ALS-Associated SOD1(G93A) Decreases SERCA Pump Levels and Increases Store-Operated Ca2+ Entry in Primary Spinal Cord Astrocytes from a Transgenic Mouse Model



Abstract
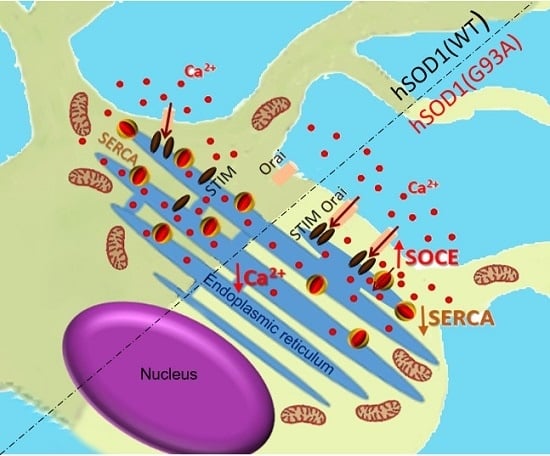
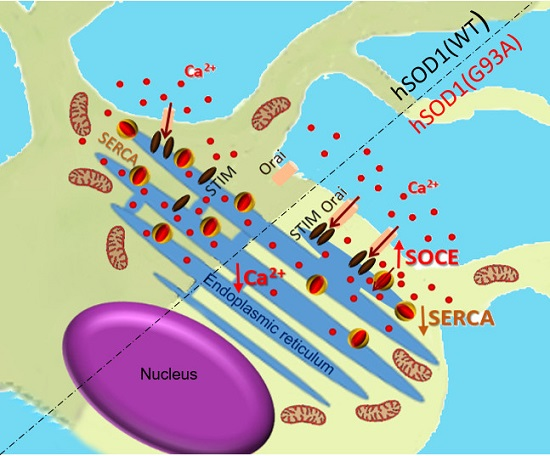
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
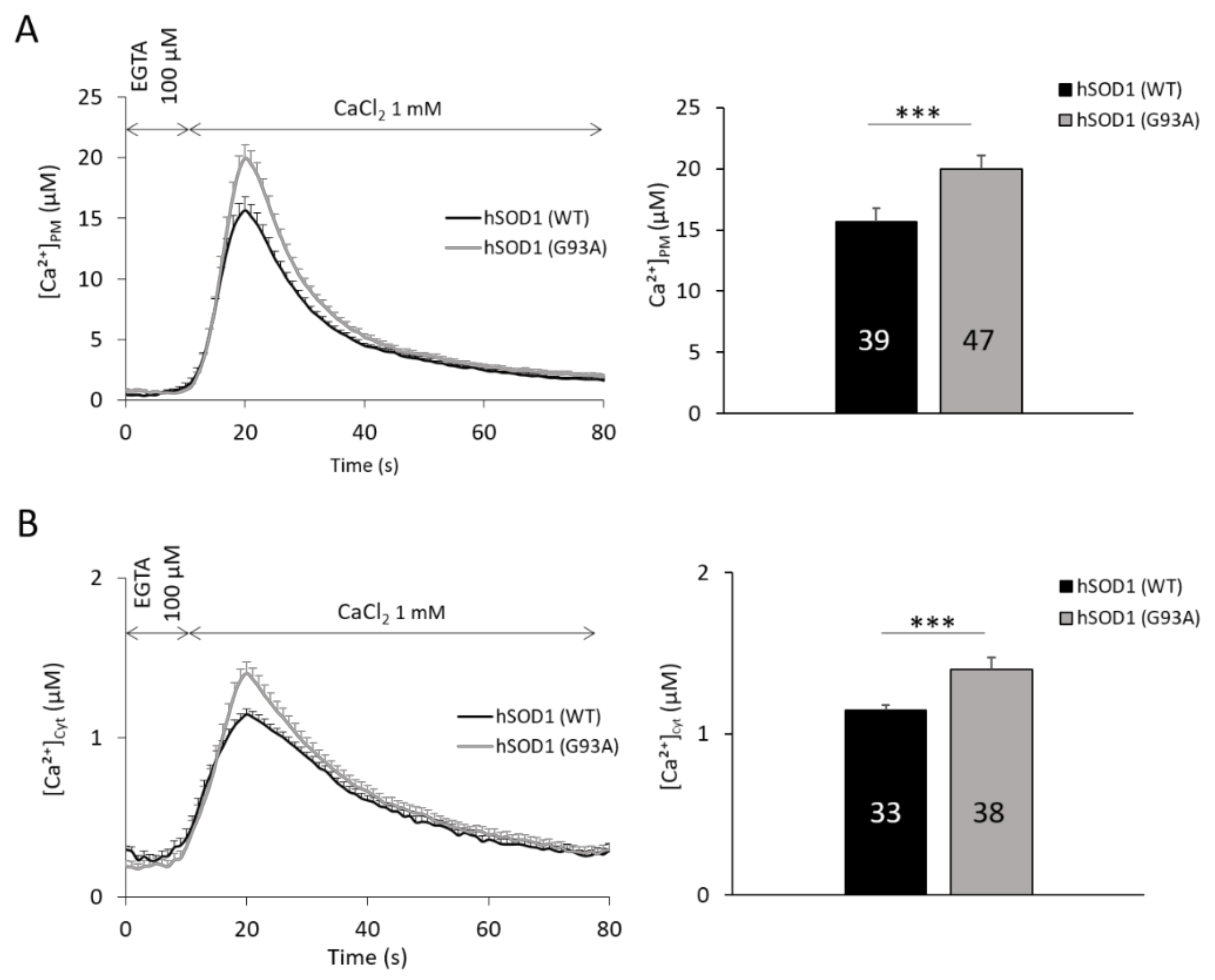
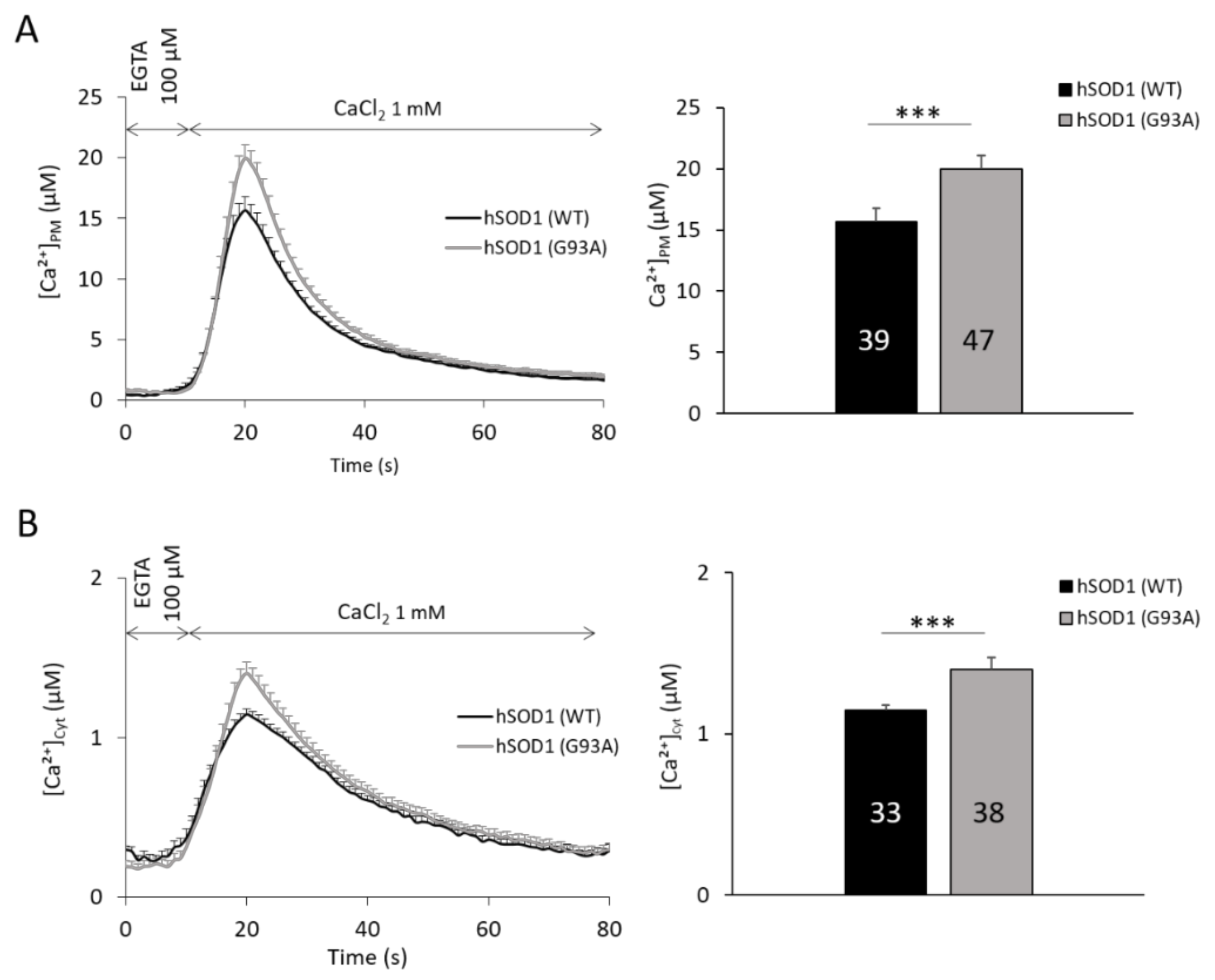
2.1. hSOD1(G93A)-Expressing Astrocytes Have Enhanced SOCE-Mediated Ca2+ Influx
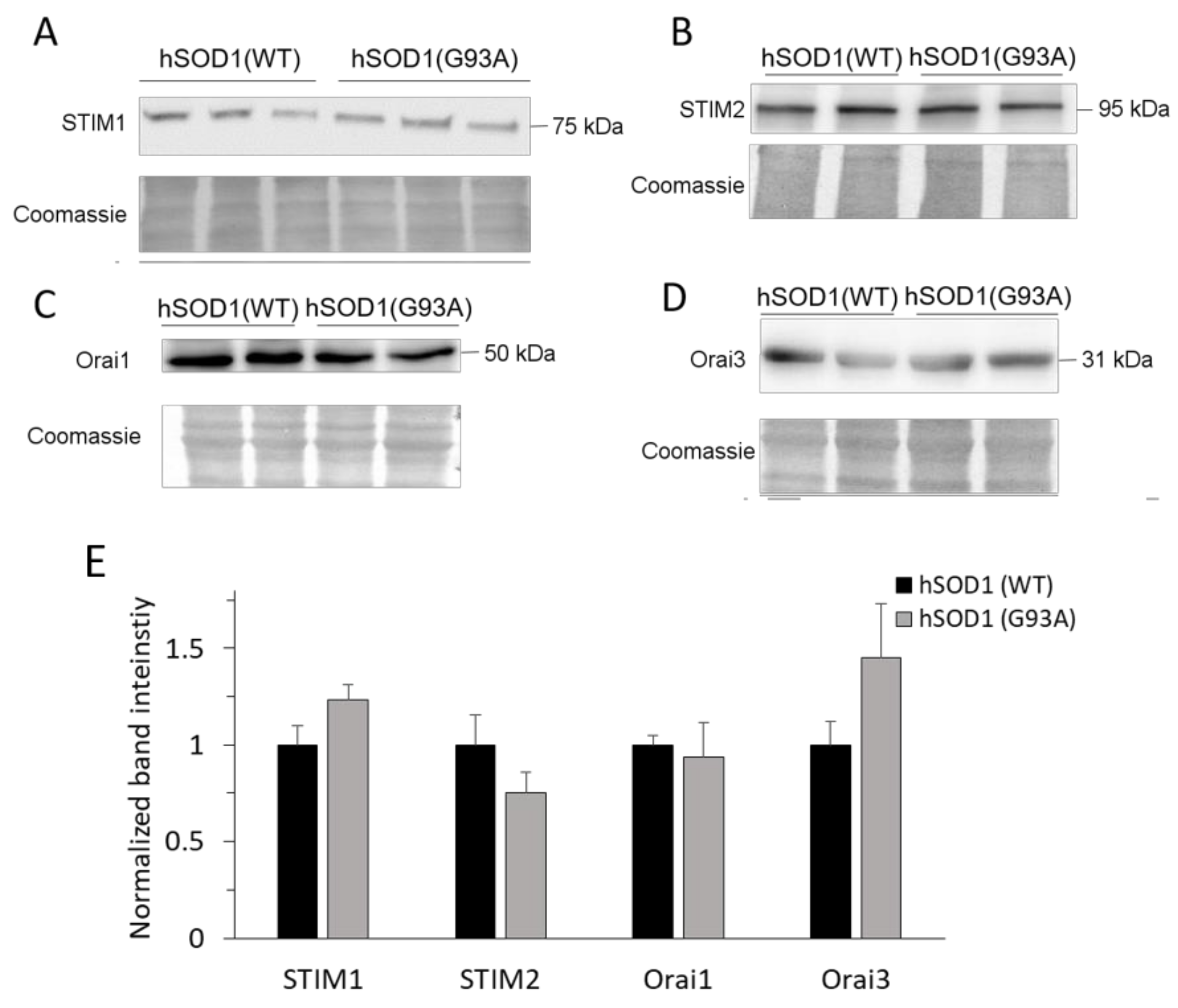
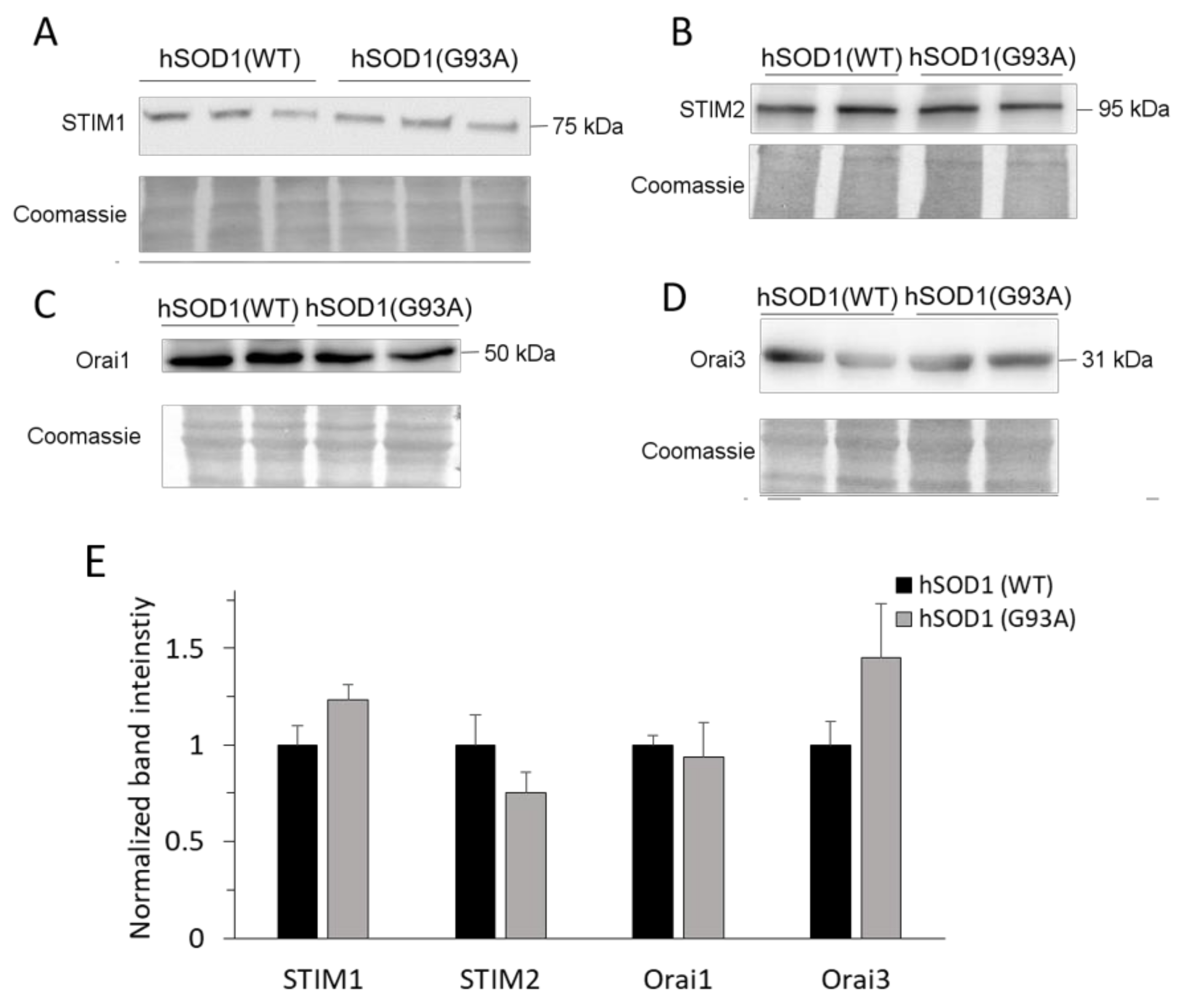
2.2. SOCE-Mediating Proteins Are Equally Present in hSOD1(WT) and hSOD1(G93A) Primary Astrocytes
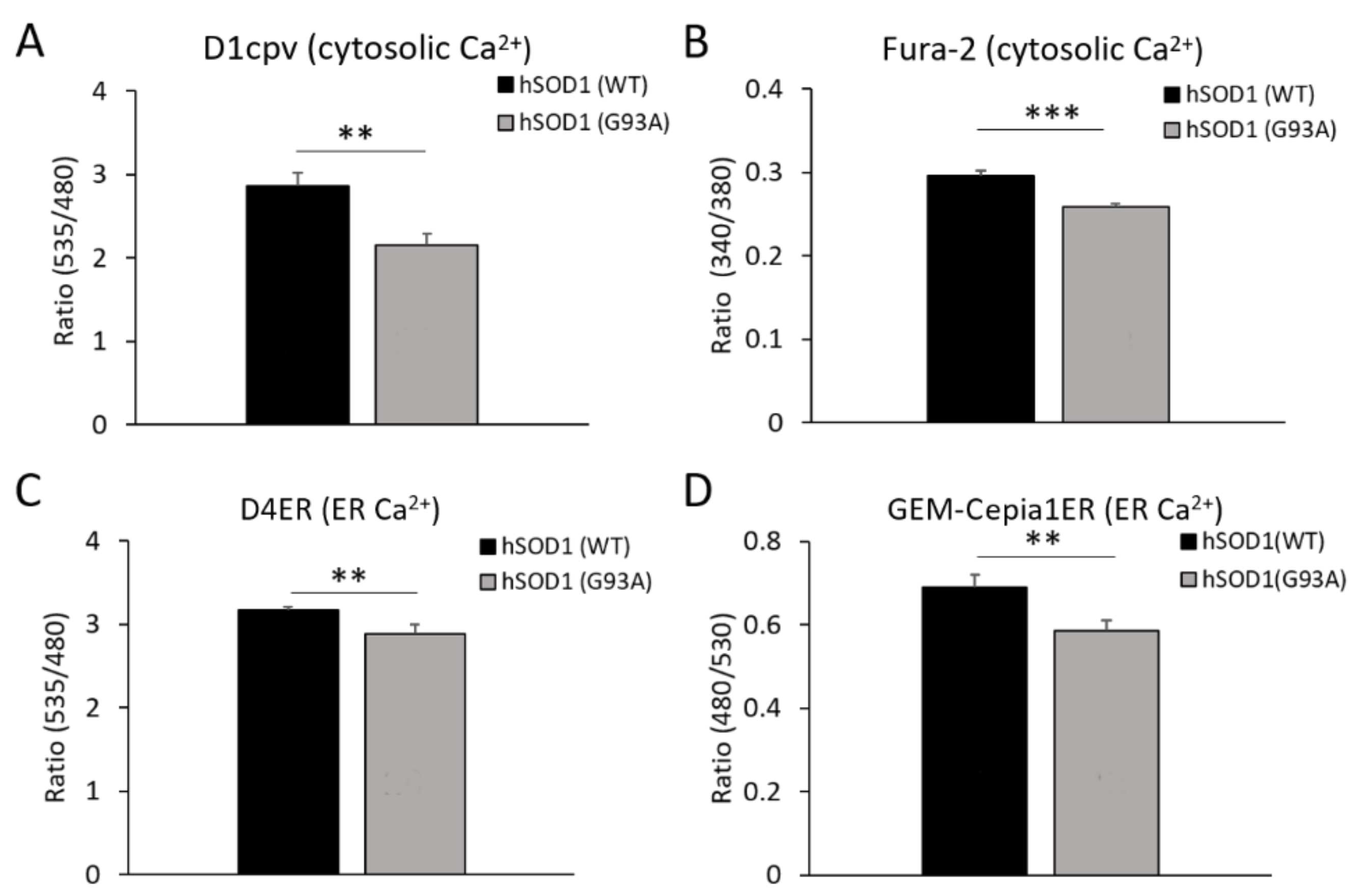
2.3. hSOD1(G93A) Astrocytes Have Reduced basal Ca2+ Levels in the Cytosol and the ER Lumen Compared to Control Astrocytes
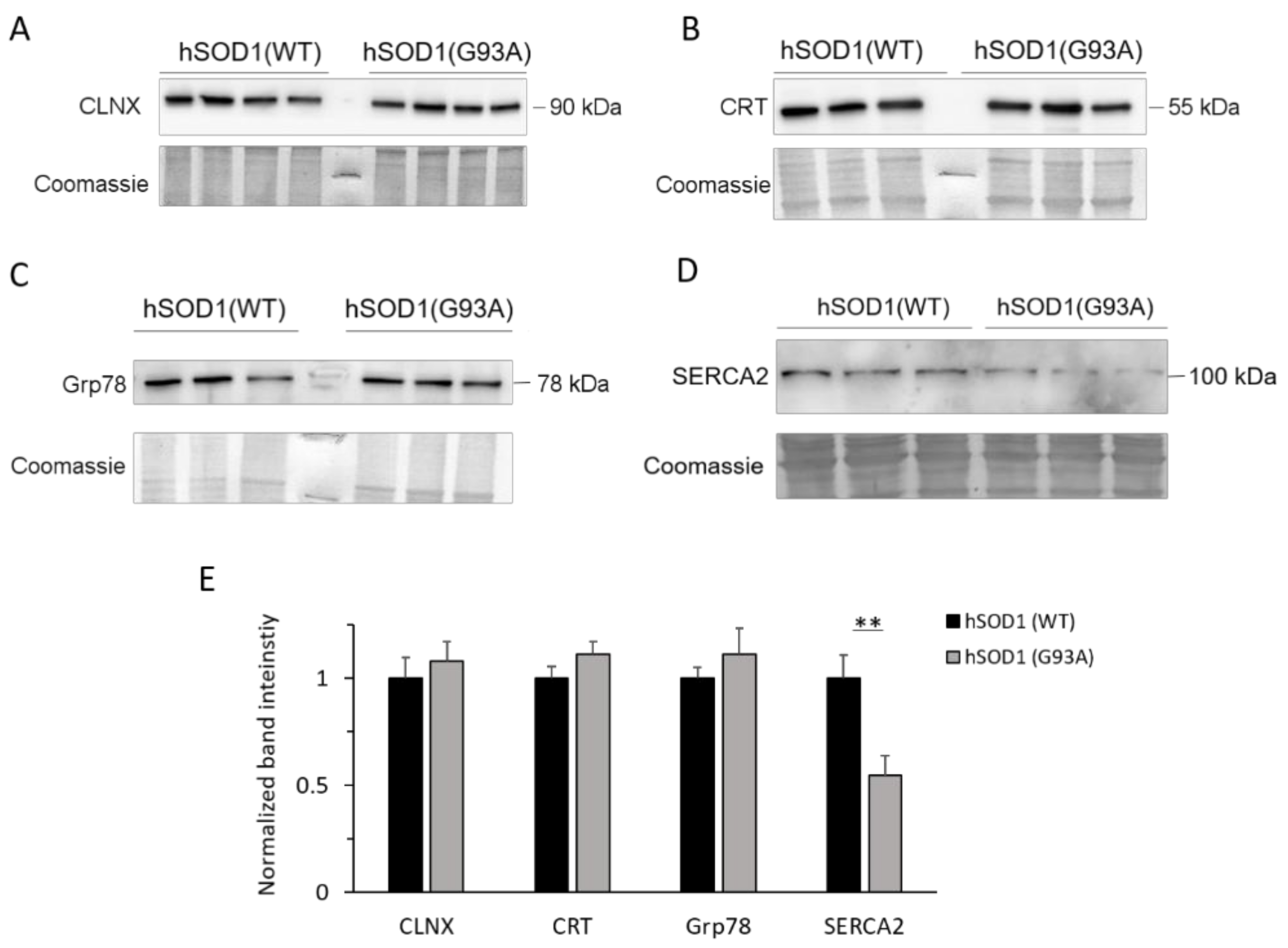
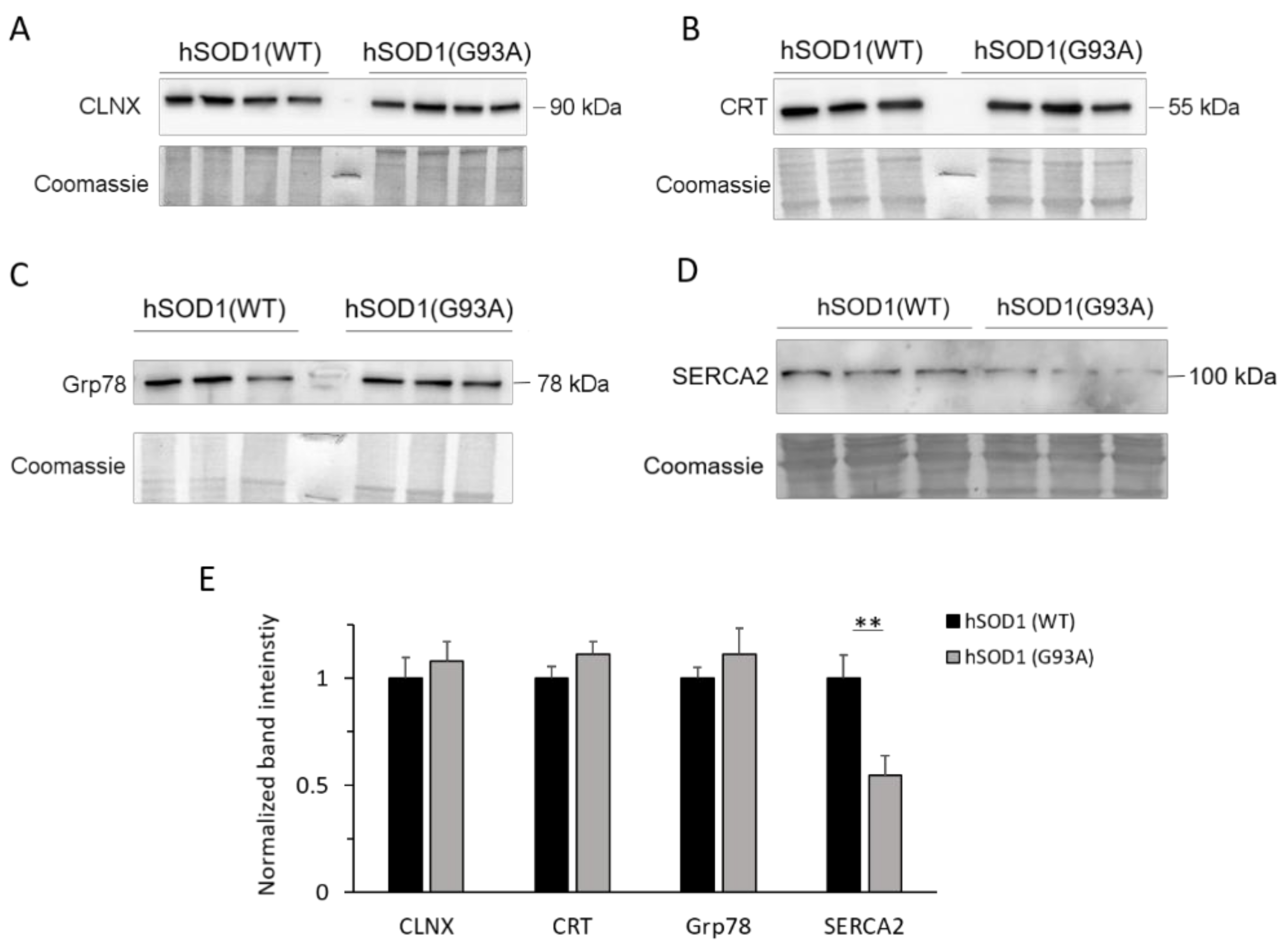
2.4. SERCA2 Is Down-regulated in hSOD1(G93A) Astrocytes
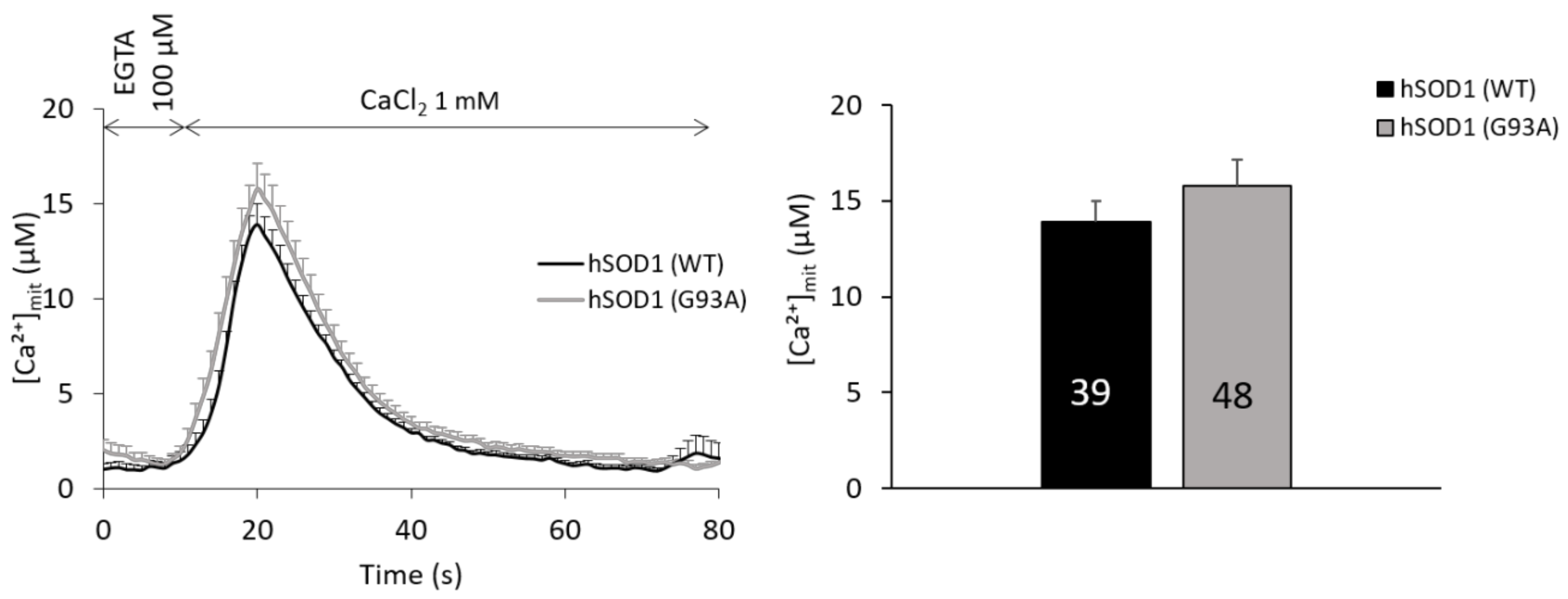
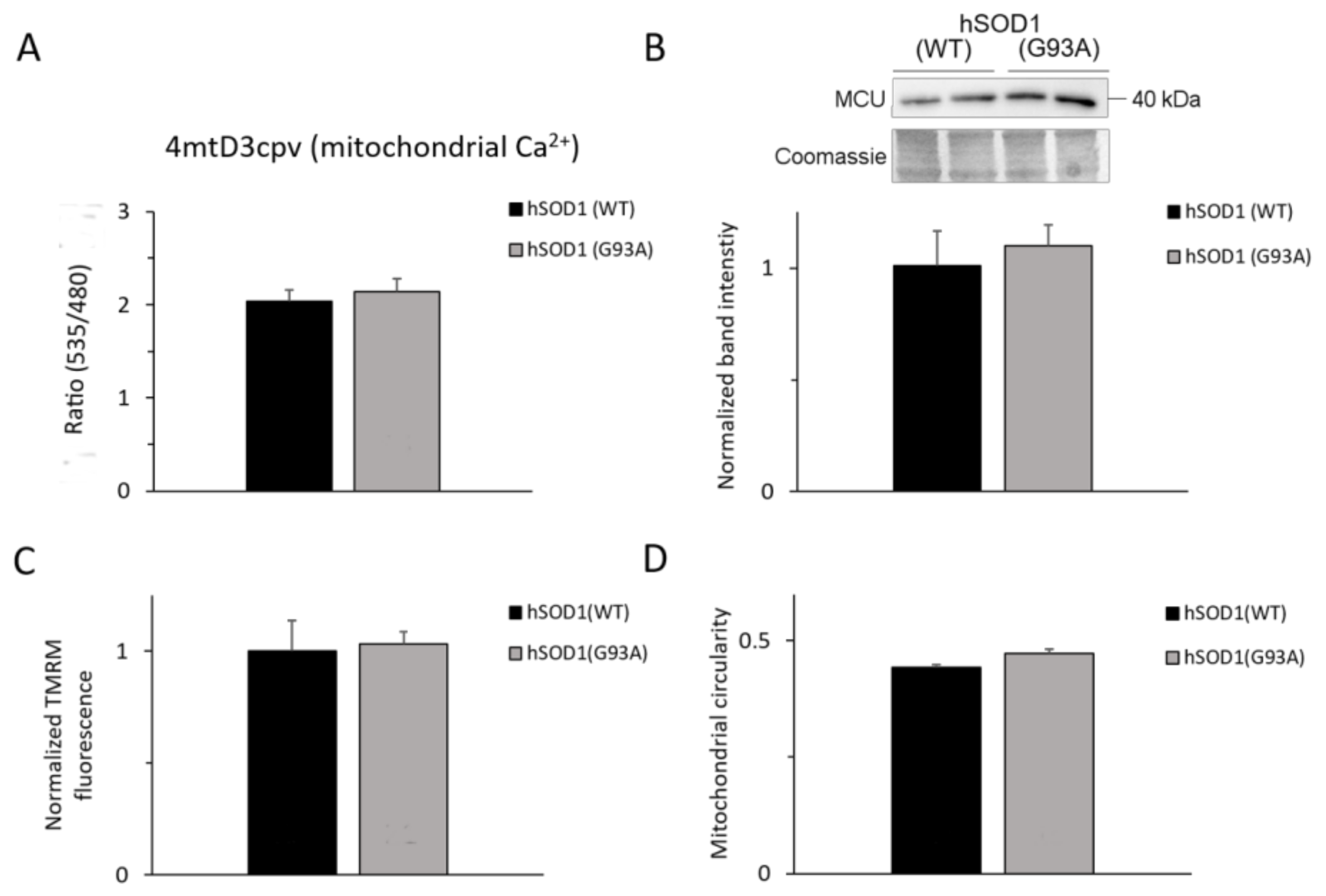
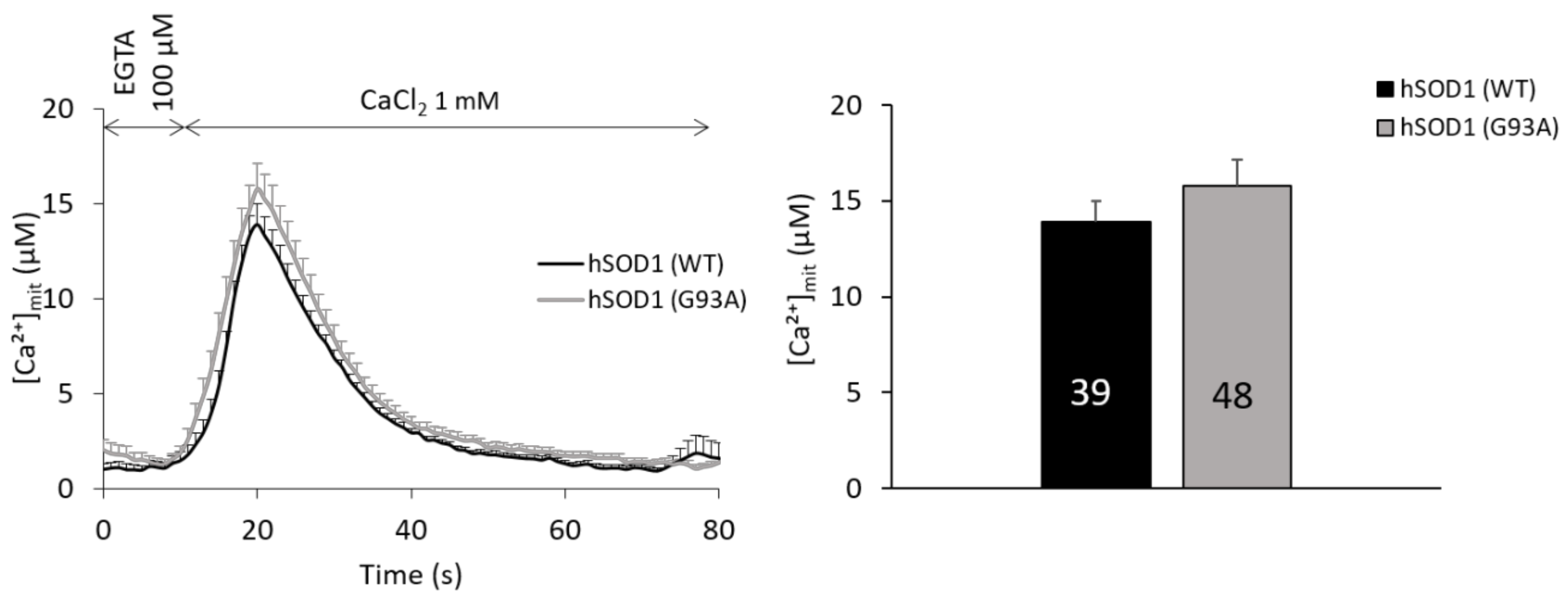
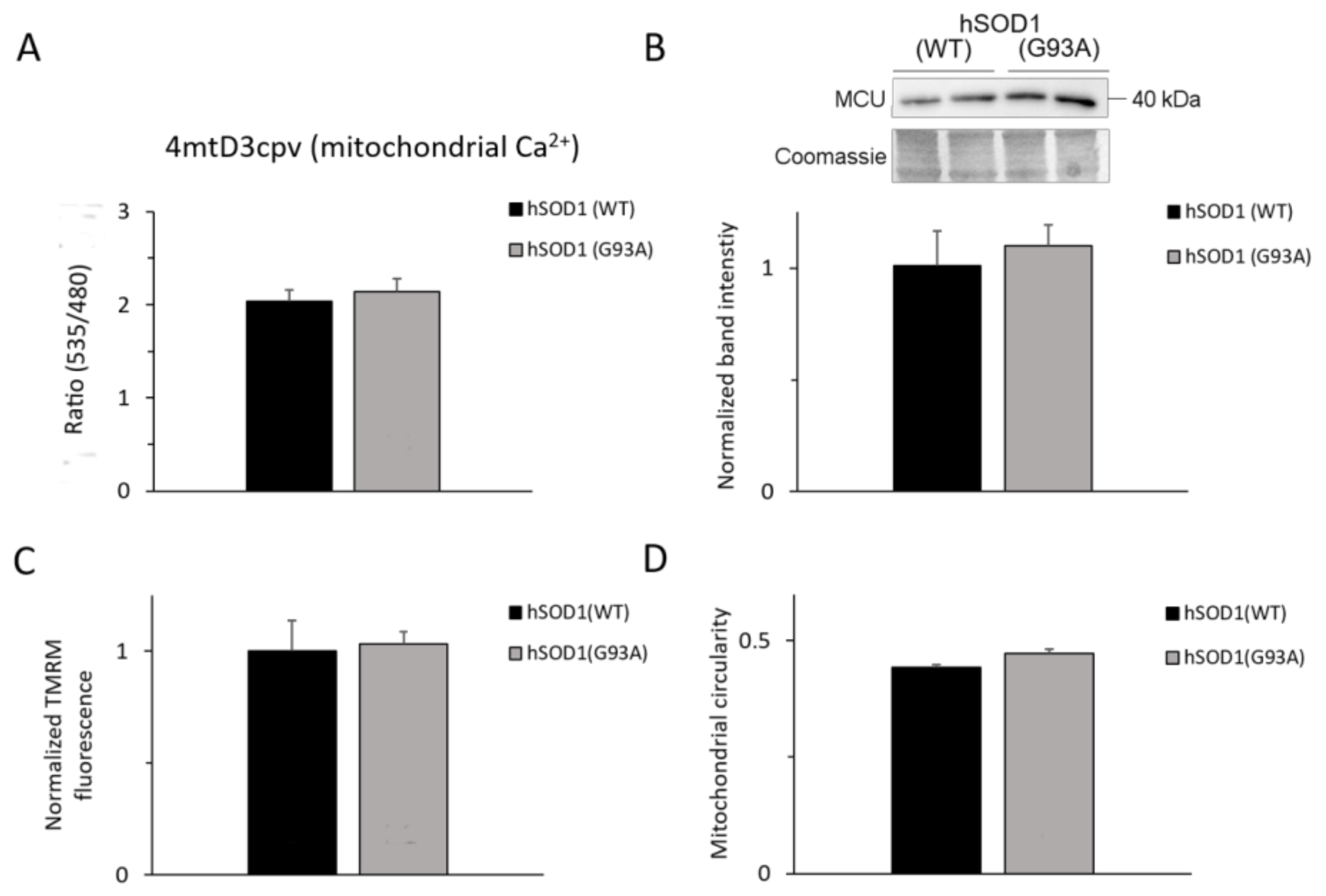
2.5. Mitochondria of hSOD1(G93A) and hSOD1(WT) Astrocytes Equally Respond to SOCE Stimulation
2.6. Ca2+-related Mitochondrial Parameters Are Comparable in hSOD1(G93A) and hSOD1(WT) Astrocytes
3. Discussion
4. Materials and Methods
4.1. Mouse Models and Breeding
4.2. Preparation of Primary Cultures of Murine Spinal Cord Astrocytes
4.3. Ca2+ Imaging
4.4. Measurement of the Mitochondrial Membrane Potential
4.5. Analysis of Mitochondrial Morphology
4.6. Western Blot Analysis
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ab | antibody |
| AEQ | aequorin |
| ALS | amyotrophic lateral sclerosis |
| BSA | bovine serum albumin |
| CLNX | calnexin |
| CRT | calreticulin |
| cyt | cytosol |
| CFP | cyan fluorescent protein |
| ER | endoplasmic reticulum |
| fALS | familial ALS |
| FCCP | carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone |
| FRET | fluorescence resonance energy transfer |
| FUS/TLS | Fused in Sarcoma/Translocated in Sarcoma |
| GECI | genetically-encoded Ca2+ indicators |
| GFAP | glial fibrillary acidic protein |
| h | human |
| KRB | Krebs-Ringer buffer |
| MCU | mitochondrial Ca2+ uniporter |
| mit | mitochondrial matrix |
| MN | motor neuron |
| mtRFP | mitochondrially-targeted red-fluorescent protein |
| n | number of replicates |
| p | polyclonal |
| PM | plasma membrane |
| PMCA | PM Ca2+ ATPase |
| NCX | Na+/Ca2+ exchanger |
| PVDF | polyvinylidene difluoride |
| ROI | region of interest |
| SDS | sodium dodecyl sulfate |
| SDS-PAGE | SDS-polyacrylamide gel electrophoresis |
| SEM | standard error of the mean |
| SERCA | sarco-endoplasmic reticulum Ca2+ ATPase |
| SOCE | store-operated Ca2+ entry |
| SOD1 | superoxide dismutase 1 |
| STIM | stromal interaction molecule |
| TBS-T | Tris-buffered saline added with Tween-20 |
| TDP-43 | TAR DNA-binding protein 43 |
| Tg | transgenic |
| TMRM | tetramethylrhodamine methyl ester |
| YFP | yellow fluorescent protein |
| WB | Western blot |
| WT | wild-type |
References
- Cleveland, D.W.; Rothstein, J.D. From charcot to lou gehrig: Deciphering selective motor neuron death in als. Nat. Rev. Neurosci. 2001, 2, 806–819. [Google Scholar] [CrossRef] [PubMed]
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H.J.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- van Damme, P.; Bogaert, E.; Dewil, M.; Hersmus, N.; Kiraly, D.; Scheveneels, W.; Bockx, I.; Braeken, D.; Verpoorten, N.; Verhoeven, K.; et al. Astrocytes regulate GluR2 expression in motor neurons and their vulnerability to excitotoxicity. Proc. Natl. Acad. Sci. USA 2007, 104, 14825–14830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, K.; Boillee, S.; Roberts, E.A.; Garcia, M.L.; McAlonis-Downes, M.; Mikse, O.R.; Cleveland, D.W.; Goldstein, L.S.B. Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc. Natl. Acad. Sci. USA 2008, 105, 7594–7599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 2009, 187, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Bylicky, M.A.; Mueller, G.P.; Day, R.M. Mechanisms of endogenous neuroprotective effects of astrocytes in brain injury. Oxid. Med. Cell. Longev. 2018, 2018, 6501031. [Google Scholar] [CrossRef]
- Bilsland, L.G.; Nirmalananthan, N.; Yip, J.; Greensmith, L.; Duchen, M.R. Expression of mutant SOD1 in astrocytes induces functional deficits in motoneuron mitochondria. J. Neurochem. 2008, 107, 1271–1283. [Google Scholar] [CrossRef]
- Tong, J.; Huang, C.; Bi, F.; Wu, Q.; Huang, B.; Liu, X.; Li, F.; Zhou, H.; Xia, X.-G. Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. EMBO J. 2013, 32, 1917–1926. [Google Scholar] [CrossRef]
- Cassina, P.; Cassina, A.; Pehar, M.; Castellanos, R.; Gandelman, M.; de Leon, A.; Robinson, K.M.; Mason, R.P.; Beckman, J.S.; Barbeito, L.; et al. Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: Prevention by mitochondrial-targeted antioxidants. J. Neurosci. 2008, 28, 4115–4122. [Google Scholar] [CrossRef] [PubMed]
- Martorana, F.; Brambilla, L.; Valori, C.F.; Bergamaschi, C.; Roncoroni, C.; Aronica, E.; Volterra, A.; Bezzi, P.; Rossi, D. The BH4 domain of Bcl-XL rescues astrocyte degeneration in amyotrophic lateral sclerosis by modulating intracellular calcium signals. Hum. Mol. Genet. 2012, 21, 826–840. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, H.; Ng, S.K.; Diaz, N.; Burstein, S.; Morel, L.; Osgood, A.; Sider, B.; Higashimori, H.; Haydon, P.G.; Manfredi, G.; et al. Abnormal intracellular calcium signaling and SNARE-dependent exocytosis contributes to SOD1G93A astrocyte-mediated toxicity in amyotrophic lateral sclerosis. J. Neurosci. 2014, 34, 2331–2348. [Google Scholar] [CrossRef] [PubMed]
- Fritz, E.; Izaurieta, P.; Weiss, A.; Mir, F.R.; Rojas, P.; Gonzalez, D.; Rojas, F.; Brown, R.H.J.; Madrid, R.; van Zundert, B. Mutant SOD1-expressing astrocytes release toxic factors that trigger motoneuron death by inducing hyperexcitability. J. Neurophysiol. 2013, 109, 2803–2814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madill, M.; McDonagh, K.; Ma, J.; Vajda, A.; McLoughlin, P.; O’Brien, T.; Hardiman, O.; Shen, S. Amyotrophic lateral sclerosis patient iPSC-derived astrocytes impair autophagy via non-cell autonomous mechanisms. Mol. Brain 2017, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.; Rodriguez-Muela, N.; Klim, J.R.; de Boer, A.S.; Agrawal, S.; Sandoe, J.; Lopes, C.S.; Ogliari, K.S.; Williams, L.A.; Shear, M.; et al. Reactive astrocytes promote ALS-like degeneration and intracellular protein aggregation in human motor neurons by disrupting autophagy through TGF-β1. Stem Cell Rep. 2017, 9, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Komine, O. The multi-dimensional roles of astrocytes in ALS. Neurosci. Res. 2018, 126, 31–38. [Google Scholar] [CrossRef]
- Zonta, M.; Carmignoto, G. Calcium oscillations encoding neuron-to-astrocyte communication. J. Physiol. Paris 2002, 96, 193–198. [Google Scholar] [CrossRef]
- Scemes, E.; Giaume, C. Astrocyte calcium waves: What they are and what they do. Glia 2006, 54, 716–725. [Google Scholar] [CrossRef] [Green Version]
- Zorec, R.; Araque, A.; Carmignoto, G.; Haydon, P.G.; Verkhratsky, A.; Parpura, V. Astroglial excitability and gliotransmission: An appraisal of Ca2+ as a signalling route. ASN Neuro 2012, 4, AN20110061. [Google Scholar] [CrossRef]
- Verkhratsky, A. Astroglial calcium signaling in aging and Alzheimer’s disease. Cold Spring Harb. Perspect. Biol. 2019, 11, a035188. [Google Scholar] [CrossRef] [PubMed]
- Parekh, A.B.; Putney, J.W.J. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Parpura, V. Store-operated calcium entry in neuroglia. Neurosci. Bull. 2014, 30, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Michelucci, A.; Garcia-Castaneda, M.; Boncompagni, S.; Dirksen, R.T. Role of STIM1/ORAI1-mediated store-operated Ca2+ entry in skeletal muscle physiology and disease. Cell Calcium 2018, 76, 101–115. [Google Scholar] [CrossRef]
- Rosenberg, P.; Katz, D.; Bryson, V. SOCE and STIM1 signaling in the heart: Timing and location matter. Cell Calcium 2019, 77, 20–28. [Google Scholar] [CrossRef]
- Parekh, A.B. Store-operated CRAC channels: Function in health and disease. Nat. Rev. Drug Discov. 2010, 9, 399–410. [Google Scholar] [CrossRef]
- Soboloff, J.; Rothberg, B.S.; Madesh, M.; Gill, D.L. STIM proteins: Dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol. 2012, 13, 549–565. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.; van den Abeele, F.; Lehen’kyi, V.; Gkika, D.; Guarmit, B.; Lepage, G.; Slomianny, C.; Borowiec, A.S.; Bidaux, G.; Benahmed, M.; et al. Remodeling of channel-forming ORAI proteins determines an oncogenic switch in prostate cancer. Cancer Cell 2014, 26, 19–32. [Google Scholar] [CrossRef]
- Lioudyno, M.I.; Kozak, J.A.; Penna, A.; Safrina, O.; Zhang, S.L.; Sen, D.; Roos, J.; Stauderman, K.A.; Cahalan, M.D. Orai1 and STIM1 move to the immunological synapse and are up-regulated during T cell activation. Proc. Natl. Acad. Sci. USA 2008, 105, 2011–2016. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Finch, E.A.; Graham, V.; Zhang, Z.-S.; Ding, J.-D.; Burch, J.; Oh-hora, M.; Rosenberg, P. STIM1-Ca2+ signaling is required for the hypertrophic growth of skeletal muscle in mice. Mol. Cell. Biol. 2012, 32, 3009–3017. [Google Scholar] [CrossRef]
- Majewski, L.; Kuznicki, J. SOCE in neurons: Signaling or just refilling? Biochim. Biophys. Acta Mol. Cell Res. 2014, 1853, 1940–1952. [Google Scholar] [CrossRef] [PubMed]
- Wegierski, T.; Kuznicki, J. Neuronal calcium signaling via store-operated channels in health and disease. Cell Calcium 2018, 74, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Feske, S. CRAC channelopathies. Pflug. Arch. Eur. J. Physiol. 2010, 460, 417–435. [Google Scholar] [CrossRef] [PubMed]
- Lacruz, R.S.; Feske, S. Diseases caused by mutations in ORAI1 and STIM1. Ann. N. Y. Acad. Sci. 2015, 1356, 45–79. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-F.; Lin, P.-C.; Yeh, Y.-M.; Chen, L.-H.; Shen, M.-R. Store-operated Ca2+ entry in tumor progression: From molecular mechanisms to clinical implications. Cancers 2019, 11, 899. [Google Scholar] [CrossRef]
- Moccia, F.; Zuccolo, E.; Soda, T.; Tanzi, F.; Guerra, G.; Mapelli, L.; Lodola, F.; D’Angelo, E. Stim and Orai proteins in neuronal Ca2+ signaling and excitability. Front. Cell. Neurosci. 2015, 9, 153. [Google Scholar] [CrossRef]
- Putney, J.W. Forms and functions of store-operated calcium entry mediators, STIM and Orai. Adv. Biol. Regul. 2018, 68, 88–96. [Google Scholar] [CrossRef]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 2007, 131, 1327–1339. [Google Scholar] [CrossRef]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E.J.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef]
- Kraft, R. STIM and ORAI proteins in the nervous system. Channels 2015, 9, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; An, H.; Sa, M.; Won, J.; Shin, J.I.; Lee, C.J. Orai1 and Orai3 in combination with Stim1 mediate the majority of store-operated calcium entry in astrocytes. Exp. Neurobiol. 2017, 26, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Corbett, E.F.; Michalak, M. Calcium, a signaling molecule in the endoplasmic reticulum? Trends Biochem. Sci. 2000, 25, 307–311. [Google Scholar] [CrossRef]
- Meldolesi, J.; Pozzan, T. The endoplasmic reticulum Ca2+ store: A view from the lumen. Trends Biochem. Sci. 1998, 23, 10–14. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pozzan, T. Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef]
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the center of cell signaling: Interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef]
- Grosskreutz, J.; van den Bosch, L.; Keller, B.U. Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium 2010, 47, 165–174. [Google Scholar] [CrossRef]
- Lautenschlaeger, J.; Prell, T.; Grosskreutz, J. Endoplasmic reticulum stress and the ER mitochondrial calcium cycle in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2012, 13, 166–177. [Google Scholar] [CrossRef]
- Tadic, V.; Prell, T.; Lautenschlaeger, J.; Grosskreutz, J. The ER mitochondria calcium cycle and ER stress response as therapeutic targets in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2014, 8, 147. [Google Scholar] [CrossRef] [Green Version]
- Baba-Aissa, F.; Raeymaekers, L.; Wuytack, F.; Dode, L.; Casteels, R. Distribution and isoform diversity of the organellar Ca2+ pumps in the brain. Mol. Chem. Neuropathol. 1998, 33, 199–208. [Google Scholar] [CrossRef]
- Rizzuto, R.; de Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef] [PubMed]
- de Stefani, D.; Rizzuto, R.; Pozzan, T. Enjoy the trip: Calcium in mitochondria back and forth. Annu. Rev. Biochem. 2016, 85, 161–192. [Google Scholar] [CrossRef] [PubMed]
- de Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coussee, E.; de Smet, P.; Bogaert, E.; Elens, I.; van Damme, P.; Willems, P.; Koopman, W.; van den Bosch, L.; Callewaert, G. G37R SOD1 mutant alters mitochondrial complex I activity, Ca2+ uptake and ATP production. Cell Calcium 2011, 49, 217–225. [Google Scholar] [CrossRef]
- Cozzolino, M.; Carri, M.T. Mitochondrial dysfunction in ALS. Prog. Neurobiol. 2012, 97, 54–66. [Google Scholar] [CrossRef]
- Dagda, R.K.; Cherra, S.J.; Kulich, S.M.; Tandon, A.; Park, D.; Chu, C.T. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 2009, 284, 13843–13855. [Google Scholar] [CrossRef]
- di Giorgio, F.P.; Boulting, G.L.; Bobrowicz, S.; Eggan, K.C. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell 2008, 3, 637–648. [Google Scholar] [CrossRef]
- Papadeas, S.T.; Kraig, S.E.; O’Banion, C.; Lepore, A.C.; Maragakis, N.J. Astrocytes carrying the superoxide dismutase 1 (SOD1G93A) mutation induce wild-type motor neuron degeneration in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 17803–17808. [Google Scholar] [CrossRef]
- Hoth, M.; Niemeyer, B.A. The neglected CRAC proteins: Orai2, Orai3, and STIM2. Curr. Top. Membr. 2013, 71, 237–271. [Google Scholar]
- Frischauf, I.; Schindl, R.; Derler, I.; Bergsmann, J.; Fahrner, M.; Romanin, C. The STIM/Orai coupling machinery ND Discovery of STIM and Orai Proteins SC RIB. Channels 2008, 2, 1–8. [Google Scholar]
- Golovina, V.A. Visualization of localized store-operated calcium entry in mouse astrocytes. Close proximity to the endoplasmic reticulum. J. Physiol. 2005, 564, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Ohara, H.; Nabika, T. A nonsense mutation of Stim1 identified in stroke-prone spontaneously hypertensive rats decreased the store-operated calcium entry in astrocytes. Biochem. Biophys. Res. Commun. 2016, 476, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, B.J.; Irrinki, K.M.; Mallilankaraman, K.; Lien, Y.-C.; Wang, Y.; Bhanumathy, C.D.; Subbiah, R.; Ritchie, M.F.; Soboloff, J.; Baba, Y.; et al. S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J. Cell Biol. 2010, 190, 391–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, H.F.; Hensley, K.; Thongboonkerd, V.; Merchant, M.L.; Lynn, B.C.; Pierce, W.M.; Klein, J.B.; Calabrese, V.; Butterfield, D.A. Redox proteomics analysis of oxidatively modified proteins in G93A-SOD1 transgenic mice—A model of familial amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2005, 39, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Prell, T.; Lautenschlager, J.; Grosskreutz, J. Calcium-dependent protein folding in amyotrophic lateral sclerosis. Cell Calcium 2013, 54, 132–143. [Google Scholar] [CrossRef]
- Grolla, A.A.; Sim, J.A.; Lim, D.; Rodriguez, J.J.; Genazzani, A.A.; Verkhratsky, A. Amyloid-β and Alzheimer’s disease type pathology differentially affects the calcium signalling toolkit in astrocytes from different brain regions. Cell Death Dis. 2013, 4, e623. [Google Scholar] [CrossRef]
- Morel, L.; Chiang, M.S.R.; Higashimori, H.; Shoneye, T.; Iyer, L.K.; Yelick, J.; Tai, A.; Yang, Y. Molecular and functional properties of regional astrocytes in the adult brain. J. Neurosci. 2017, 37, 8706–8717. [Google Scholar] [CrossRef]
- Valori, C.F.; Brambilla, L.; Martorana, F.; Rossi, D. The multifaceted role of glial cells in amyotrophic lateral sclerosis. Cell. Mol. Life Sci. 2014, 71, 287–297. [Google Scholar] [CrossRef]
- Rossi, D.; Brambilla, L.; Valori, C.F.; Roncoroni, C.; Crugnola, A.; Yokota, T.; Bredesen, D.E.; Volterra, A. Focal degeneration of astrocytes in amyotrophic lateral sclerosis. Cell Death Differ. 2008, 15, 1691–1700. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Amarilla, P.; Olivera-Bravo, S.; Trias, E.; Cragnolini, A.; Martinez-Palma, L.; Cassina, P.; Beckman, J.; Barbeito, L. Phenotypically aberrant astrocytes that promote motoneuron damage in a model of inherited amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2011, 108, 18126–18131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- dal Canto, M.C.; Gurney, M.E. Neuropathological changes in two lines of mice carrying a transgene for mutant human Cu, Zn SOD, and in mice overexpressing wild type human SOD: A model of familial amyotrophic lateral sclerosis (FALS). Brain Res. 1995, 676, 25–40. [Google Scholar] [CrossRef]
- Norante, R.P.; Massimino, M.L.; Lorenzon, P.; de Mario, A.; Peggion, C.; Vicario, M.; Albiero, M.; Sorgato, M.C.; Lopreiato, R.; Bertoli, A. Generation and validation of novel adeno-associated viral vectors for the analysis of Ca2+ homeostasis in motor neurons. Sci. Rep. 2017, 7, 6521. [Google Scholar] [CrossRef] [PubMed]
- Lazzari, C.; Peggion, C.; Stella, R.; Massimino, M.L.; Lim, D.; Bertoli, A.; Sorgato, M.C. Cellular prion protein is implicated in the regulation of local Ca2+ movements in cerebellar granule neurons. J. Neurochem. 2011, 116, 881–890. [Google Scholar] [CrossRef]
- de Mario, A.; Castellani, A.; Peggion, C.; Massimino, M.L.; Lim, D.; Hill, A.F.; Sorgato, M.C.; Bertoli, A. The prion protein constitutively controls neuronal store-operated Ca2+ entry through Fyn kinase. Front. Cell. Neurosci. 2015, 9, 416. [Google Scholar] [CrossRef]
- Lim, D.; Bertoli, A.; Sorgato, M.C.; Moccia, F. Generation and usage of aequorin lentiviral vectors for Ca2+ measurement in sub-cellular compartments of hard-to-transfect cells. Cell Calcium 2016, 59, 228–239. [Google Scholar] [CrossRef]
- Brini, M.; Marsault, R.; Bastianutto, C.; Alvarez, J.; Pozzan, T.; Rizzuto, R. Transfected aequorin in the measurement of cytosolic Ca2+ concentration ([Ca2+]c). A critical evaluation. J. Biol. Chem. 1995, 270, 9896–9903. [Google Scholar] [CrossRef]
- Palmer, A.E.; Giacomello, M.; Kortemme, T.; Hires, S.A.; Lev-Ram, V.; Baker, D.; Tsien, R.Y. Ca2+ indicators based on computationally redesigned calmodulin-peptide pairs. Chem. Biol. 2006, 13, 521–530. [Google Scholar] [CrossRef]
- Palmer, A.E.; Tsien, R.Y. Measuring calcium signaling using genetically targetable fluorescent indicators. Nat. Protoc. 2006, 1, 1057–1065. [Google Scholar] [CrossRef]
- Greotti, E.; Wong, A.; Pozzan, T.; Pendin, D.; Pizzo, P. Characterization of the ER-targeted low affinity Ca2+ probe D4ER. Sensors 2016, 16, 1419. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Kanemaru, K.; Ishii, K.; Ohkura, M.; Okubo, Y.; Iino, M. Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat. Commun. 2014, 5, 4153. [Google Scholar] [CrossRef] [PubMed]
- Gazda, K.; Kuznicki, J.; Wegierski, T. Knockdown of amyloid precursor protein increases calcium levels in the endoplasmic reticulum. Sci. Rep. 2017, 7, 14512. [Google Scholar] [CrossRef] [PubMed]
- Joshi, D.C.; Bakowska, J.C. Determination of mitochondrial membrane potential and reactive oxygen species in live rat cortical neurons. J. Vis. Exp. 2011, 51, e2704. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, A.; Cottet-Rousselle, C.; Arnaud, J.; Julien Saint Amand, K.; Ben Messaoud, R.; Lenon, M.; Demeilliers, C.; Moulis, J.-M. Mitochondrial morphology and function of the pancreatic β-cells INS-1 model upon chronic exposure to sub-lethal cadmium doses. Toxics 2018, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Welinder, C.; Ekblad, L. Coomassie staining as loading control in Western blot analysis. J. Proteome Res. 2011, 10, 1416–1419. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Norante, R.P.; Peggion, C.; Rossi, D.; Martorana, F.; De Mario, A.; Lia, A.; Massimino, M.L.; Bertoli, A. ALS-Associated SOD1(G93A) Decreases SERCA Pump Levels and Increases Store-Operated Ca2+ Entry in Primary Spinal Cord Astrocytes from a Transgenic Mouse Model. Int. J. Mol. Sci. 2019, 20, 5151. https://doi.org/10.3390/ijms20205151
Norante RP, Peggion C, Rossi D, Martorana F, De Mario A, Lia A, Massimino ML, Bertoli A. ALS-Associated SOD1(G93A) Decreases SERCA Pump Levels and Increases Store-Operated Ca2+ Entry in Primary Spinal Cord Astrocytes from a Transgenic Mouse Model. International Journal of Molecular Sciences. 2019; 20(20):5151. https://doi.org/10.3390/ijms20205151
Chicago/Turabian StyleNorante, Rosa Pia, Caterina Peggion, Daniela Rossi, Francesca Martorana, Agnese De Mario, Annamaria Lia, Maria Lina Massimino, and Alessandro Bertoli. 2019. "ALS-Associated SOD1(G93A) Decreases SERCA Pump Levels and Increases Store-Operated Ca2+ Entry in Primary Spinal Cord Astrocytes from a Transgenic Mouse Model" International Journal of Molecular Sciences 20, no. 20: 5151. https://doi.org/10.3390/ijms20205151
APA StyleNorante, R. P., Peggion, C., Rossi, D., Martorana, F., De Mario, A., Lia, A., Massimino, M. L., & Bertoli, A. (2019). ALS-Associated SOD1(G93A) Decreases SERCA Pump Levels and Increases Store-Operated Ca2+ Entry in Primary Spinal Cord Astrocytes from a Transgenic Mouse Model. International Journal of Molecular Sciences, 20(20), 5151. https://doi.org/10.3390/ijms20205151