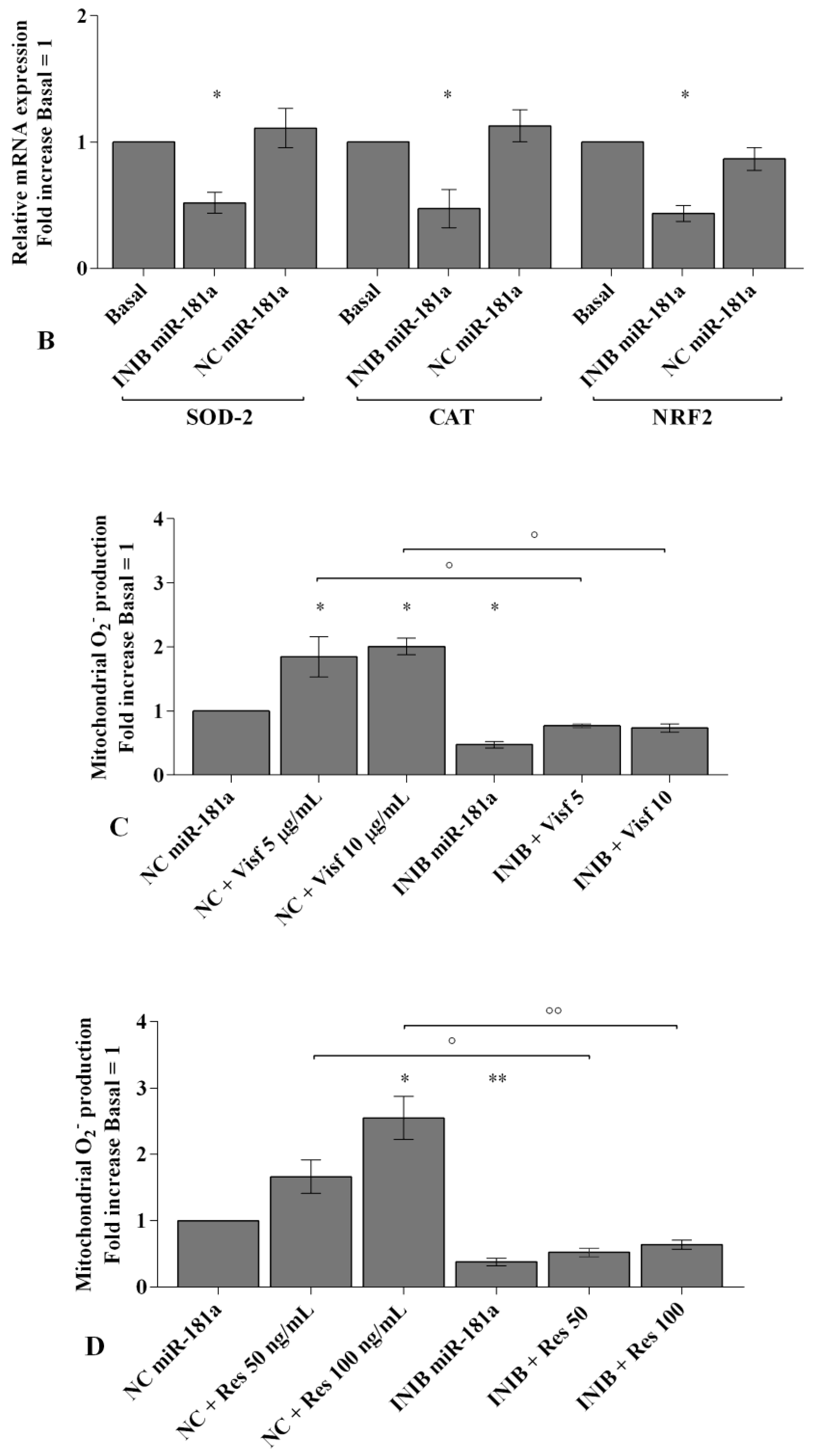
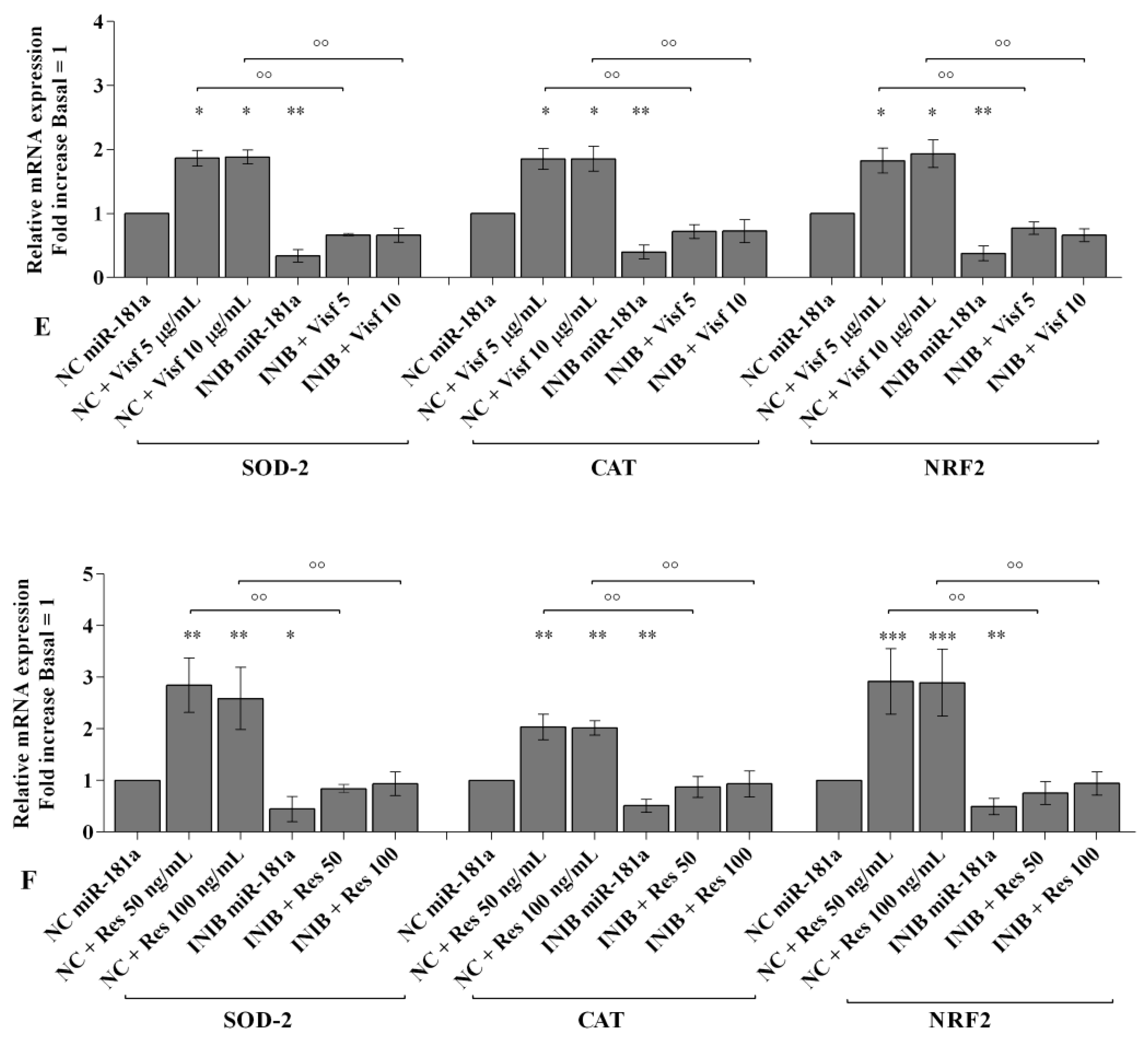
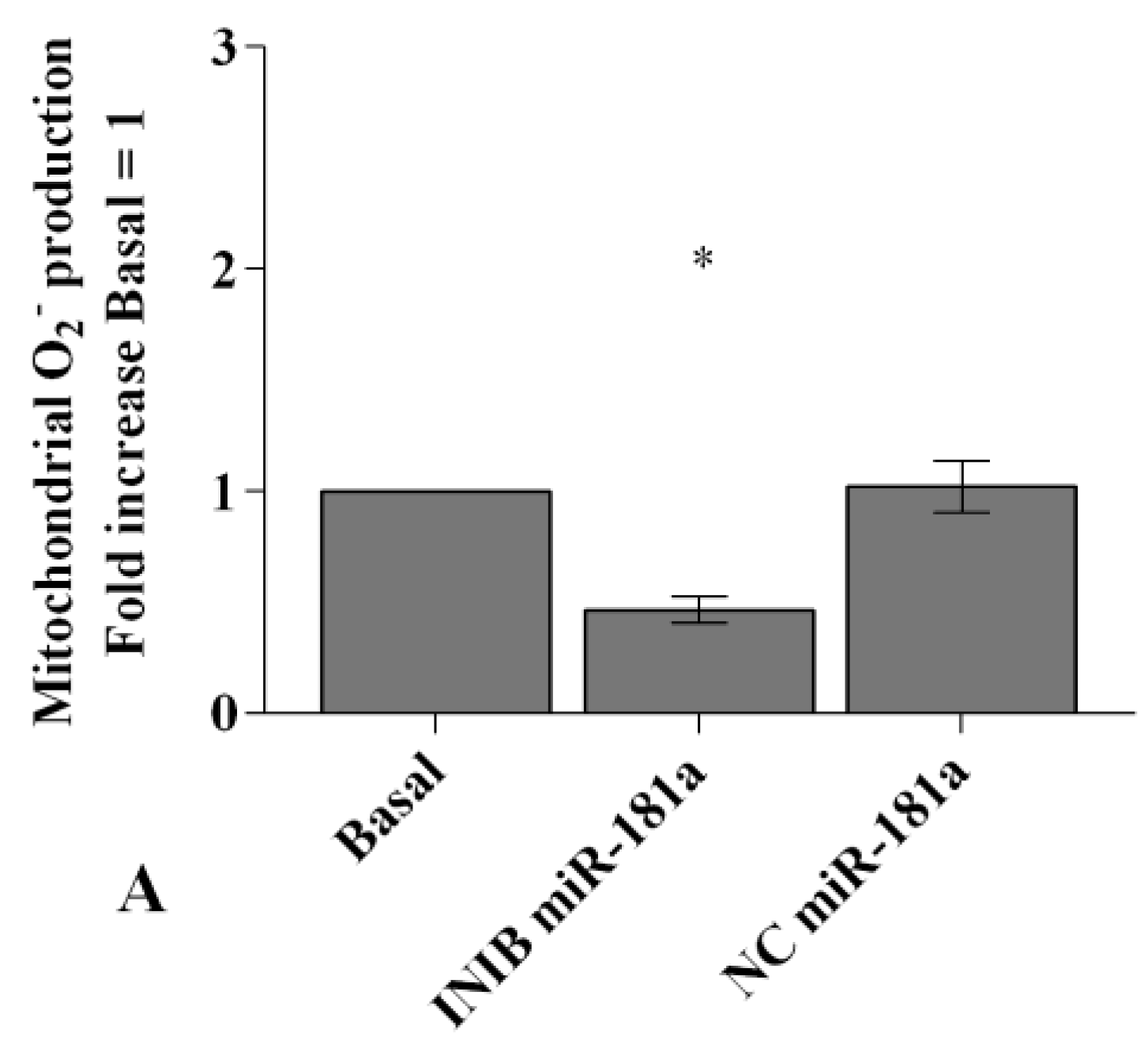
2.6. MiRNA Regulate Oxidative Stress Induced by Visfatin and Resistin
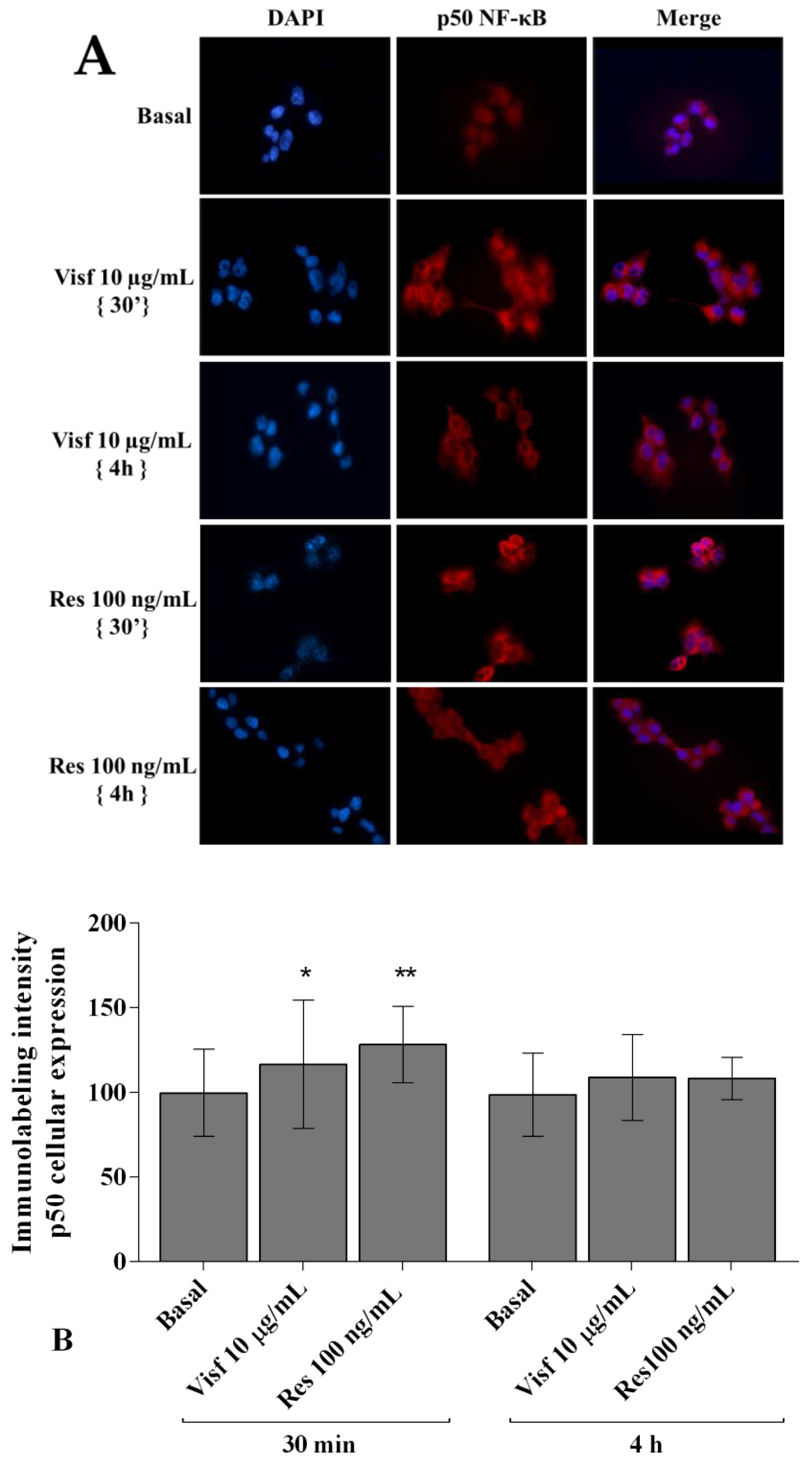
2.7. Visfatin and Resistin Activate NF-κB Signaling Pathway
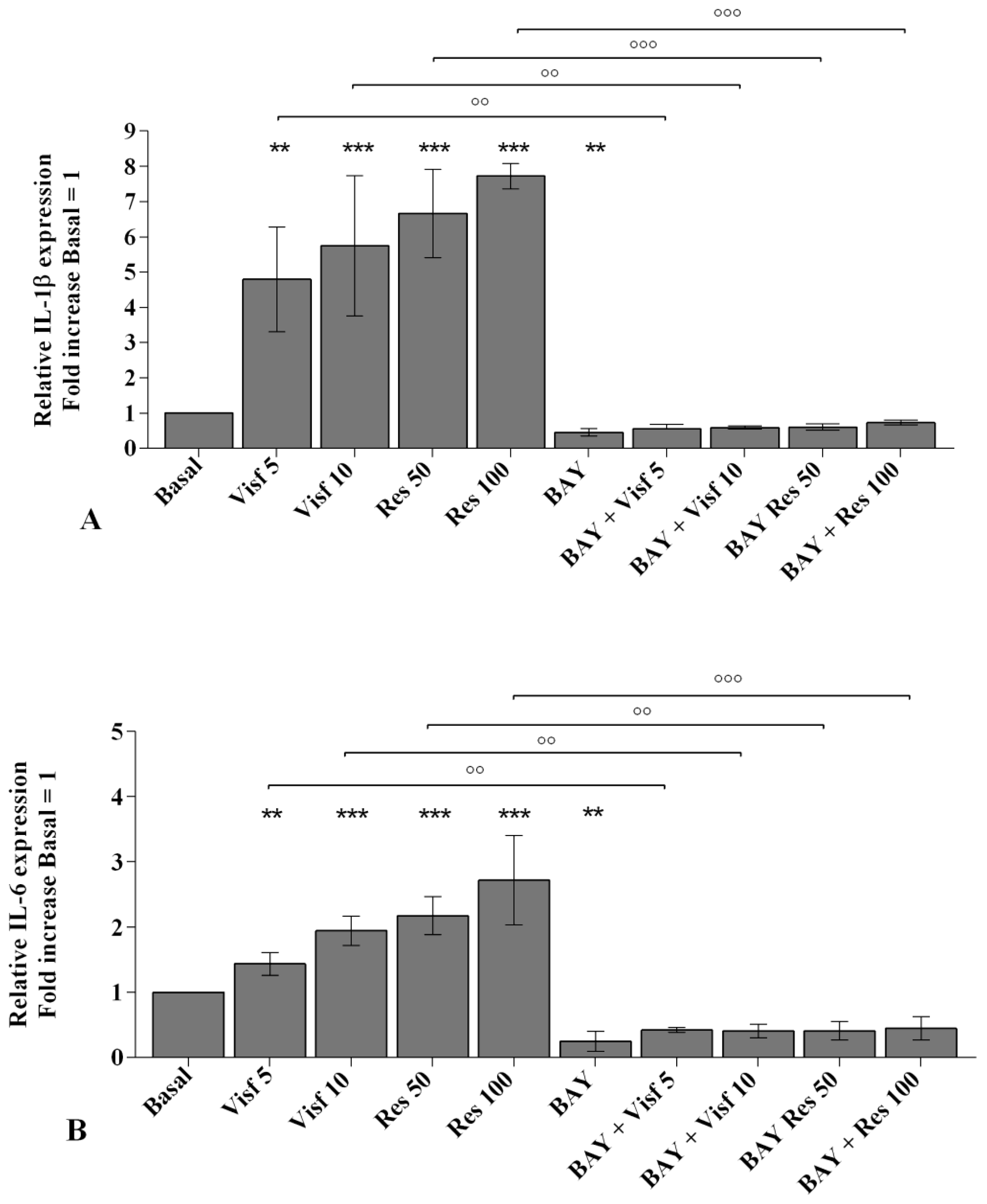
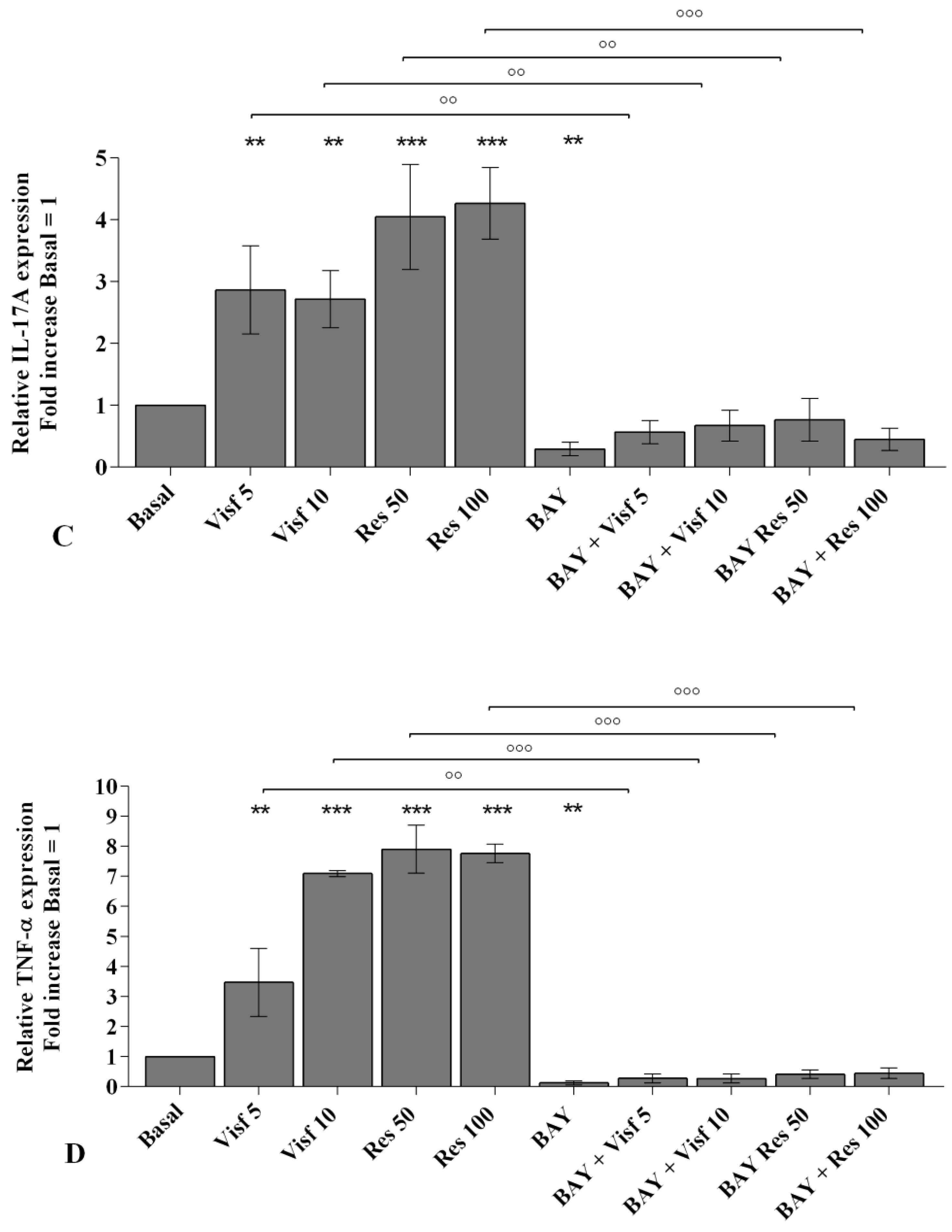
2.8. NF-κB Signaling Pathway Inhibits Visfatin and Resistin Effects
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Cell Culture
4.2. Stimulus of Synovial Cell Cultures
4.3. MTT Assay
4.4. Transfection of Synovial Cells
4.5. Quantitative Real-Time PCR of mRNA and miRNA
4.6. Apoptosis Detection
4.7. Mitochondrial Superoxide Anion (•O2-) Production
4.8. Immunofluorescence Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vos, T.; Allen, C.; Arora, M.; Barber, R.M.; Bhutta, Z.A.; Brown, A.; Cater, A.; Casey, C.C.; Charlson, J.F.; Chen, Z.A.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1545–1602. [Google Scholar] [CrossRef]
- Poulet, B.; Staines, K.A. New developments in osteoarthritis and cartilage biology. Curr. Opin. Pharmacol. 2016, 28, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellam, J.; Berenbaum, F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat. Rev. Rheumatol. 2010, 6, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Scanzello, C.R.; Goldring, S.R. The role of synovitis in osteoarthritis pathogenesis. Bone 2012, 51, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Sachdeva, M.; Aggarwal, A.; Sharma, R.; Randhawa, A.; Sahni, D.; Jacob, J.; Sharma, V.; Aggarwal, A. Chronic inflammation during osteoarthritis is associated with an increased expression of CD161 during advanced stage. Scand J. Immunol 2019, e12770. [Google Scholar] [CrossRef]
- Neumann, E.; Junker, S.; Schett, G.; Frommer, K.; Müller-Ladner, U. Adipokines in bone disease. Nat. Rev. Rheumatol. 2016, 12, 296–302. [Google Scholar] [CrossRef]
- Azamar-Llamas, D.; Hernández-Molina, G.; Ramos-Ávalos, B.; Furuzawa-Carballeda, J. Adipokine Contribution to the Pathogenesis of Osteoarthritis. Mediators Inflamm. 2017, 2017, 5468023. [Google Scholar] [CrossRef]
- Tenti, S.; Palmitesta, P.; Giordano, N.; Galeazzi, M.; Fioravanti, A. Increased serum leptin and visfatin levels in patients with diffuse idiopathic skeletal hyperostosis: A comparative study. Scand J. Rheumatol 2017, 46, 156–158. [Google Scholar] [CrossRef]
- Fioravanti, A.; Cheleschi, S.; De Palma, A.; Addimanda, O.; Mancarella, L.; Pignotti, E.; Pulsatelli, L.; Galezzi, M.; Meliconi, R. Can adipokines serum levels be used as biomarkers of hand osteoarthritis? Biomarkers 2018, 23, 265–270. [Google Scholar] [CrossRef]
- Carrión, M.; Frommer, K.W.; Pérez-García, S.; Müller-Ladner, U.; Gomariz, R.P.; Neumann, E. The Adipokine Network in Rheumatic Joint Diseases. Int. J. Mol. Sci. 2019, 20, 4091. [Google Scholar] [CrossRef]
- Sun, Z.; Lei, H.; Zhang, Z. Pre-B cell colony enhancing factor (PBEF), a cytokine with multiple physiological functions. Cytokine Growth F. R. 2013, 24, 433–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senolt, L.; Housa, D.; Vernerová, Z.; Jirásek, T.; Svobodová, R.; Veigl, D.; Anderlova, K.; Muller-Ladner, U.; Pavelka, K.; Haluzik, M. Resistin in rheumatoid arthritis synovial tissue, synovial fluid and serum. Ann. Rheum. Dis. 2007, 66, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Fioravanti, A.; Giannitti, C.; Cheleschi, S.; Simpatico, A.; Pascarelli, N.A.; Galeazzi, M. Circulating levels of adiponectin, resistin, and visfatin after mud-bath therapy in patients with bilateral knee osteoarthritis. Int. J. Biometeorol. 2015, 59, 1691–1700. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Chen, Y.; Wang, W. The current progress in understanding the molecular functions and mechanisms of visfatin in osteoarthritis. J. Bone Miner. Metab. 2016, 34, 485–490. [Google Scholar] [CrossRef]
- Calvet, J.; Orellana, C.; Gratacós, J.; Berenguer-Llergo, A.; Caixàs, A.; Chillarón, J.J.; Pedro-Botet, J.; Garcia-Manrique, M.; Navarro, N.; Larrosa, M. Synovial fluid adipokines are associated with clinical severity in knee osteoarthritis: A cross-sectional study in female patients with joint effusion. Arthritis Res. Ther. 2016, 18, 207. [Google Scholar] [CrossRef]
- Zhang, Z.; Xing, X.; Hensley, G.; Chang, L.W.; Liao, W.; Abu-Amer, Y.; Sandell, L.J. Resistin induces expression of proinflammatory cytokines and chemokines in human articular chondrocytes via transcription and messenger RNA stabilization. Arthritis Rheum. 2010, 62, 1993–2003. [Google Scholar] [CrossRef]
- Cheleschi, S.; Giordano, N.; Volpi, N.; Tenti, S.; Gallo, I.; Di Meglio, M.; Giannotti, S.; Fioravanta, A. A Complex Relationship between Visfatin and Resistin and microRNA: An In Vitro Study on Human Chondrocyte Cultures. Int. J. Mol. Sci. 2018, 19, 3909. [Google Scholar] [CrossRef]
- Wu, M.H.; Tsai, C.H.; Huang, Y.L.; Fong, Y.C.; Tang, C.H. Visfatin Promotes IL-6 and TNF-α Production in Human Synovial Fibroblasts by Repressing miR-199a-5p through ERK, p38 and JNK Signaling Pathways. Int. J. Mol. Sci. 2018, 19, 190. [Google Scholar] [CrossRef]
- Chen, W.C.; Wang, S.W.; Lin, C.Y.; Tsai, C.H.; Fong, Y.C.; Lin, T.Y.; Wang, S.L.; Huang, H.D.; Liao, K.W.; Tang, C.H. Resistin Enhances Monocyte Chemoattractant Protein-1 Production in Human Synovial Fibroblasts and Facilitates Monocyte Migration. Cell Physiol. Biochem. 2019, 52, 408–420. [Google Scholar] [CrossRef]
- Gerin, I.; Bommer, G.T.; McCoin, C.S.; Sousa, K.M.; Krishnan, V.; MacDougald, O.A. (2010) Roles for miRNA-378/378* in adipocyte gene expression and lipogenesis. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E198–E206. [Google Scholar] [CrossRef]
- Maurizi, G.; Babini, L.; Della Guardia, L. Potential role of microRNAs in the regulation of adipocytes liposecretion and adipose tissue physiology. J. Cell Physiol. 2018, 233, 9077–9086. [Google Scholar] [CrossRef] [PubMed]
- Malemud, C.J. MicroRNAs and Osteoarthritis. Cells 2018, 7, 92. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Prado, S.; Cicione, C.; Muiños-López, E.; Hermida-Gómez, T.; Oreiro, N.; Fernández-López, C.; Blanco, F.J. Characterization of microRNA expression profiles in normal and osteoarthritic human chondrocytes. BMC Musculoskelet. Disord. 2012, 13, 144. [Google Scholar] [CrossRef] [PubMed]
- De Palma, A.; Cheleschi, S.; Pascarelli, N.A.; Tenti, S.; Galeazzi, M.; Fioravanti, A. Do MicroRNAs have a key epigenetic role in osteoarthritis and in mechanotransduction? Clin. Exp. Rheumatol. 2017, 35, 518–526. [Google Scholar]
- Cheleschi, S.; De Palma, A.; Pecorelli, A.; Pascarelli, N.A.; Valacchi, G.; Belmonte, G.; Carta, S.; Galeazzi, M.; Fioravanti, A. Hydrostatic Pressure Regulates MicroRNA Expression Levels in Osteoarthritic Chondrocyte Cultures via the Wnt/β-Catenin Pathway. Int. J. Mol. Sci. 2017, 18, 133. [Google Scholar] [CrossRef]
- Fathollahi, A.; Aslani, S.; Jamshidi, A.; Mahmoudi, M. Epigenetics in osteoarthritis: Novel spotlight. J. Cell Physiol. 2019, 234, 12309–12324. [Google Scholar] [CrossRef]
- Cheleschi, S.; De Palma, A.; Pascarelli, N.A.; Giordano, N.; Galeazzi, M.; Tenti, S.; Fioravanti, A. Could Oxidative Stress Regulate the Expression of MicroRNA-146a and MicroRNA-34a in Human Osteoarthritic Chondrocyte Cultures? Int. J. Mol. Sci. 2017, 18, 2660. [Google Scholar] [CrossRef]
- Bu, H.; Wedel, S.; Cavinato, M.; Jansen-Dürr, P. MicroRNA Regulation of Oxidative Stress-Induced Cellular Senescence. Oxid. Med. Cell Longev. 2017, 2017, 2398696. [Google Scholar] [CrossRef]
- Machado, C.R.L.; Resende, G.G.; Macedo, R.B.V.; do Nascimento, V.C.; Branco, A.S.; Kakehasi, A.M.; Andrade, M.V. Fibroblast-like synoviocytes from fluid and synovial membrane from primary osteoarthritis demonstrate similar production of interleukin 6, and metalloproteinases 1 and 3. Clin. Exp. Rheumatol. 2019, 37, 306–309. [Google Scholar]
- Ioan-Facsinay, A.; Kloppenburg, M. An emerging player in knee osteoarthritis: The infrapatellar fat pad. Arthritis Res. Ther. 2013, 15, 225. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Francin, P.J.; Abot, A.; Guillaume, C.; Moulin, D.; Bianchi, A.; Gegout-Pottie, P.; Jouzeau, J.-Y.; Mainard, D.; Presle, D. Association between adiponectin and cartilage degradation in human osteoarthritis. Osteoarthr. Cartilage 2014, 22, 519–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.P.; Chen, C.N.; Chang, H.I.; Huang, K.C.; Cheng, C.C.; Chiu, F.Y.; Lee, K.C.; Lo, C.M.; Chang, S.F. Low Shear Stress Attenuates COX-2 Expression Induced by Resistin in Human Osteoarthritic Chondrocytes. J. Cell Physiol. 2017, 232, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- Laiguillon, M.C.; Houard, X.; Bougault, C.; Gosset, M.; Nourissat, G.; Sautet, A.; Jacques, C.; Berenbaum, F.; Sellam, J. Expression and function of visfatin (Nampt), an adipokine-enzyme involved in inflammatory pathways of osteoarthritis. Arthritis Res. Ther. 2014, 16, R38. [Google Scholar] [CrossRef]
- Wang, T.; He, C. Pro-inflammatory cytokines: The link between obesity and osteoarthritis. Cytokine Growth F. R. 2018, 44, 38–50. [Google Scholar] [CrossRef]
- Pelletier, J.P.; McCollum, R.; Cloutier, J.M.; Martel-Pelletier, J. Synthesis of metalloproteases and interleukin 6 (IL-6) in human osteoarthritic synovial membrane is an IL-1 mediated process. J. Rheumatol. Suppl. 1995, 43, 109–114. [Google Scholar]
- Altobelli, E.; Angeletti, P.M.; Piccolo, D.; De Angelis, R. Synovial Fluid and Serum Concentrations of Inflammatory Markers in Rheumatoid Arthritis, Psoriatic Arthritis and Osteoarthitis: A Systematic Review. Curr. Rheumatol. Rev. 2017, 13, 170–179. [Google Scholar] [CrossRef]
- Sato, H.; Muraoka, S.; Kusunoki, N.; Masuoka, S.; Yamada, S.; Ogasawara, H.; Imai, T.; Akasaka, Y.; Tochigi, N.; Takahashi, H.; et al. Resistin upregulates chemokine production by fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis Res. Ther. 2017, 19, 263. [Google Scholar] [CrossRef]
- Cawston, T.E.; Young, D.A. Proteinases involved in matrix turnover during cartilage and bone breakdown. Cell Tissue Res. 2010, 339, 221–235. [Google Scholar] [CrossRef]
- Troeberg, L.; Nagase, H. Proteases involved in cartilage matrix degradation in osteoarthritis. Biochim Biophys Acta 2012, 1824, 133–145. [Google Scholar] [CrossRef] [Green Version]
- Pérez-García, S.; Carrión, M.; Jimeno, R.; Ortiz, A.M.; González-Álvaro, I.; Fernández, J.; Gomariz, R.P.; Juarranz, Y. Urokinase plasminogen activator system in synovial fibroblasts from osteoarthritis patients: Modulation by inflammatory mediators and neuropeptides. J. Mol. Neurosci. 2014, 52, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Pérez-García, S.; Gutiérrez-Cañas, I.; Seoane, I.V.; Fernández, J.; Mellado, M.; Leceta, J.; Tío, L.; Villanueva-Romero, R.; Juarranz, Y.; Gomariz, R.P. Healthy and Osteoarthritic Synovial Fibroblasts Produce a Disintegrin and Metalloproteinase with Thrombospondin Motifs 4, 5, 7, and 12: Induction by IL-1β and Fibronectin and Contribution to Cartilage Damage. Am. J. Pathol. 2016, 186, 2449–2461. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Otero, M. Inflammation in osteoarthritis. Curr. Opin. Rheumatol. 2011, 23, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Meier, F.M.; Frommer, K.W.; Peters, M.A.; Brentano, F.; Lefèvre, S.; Schröder, D.; Kyburz, D.; Steinmeyer, J.; Rehart, S.; Gay, S.; et al. Visfatin/pre-B-cell colony-enhancing factor (PBEF), a proinflammatory and cell motility-changing factor in rheumatoid arthritis. J. Biol. Chem. 2012, 287, 28378–28385. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.S.; Kim, H.A. Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis. Int. J. Mol. Sci. 2015, 16, 26035–26054. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Lu, H.S.; Guan, Z.P.; Sun, T.Z.; Chen, Y.Y.; Ruan, G.R.; Chen, Z.K.; Jiang, J.; Bai, C.J. Involvement of PDCD5 in the regulation of apoptosis in fibroblast-like synoviocytes of rheumatoid arthritis. Apoptosis 2007, 12, 1433–1441. [Google Scholar] [CrossRef]
- Feng, L.; Precht, P.; Balakir, R.; Horton, W.E., Jr. Evidence of a direct role for Bcl-2 in the regulation of articular chondrocyte apoptosis under the conditions of serum withdrawal and retinoic acid treatment. J. Cell Biochem. 1998, 71, 302–309. [Google Scholar] [CrossRef]
- Jiao, Y.; Ding, H.; Huang, S.; Liu, Y.; Sun, X.; Wei, W.; Ma, J.; Zheng, F. Bcl-XL and Mcl-1 upregulation by calreticulin promotes apoptosis resistance of fibroblast-like synoviocytes via activation of PI3K/Akt and STAT3 pathways in rheumatoid arthritis. Clin. Exp. Rheumatol. 2018, 36, 841–849. [Google Scholar]
- Sun, L.; Chen, S.; Gao, H.; Ren, L.; Song, G. Visfatin induces the apoptosis of endothelial progenitor cells via the induction of pro-inflammatory mediators through the NF-κB pathway. Int. J. Mol. Med. 2017, 40, 637–646. [Google Scholar] [CrossRef]
- Marchev, A.S.; Dimitrova, P.A.; Burns, A.J.; Kostov, R.V.; Dinkova-Kostova, A.T.; Georgiev, M.I. Oxidative stress and chronic inflammation in osteoarthritis: Can NRF2 counteract these partners in crime? Ann. N Y Acad Sci 2017, 1401, 114–135. [Google Scholar] [CrossRef]
- Marampon, F.; Codenotti, S.; Megiorni, F.; Del Fattore, A.; Camero, S.; Gravina, G.L.; Festuccia, C.; Musio, D.; Felice, F.D.; Nardone, V.; et al. NRF2 orchestrates the redox regulation induced by radiation therapy, sustaining embryonal and alveolar rhabdomyosarcoma cells radioresistance. J. Cancer Res. Clin. Oncol. 2019, 145, 881–893. [Google Scholar] [CrossRef]
- Huang, M.L.; Chiang, S.; Kalinowski, D.S.; Bae, D.H.; Sahni, S.; Richardson, D.R. The Role of the Antioxidant Response in Mitochondrial Dysfunction in Degenerative Diseases: Cross-Talk between Antioxidant Defense, Autophagy, and Apoptosis. Oxid. Med. Cell Longev. 2019, 2019, 6392763. [Google Scholar] [CrossRef]
- Raghuraman, G.; Zuniga, M.C.; Yuan, H.; Zhou, W. PKCε mediates resistin-induced NADPH oxidase activation and inflammation leading to smooth muscle cell dysfunction and intimal hyperplasia. Atherosclerosis 2016, 253, 29–37. [Google Scholar] [CrossRef]
- Teixeira, T.M.; da Costa, D.C.; Resende, A.C.; Soulage, C.O.; Bezerra, F.F.; Daleprane, J.B. Activation of Nrf2-Antioxidant Signaling by 1,25-Dihydroxycholecalciferol Prevents Leptin-Induced Oxidative Stress and Inflammation in Human Endothelial Cells. J. Nutr. 2017, 147, 506–513. [Google Scholar] [CrossRef]
- Lin, Y.T.; Chen, L.K.; Jian, D.Y.; Hsu, T.C.; Huang, W.C.; Kuan, T.T.; Wu, S.Y.; Kwok, C.F.; Ho, L.T.; Juan, C.C. Visfatin Promotes Monocyte Adhesion by Upregulating ICAM-1 and VCAM-1 Expression in Endothelial Cells via Activation of p38-PI3K-Akt Signaling and Subsequent ROS Production and IKK/NF-κB Activation. Cell Physiol. Biochem. 2019, 52, 1398–1411. [Google Scholar] [CrossRef]
- Cheleschi, S.; Tenti, S.; Mondanelli, N.; Corallo, C.; Barbarino, M.; Giannotti, S.; Gallo, I.; Giordano, A.; Fioravanti, A. MicroRNA-34a and MicroRNA-181a Mediate Visfatin-Induced Apoptosis and Oxidative Stress via NF-κB Pathway in Human Osteoarthritic Chondrocytes. Cells 2019, 8, 874. [Google Scholar] [CrossRef]
- Lin, Y.H. MicroRNA Networks Modulate Oxidative Stress in Cancer. Int. J. Mol. Sci. 2019, 20, 4497. [Google Scholar] [CrossRef]
- Yan, S.; Wang, M.; Zhao, J.; Zhang, H.; Zhou, C.; Jin, L.; Zhang, Y.; Qiu, X.; Ma, B.; Fan, Q. MicroRNA-34a affects chondrocyte apoptosis and proliferation by targeting the SIRT1/p53 signaling pathway during the pathogenesis of osteoarthritis. Int. J. Mol. Med. 2016, 38, 201–209. [Google Scholar] [CrossRef] [Green Version]
- Abouheif, M.M.; Nakasa, T.; Shibuya, H.; Niimoto, T.; Kongcharoensombat, W.; Ochi, M. Silencing microRNA-34a inhibits chondrocyte apoptosis in a rat osteoarthritis model in vitro. Rheumatology 2010, 49, 2054–2060. [Google Scholar] [CrossRef] [Green Version]
- Niederer, F.; Trenkmann, M.; Ospelt, C.; Karouzakis, E.; Neidhart, M.; Stanczyk, J.; Kolling, C.; Gay, R.E.; Detmar, M.; Gay, S.; et al. Down-regulation of microRNA-34a* in rheumatoid arthritis synovial fibroblasts promotes apoptosis resistance. Arthritis Rheum. 2012, 64, 1771–1779. [Google Scholar] [CrossRef]
- Zhong, X.; Li, P.; Li, J.; He, R.; Cheng, G.; Li, Y. Downregulation of microRNA-34a inhibits oxidized low-density lipoprotein-induced apoptosis and oxidative stress in human umbilical vein endothelial cells. Int. J. Mol. Med. 2018, 42, 1134–1144. [Google Scholar] [CrossRef]
- Okuhara, A.; Nakasa, T.; Shibuya, H.; Niimoto, T.; Adachi, N.; Deie, M.; Ochi, M. Changes in microRNA expression in peripheral mononuclear cells according to the progression of osteoarthritis. Mod. Rheumatol. 2012, 22, 446–457. [Google Scholar] [CrossRef]
- Zheng, H.; Liu, J.; Tycksen, E.; Nunley, R.; McAlinden, A. MicroRNA-181a/b-1 over-expression enhances osteogenesis by modulating PTEN/PI3K/AKT signaling and mitochondrial metabolism. Bone 2019, 123, 92–102. [Google Scholar] [CrossRef]
- Chen, K.L.; Fu, Y.Y.; Shi, M.Y.; Li, H.X. Down-regulation of miR-181a can reduce heat stress damage in PBMCs of Holstein cows. In Vitro Cell Dev. Biol. Anim. 2016, 52, 864–871. [Google Scholar] [CrossRef]
- Feng, X.; Zhang, C.; Yang, Y.; Hou, D.; Zhu, A. Role of miR-181a in the process of apoptosis of multiple malignant tumors: A literature review. Adv. Clin. Exp. Med. 2018, 27, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, K.; Nakasa, T.; Miyaki, S.; Ishikawa, M.; Deie, M.; Adachi, N.; Yasunaga, Y.; Asahara, H.; Ochi, M. Expression of microRNA-146a in osteoarthritis cartilage. Arthritis Rheumatol. 2009, 60, 1035–1041. [Google Scholar] [CrossRef]
- Li, L.; Chen, X.P.; Li, Y.J. MicroRNA-146a and human disease. Scand. J. Immunol. 2010, 71, 227–231. [Google Scholar] [CrossRef]
- Xie, Y.; Chu, A.; Feng, Y.; Chen, L.; Shao, Y.; Luo, Q.; Deng, X.; Wu, M.; Shi, X.; Chen, Y. MicroRNA-146a: A Comprehensive Indicator of Inflammation and Oxidative Stress Status Induced in the Brain of Chronic T2DM Rats. Front. Pharmacol. 2018, 9, 478. [Google Scholar] [CrossRef]
- Li, J.; Huang, J.; Dai, L.; Yu, D.; Chen, Q.; Zhang, X.; Dai, K. miR-146a, an IL-1β responsive miRNA, induces vascular endothelial growth factor and chondrocyte apoptosis by targeting Smad4. Arthritis Res. Ther. 2012, 14, R75. [Google Scholar] [CrossRef]
- Wang, J.H.; Shih, K.S.; Wu, Y.W.; Wang, A.W.; Yang, C.R. Histone deacetylase inhibitors increase microRNA-146a expression and enhance negative regulation of interleukin-1β signaling in osteoarthritis fibroblast-like synoviocytes. Osteoarthr. Cartilage 2013, 21, 1987–1996. [Google Scholar] [CrossRef]
- Wen, F.; Li, B.; Huang, C.; Wei, Z.; Zhou, Y.; Liu, J.; Zhang, H. MiR-34a is Involved in the Decrease of ATP Contents Induced by Resistin Through Target on ATP5S in HepG2 Cells. Biochem. Genet. 2015, 53, 301–309. [Google Scholar] [CrossRef]
- Gong, Y.Y.; Luo, J.Y.; Wang, L.; Huang, Y. MicroRNAs regulating reactive oxygen species in cardiovascular diseases. Antioxid. Redox Signal. 2018, 29, 1092–1107. [Google Scholar] [CrossRef]
- D’Adamo, S.; Cetrullo, S.; Guidotti, S.; Borzì, R.M.; Flamigni, F. Hydroxytyrosol modulates the levels of microRNA-9 and its target sirtuin-1 thereby counteracting oxidative stress-induced chondrocyte death. Osteoarthr. Cartilage 2017, 25, 600–610. [Google Scholar] [CrossRef] [Green Version]
- Ji, G.; Lv, K.; Chen, H.; Wang, T.; Wang, Y.; Zhao, D.; Qu, L.; Li, Y. MiR-146a regulates SOD2 expression in H2O2 stimulated PC12 cells. PLoS ONE 2013, 8, e69351. [Google Scholar] [CrossRef]
- Wang, L.; Huang, H.; Fan, Y.; Kong, B.; Hu, H.; Hu, K.; Guo, J.; Mei, Y.; Liu, W.L. Effects of downregulation of microRNA-181a on H2O2-induced H9c2 cell apoptosis via the mitochondrial apoptotic pathway. Oxid. Med. Cell Longev. 2014, 2014, 960362. [Google Scholar] [CrossRef]
- Baker, J.R.; Vuppusetty, C.; Colley, T.; Papaioannou, A.I.; Fenwick, P.; Donnelly, L.; Ito, K.; Barnes, P.J. Oxidative stress dependent microRNA-34a activation via PI3Kα reduces the expression of sirtuin-1 and sirtuin-6 in epithelial cells. Sci. Rep. 2016, 6, 35871. [Google Scholar] [CrossRef]
- Huang, X.; Gao, Y.; Qin, J.; Lu, S. The role of miR-34a in the hepatoprotective effect of hydrogen sulfide on ischemia/reperfusion injury in young and old rats. PLoS ONE 2014, 9, e113305. [Google Scholar] [CrossRef]
- Rigoglou, S.; Papavassiliou, A.G. The NF-κB signaling pathway in osteoarthritis. Int. J. Biochem. Cell Biol. 2013, 45, 2580–2584. [Google Scholar] [CrossRef]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A blossoming of relevance to human pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Z.; Kang, Y.; Hou, C.; Duan, X.; Sheng, P.; Sandell, L.J.; Liao, W. Resistin stimulates expression of chemokine genes in chondrocytes via combinatorial regulation of C/EBPβ and NF-κB. Int. J. Mol. Sci. 2014, 15, 17242–17255. [Google Scholar] [CrossRef]
- Aslani, M.R.; Keyhanmanesh, R.; Alipour, M.R. Increased Visfatin Expression Is Associated with Nuclear Factor-κB in Obese Ovalbumin-Sensitized Male Wistar Rat Tracheae. Med. Princ. Pract. 2017, 26, 351–358. [Google Scholar] [CrossRef]
- Xu, B.; Li, Y.Y.; Ma, J.; Pei, F.X. Roles of microRNA and signaling pathway in osteoarthritis pathogenesis. J. Zhejiang Univ. Sci. B 2016, 17, 200–208. [Google Scholar] [CrossRef]
- Altman, R.; Asch, E.; Bloch, D.; Bole, G.; Borenstein, D.; Brandt, K.; Christy, W.; Cooke, T.D.; Greenwald, R.; Hochberg, M.; et al. Development of criteria for the classification and reporting of osteoarthritis. Classification of osteoarthritis of the knee. Diagnostic and Therapeutic Criteria Committee of the American Rheumatism Association. Arthritis Rheum. 1986, 29, 1039–1049. [Google Scholar] [CrossRef]
- Gosset, M.; Berenbaum, F.; Salvat, C.; Sautet, A.; Pigenet, A.; Tahiri, K.; Jacques, C. Crucial role of visfatin/pre-B cell colony-enhancing factor in matrix degradation and prostaglandin E2 synthesis in chondrocytes: Possible influence on osteoarthritis. Arthritis Rheum. 2008, 58, 1399–1409. [Google Scholar] [CrossRef]
- Ramakers, C.; Ruijter, J.M.; Deprez, R.H.; Moorman, A.F. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci. Lett 2003, 339, 62–66. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real RT-PCR. Nucleic Acid Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Vandesompele, J.; de Preter, K.; Pattyn, F.; Poppe, B.; van Roy, N.; de Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034.1. [Google Scholar] [CrossRef]
- Cheleschi, S.; Calamia, V.; Fernandez-Moreno, M.; Biava, M.; Giordani, A.; Fioravanti, A.; Anzini, M.; Blanco, F. In vitro comprehensive analysis of VA692 a new chemical entity for the treatment of osteoarthritis. Int. Immunopharmacol. 2018, 64, 86–100. [Google Scholar] [CrossRef]
- Cheleschi, S.; Fioravanti, A.; De Palma, A.; Corallo, C.; Franci, D.; Volpi, N.; Bedogni, G.; Giannotti, S.; Giordano, N. Methylsulfonylmethane and mobilee prevent negative effect of IL-1β in human chondrocyte cultures via NF-κB signaling pathway. Int. Immunopharmacol. 2018, 65, 129–139. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA Genes | Cat. No. (Qiagen) |
| miR-34a | MS00003318 |
| miR-146a | MS00003535 |
| miR-181a | MS00006692 |
| SNORD-25 | MS00014007 |
| Target Genes | Cat. No. (Qiagen) |
| IL-1β | QT00021385 |
| IL-6 | QT00083720 |
| IL-17A | QT00009233 |
| TNF-α | QT00029162 |
| MMP-1 | QT00014581 |
| MMP-13 | QT00001764 |
| Col2a1 | QT00049518 |
| BCL2 | QT00000721 |
| SOD-2 | QT01008693 |
| CAT | QT00079674 |
| NRF2 | QT00027384 |
| ACTB | QT00095431 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheleschi, S.; Gallo, I.; Barbarino, M.; Giannotti, S.; Mondanelli, N.; Giordano, A.; Tenti, S.; Fioravanti, A. MicroRNA Mediate Visfatin and Resistin Induction of Oxidative Stress in Human Osteoarthritic Synovial Fibroblasts Via NF-κB Pathway. Int. J. Mol. Sci. 2019, 20, 5200. https://doi.org/10.3390/ijms20205200
Cheleschi S, Gallo I, Barbarino M, Giannotti S, Mondanelli N, Giordano A, Tenti S, Fioravanti A. MicroRNA Mediate Visfatin and Resistin Induction of Oxidative Stress in Human Osteoarthritic Synovial Fibroblasts Via NF-κB Pathway. International Journal of Molecular Sciences. 2019; 20(20):5200. https://doi.org/10.3390/ijms20205200
Chicago/Turabian StyleCheleschi, Sara, Ines Gallo, Marcella Barbarino, Stefano Giannotti, Nicola Mondanelli, Antonio Giordano, Sara Tenti, and Antonella Fioravanti. 2019. "MicroRNA Mediate Visfatin and Resistin Induction of Oxidative Stress in Human Osteoarthritic Synovial Fibroblasts Via NF-κB Pathway" International Journal of Molecular Sciences 20, no. 20: 5200. https://doi.org/10.3390/ijms20205200