From Crosstalk between Immune and Bone Cells to Bone Erosion in Infection


Abstract
1. Introduction
2. Epidemiology
3. Osteoblasts and Osteoclasts
4. Bone Formation and Remodelling
5. Calcineurin/Nuclear Factor of Activated T Cells: An Important Signaling Pathway Associated with Osteoclastogenesis and Regulation of Immune Cells
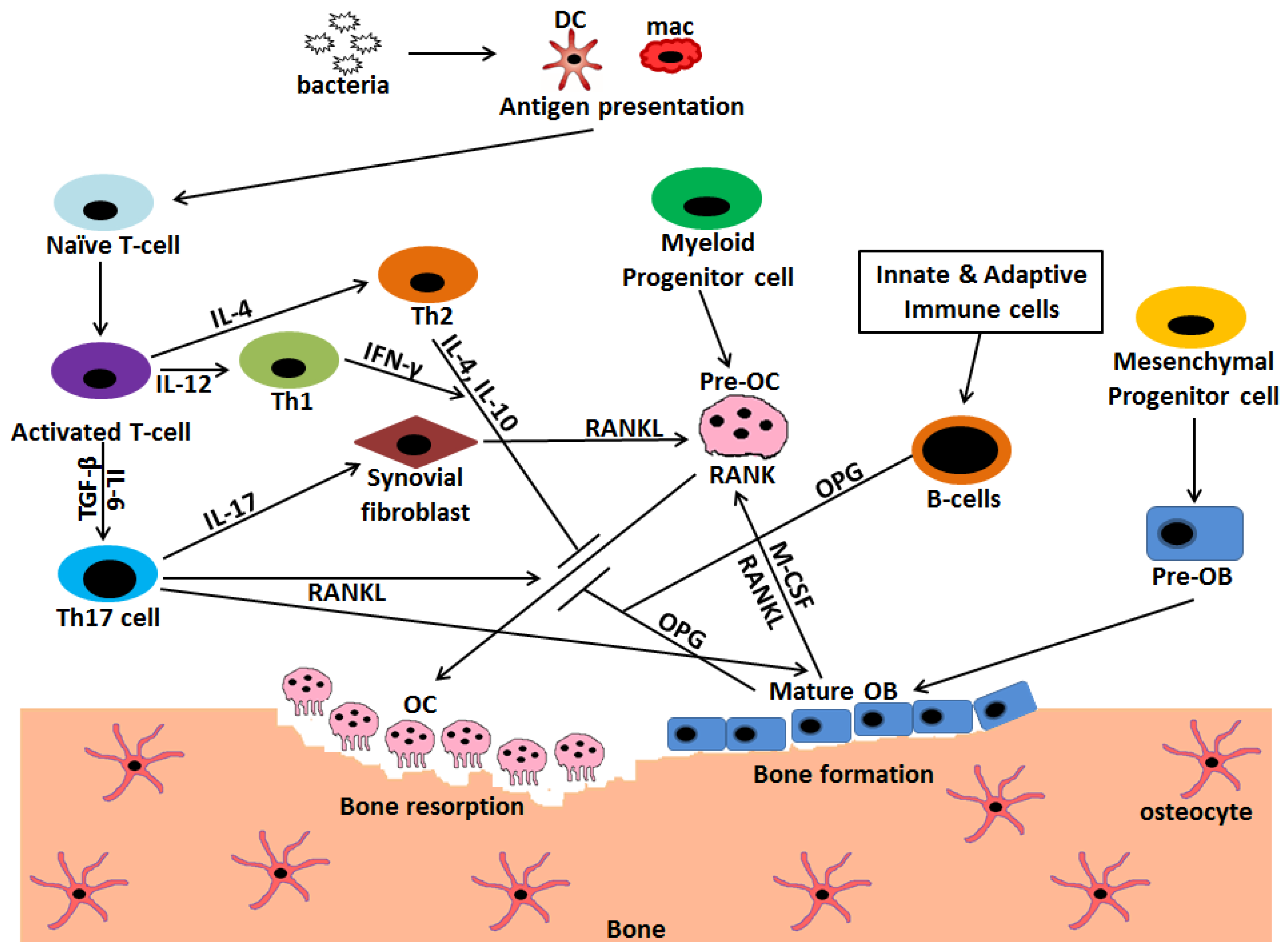
6. Infection Initiates the Cross-Talk between Osteoclasts, Osteoblast, and Immune Cells
7. Biofilm and Host Cell Interaction Leads to Loosening of Implants
8. Mimicking Function, a Strong Support Favoring Osteoimmunology
9. Innate and Adaptive Immune Cells Modulate Bone Resorption during Infection
9.1. T Cells
9.1.1. Tregs
9.1.2. Th17 Cells
9.1.3. T Cells Alter Costimulatory Molecules and Play a Protective Role in Bone Infection in Humans
9.2. B Cells
9.3. Neutrophils
9.4. Dendritic Cells
9.5. Macrophages
10. Treatment Approach for Bone Infection Is Limited
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Terashima, A.; Takayanagi, H. The role of bone cells in immune regulation during the course of infection. Semin Immunopathol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Gwyer, E.; Snelgrove, R.; Hussell, T. The therapeutic potential of positive and negative immune cell co-stimulation during inflammation. Biochem. Soc. Trans. 2006, 34, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Arron, J.R.; Choi, Y. Bone versus immune system. Nature 2000, 408, 535–536. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, M.; Takayanagi, H. Osteoimmunology: Evolving concepts in bone-immune interactions in health and disease. Nat. Rev. Immunol. 2019, 19, 626–642. [Google Scholar] [CrossRef]
- Takayanagi, H. Osteoimmunology and the effects of the immune system on bone. Nat. Rev. Rheumatol. 2009, 5, 667–676. [Google Scholar] [CrossRef]
- Bar-Shavit, Z. Taking a toll on the bones: Regulation of bone metabolism by innate immune regulators. Autoimmunity 2008, 41, 195–203. [Google Scholar] [CrossRef]
- Dapunt, U.; Radzuweit-Mihaljevic, S.; Lehner, B.; Haensch, G.M.; Ewerbeck, V. Bacterial Infection and Implant Loosening in Hip and Knee Arthroplasty: Evaluation of 209 Cases. Materials 2016, 9. [Google Scholar] [CrossRef]
- Sanderson, P.J. Infection in orthopaedic implants. J. Hosp. Infect. 1991, 18 (Suppl. A), 367–375. [Google Scholar] [CrossRef]
- Dudareva, M.; Hotchen, A.J.; Ferguson, J.; Hodgson, S.; Scarborough, M.; Atkins, B.L.; McNally, M.A. The microbiology of chronic osteomyelitis: Changes over ten years. J. Infect. 2019, 79, 189–198. [Google Scholar] [CrossRef]
- Darley, E.S.; MacGowan, A.P. Antibiotic treatment of gram-positive bone and joint infections. J. Antimicrob. Chemother. 2004, 53, 928–935. [Google Scholar] [CrossRef]
- Yang, D.; Wijenayaka, A.R.; Solomon, L.B.; Pederson, S.M.; Findlay, D.M.; Kidd, S.P.; Atkins, G.J. Novel Insights into Staphylococcus aureus Deep Bone Infections: The Involvement of Osteocytes. Microbiology 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Lew, D.P.; Waldvogel, F.A. Osteomyelitis. Lancet 2004, 364, 369–379. [Google Scholar] [CrossRef]
- Dojode, C.M.R.; Hemingway, J.S.; Damodaran, P.; Shah, N.N. Total hip arthroplasty infection caused by an unusual organism, Salmonella; its successful management and literature review. Bmj. Case Rep. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.J.; Glatt, A.E. Salmonella osteomyelitis and arthritis in sickle cell disease. Semin Arthritis Rheum. 1994, 24, 211–221. [Google Scholar] [CrossRef]
- Phillips, J.E.; Crane, T.P.; Noy, M.; Elliott, T.S.; Grimer, R.J. The incidence of deep prosthetic infections in a specialist orthopaedic hospital: A 15-year prospective survey. J. Bone Jt. Surg. Br. Vol. 2006, 88, 943–948. [Google Scholar] [CrossRef]
- Lentino, J.R. Prosthetic joint infections: Bane of orthopedists, challenge for infectious disease specialists. Clin. Infect. Dis. 2003, 36, 1157–1161. [Google Scholar] [CrossRef]
- Adhami, M.D.; Rashid, H.; Chen, H.; Clarke, J.C.; Yang, Y.; Javed, A. Loss of Runx2 in committed osteoblasts impairs postnatal skeletogenesis. J. Bone Miner. Res. 2015, 30, 71–82. [Google Scholar] [CrossRef]
- Komori, T.; Yagi, H.; Nomura, S.; Yamaguchi, A.; Sasaki, K.; Deguchi, K.; Shimizu, Y.; Bronson, R.T.; Gao, Y.H.; Inada, M.; et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997, 89, 755–764. [Google Scholar] [CrossRef]
- Kylmaoja, E.; Nakamura, M.; Tuukkanen, J. Osteoclasts and Remodeling Based Bone Formation. Curr. Stem Cell Res. 2016, 11, 626–633. [Google Scholar] [CrossRef]
- Teitelbaum, S.L.; Ross, F.P. Genetic regulation of osteoclast development and function. Nat. Rev. Genet. 2003, 4, 638–649. [Google Scholar] [CrossRef]
- Boyce, B.F. Advances in the regulation of osteoclasts and osteoclast functions. J. Dent. Res. 2013, 92, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Lacey, D.L.; Timms, E.; Tan, H.L.; Kelley, M.J.; Dunstan, C.R.; Burgess, T.; Elliott, R.; Colombero, A.; Elliott, G.; Scully, S.; et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998, 93, 165–176. [Google Scholar] [CrossRef]
- Raggatt, L.J.; Partridge, N.C. Cellular and molecular mechanisms of bone remodeling. J. Biol. Chem. 2010, 285, 25103–25108. [Google Scholar] [CrossRef] [PubMed]
- Teti, A. Bone development: Overview of bone cells and signaling. Curr. Osteoporos Rep. 2011, 9, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Klein-Nulend, J.; Bakker, A.D.; Bacabac, R.G.; Vatsa, A.; Weinbaum, S. Mechanosensation and transduction in osteocytes. Bone 2013, 54, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.P.; Utz, P.J.; Durand, D.B.; Toole, J.J.; Emmel, E.A.; Crabtree, G.R. Identification of a putative regulator of early T cell activation genes. Science 1988, 241, 202–205. [Google Scholar] [CrossRef]
- Aliprantis, A.O.; Ueki, Y.; Sulyanto, R.; Park, A.; Sigrist, K.S.; Sharma, S.M.; Ostrowski, M.C.; Olsen, B.R.; Glimcher, L.H. NFATc1 in mice represses osteoprotegerin during osteoclastogenesis and dissociates systemic osteopenia from inflammation in cherubism. J. Clin. Investig. 2008, 118, 3775–3789. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, P.; Li, W.; Liu, X.; Lu, Y.; Huang, Z.; Song, K. Calcineurin/NFAT signaling pathway mediates titanium particleinduced inflammation and osteoclast formation by inhibiting RANKL and MCSF in vitro. Mol. Med. Rep. 2017, 16, 8223–8230. [Google Scholar] [CrossRef]
- Macian, F.; Garcia-Cozar, F.; Im, S.H.; Horton, H.F.; Byrne, M.C.; Rao, A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell 2002, 109, 719–731. [Google Scholar] [CrossRef]
- Pan, M.; Winslow, M.M.; Chen, L.; Kuo, A.; Felsher, D.; Crabtree, G.R. Enhanced NFATc1 nuclear occupancy causes T cell activation independent of CD28 costimulation. J. Immunol. 2007, 178, 4315–4321. [Google Scholar] [CrossRef]
- Peng, S.L.; Gerth, A.J.; Ranger, A.M.; Glimcher, L.H. NFATc1 and NFATc2 together control both T and B cell activation and differentiation. Immunity 2001, 14, 13–20. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Deb, J.; Patra, A.K.; Thuy Pham, D.A.; Chen, W.; Vaeth, M.; Berberich-Siebelt, F.; Klein-Hessling, S.; Lamperti, E.D.; Reifenberg, K.; et al. NFATc1 affects mouse splenic B cell function by controlling the calcineurin--NFAT signaling network. J. Exp. Med. 2011, 208, 823–839. [Google Scholar] [CrossRef] [PubMed]
- Granucci, F.; Zanoni, I.; Pavelka, N.; Van Dommelen, S.L.; Andoniou, C.E.; Belardelli, F.; Degli Esposti, M.A.; Ricciardi-Castagnoli, P. A contribution of mouse dendritic cell-derived IL-2 for NK cell activation. J. Exp. Med. 2004, 200, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Jennings, C.; Kusler, B.; Jones, P.P. Calcineurin inactivation leads to decreased responsiveness to LPS in macrophages and dendritic cells and protects against LPS-induced toxicity in vivo. Innate Immun. 2009, 15, 109–120. [Google Scholar] [CrossRef]
- Lau, Y.S.; Wang, W.; Sabokbar, A.; Simpson, H.; Nair, S.; Henderson, B.; Berendt, A.; Athanasou, N.A. Staphylococcus aureus capsular material promotes osteoclast formation. Injury 2006, 37 (Suppl. 2), S41–S48. [Google Scholar] [CrossRef]
- Nair, S.; Song, Y.; Meghji, S.; Reddi, K.; Harris, M.; Ross, A.; Poole, S.; Wilson, M.; Henderson, B. Surface-associated proteins from Staphylococcus aureus demonstrate potent bone resorbing activity. J. Bone Miner. Res. 1995, 10, 726–734. [Google Scholar] [CrossRef]
- Alexander, E.H.; Bento, J.L.; Hughes, F.M., Jr.; Marriott, I.; Hudson, M.C.; Bost, K.L. Staphylococcus aureus and Salmonella enterica serovar Dublin induce tumor necrosis factor-related apoptosis-inducing ligand expression by normal mouse and human osteoblasts. Infect. Immun. 2001, 69, 1581–1586. [Google Scholar] [CrossRef]
- Bost, K.L.; Bento, J.L.; Ellington, J.K.; Marriott, I.; Hudson, M.C. Induction of colony-stimulating factor expression following Staphylococcus or Salmonella interaction with mouse or human osteoblasts. Infect. Immun. 2000, 68, 5075–5083. [Google Scholar] [CrossRef]
- Bost, K.L.; Bento, J.L.; Petty, C.C.; Schrum, L.W.; Hudson, M.C.; Marriott, I. Monocyte chemoattractant protein-1 expression by osteoblasts following infection with Staphylococcus aureus or Salmonella. J. Interferon Cytokine Res. 2001, 21, 297–304. [Google Scholar] [CrossRef]
- Kim, M.S.; Day, C.J.; Selinger, C.I.; Magno, C.L.; Stephens, S.R.; Morrison, N.A. MCP-1-induced human osteoclast-like cells are tartrate-resistant acid phosphatase, NFATc1, and calcitonin receptor-positive but require receptor activator of NFkappaB ligand for bone resorption. J. Biol. Chem. 2006, 281, 1274–1285. [Google Scholar] [CrossRef]
- Graves, D.T.; Jiang, Y.; Valente, A.J. Regulated expression of MCP-1 by osteoblastic cells in vitro and in vivo. Histol. Histopathol. 1999, 14, 1347–1354. [Google Scholar] [PubMed]
- Lampiasi, N.; Russo, R.; Zito, F. The Alternative Faces of Macrophage Generate Osteoclasts. Biomed Res. Int. 2016, 2016, 9089610. [Google Scholar] [CrossRef] [PubMed]
- Felix, R.; Cecchini, M.G.; Hofstetter, W.; Elford, P.R.; Stutzer, A.; Fleisch, H. Impairment of macrophage colony-stimulating factor production and lack of resident bone marrow macrophages in the osteopetrotic op/op mouse. J. Bone Miner. Res. 1990, 5, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Grigoriadis, A.E.; Wang, Z.Q.; Cecchini, M.G.; Hofstetter, W.; Felix, R.; Fleisch, H.A.; Wagner, E.F. c-Fos: A key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science 1994, 266, 443–448. [Google Scholar] [CrossRef]
- Tondravi, M.M.; McKercher, S.R.; Anderson, K.; Erdmann, J.M.; Quiroz, M.; Maki, R.; Teitelbaum, S.L. Osteopetrosis in mice lacking haematopoietic transcription factor PU.1. Nature 1997, 386, 81–84. [Google Scholar] [CrossRef]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef]
- Dougall, W.C.; Glaccum, M.; Charrier, K.; Rohrbach, K.; Brasel, K.; De Smedt, T.; Daro, E.; Smith, J.; Tometsko, M.E.; Maliszewski, C.R.; et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999, 13, 2412–2424. [Google Scholar] [CrossRef]
- Lo Iacono, N.; Blair, H.C.; Poliani, P.L.; Marrella, V.; Ficara, F.; Cassani, B.; Facchetti, F.; Fontana, E.; Guerrini, M.M.; Traggiai, E.; et al. Osteopetrosis rescue upon RANKL administration to Rankl(-/-) mice: A new therapy for human RANKL-dependent ARO. J. Bone Miner. Res. 2012, 27, 2501–2510. [Google Scholar] [CrossRef]
- Kong, Y.Y.; Yoshida, H.; Sarosi, I.; Tan, H.L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-dos-Santos, A.J.; Van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323. [Google Scholar] [CrossRef]
- Bucay, N.; Sarosi, I.; Dunstan, C.R.; Morony, S.; Tarpley, J.; Capparelli, C.; Scully, S.; Tan, H.L.; Xu, W.; Lacey, D.L.; et al. osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998, 12, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, A.; Kanno, T.; Hoshi, M.; Shibata, O.; Yano, K.; Fujise, N.; Kinosaki, M.; Yamaguchi, K.; Tsuda, E.; Murakami, A.; et al. Transgenic mice overexpressing soluble osteoclast differentiation factor (sODF) exhibit severe osteoporosis. J. Bone Miner. Metab. 2002, 20, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Koide, M.; Kobayashi, Y.; Ninomiya, T.; Nakamura, M.; Yasuda, H.; Arai, Y.; Okahashi, N.; Yoshinari, N.; Takahashi, N.; Udagawa, N. Osteoprotegerin-deficient male mice as a model for severe alveolar bone loss: Comparison with RANKL-overexpressing transgenic male mice. Endocrinology 2013, 154, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Del Fattore, A.; Teti, A. The tight relationship between osteoclasts and the immune system. Inflamm. Allergy Drug Targets. 2012, 11, 181–187. [Google Scholar] [CrossRef]
- Seebach, E.; Kubatzky, K.F. Chronic Implant-Related Bone Infections-Can Immune Modulation be a Therapeutic Strategy? Front. Immunol. 2019, 10, 1724. [Google Scholar] [CrossRef]
- Wright, J.A.; Nair, S.P. Interaction of staphylococci with bone. Int. J. Med. Microbiol. 2010, 300, 193–204. [Google Scholar] [CrossRef]
- Thurlow, L.R.; Hanke, M.L.; Fritz, T.; Angle, A.; Aldrich, A.; Williams, S.H.; Engebretsen, I.L.; Bayles, K.W.; Horswill, A.R.; Kielian, T. Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J. Immunol. 2011, 186, 6585–6596. [Google Scholar] [CrossRef]
- Kumar, G.; Roger, P.M.; Ticchioni, M.; Trojani, C.; Bernard de Dompsur, R.; Bronsard, N.; Carles, M.; Bernard, E. T cells from chronic bone infection show reduced proliferation and a high proportion of CD28(-) CD4 T cells. Clin. Exp. Immunol. 2014, 176, 49–57. [Google Scholar] [CrossRef]
- Wagner, C.; Heck, D.; Lautenschlager, K.; Iking-Konert, C.; Heppert, V.; Wentzensen, A.; Hansch, G.M. T lymphocytes in implant-associated posttraumatic osteomyelitis: Identification of cytotoxic T effector cells at the site of infection. Shock 2006, 25, 241–246. [Google Scholar] [CrossRef]
- Li, H.; Hong, S.; Qian, J.; Zheng, Y.; Yang, J.; Yi, Q. Cross talk between the bone and immune systems: Osteoclasts function as antigen-presenting cells and activate CD4+ and CD8+ T cells. Blood 2010, 116, 210–217. [Google Scholar] [CrossRef]
- Kiesel, J.R.; Buchwald, Z.S.; Aurora, R. Cross-presentation by osteoclasts induces FoxP3 in CD8+ T cells. J. Immunol. 2009, 182, 5477–5487. [Google Scholar] [CrossRef] [PubMed]
- Buchwald, Z.S.; Kiesel, J.R.; Yang, C.; DiPaolo, R.; Novack, D.V.; Aurora, R. Osteoclast-induced Foxp3+ CD8 T-cells limit bone loss in mice. Bone 2013, 56, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Axmann, R.; Herman, S.; Zaiss, M.; Franz, S.; Polzer, K.; Zwerina, J.; Herrmann, M.; Smolen, J.; Schett, G. CTLA-4 directly inhibits osteoclast formation. Ann. Rheum. Dis. 2008, 67, 1603–1609. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.P.; Deng, T.Z.; Witherden, D.A.; Goldrath, A.W. Origins of CD4(+) circulating and tissue-resident memory T-cells. Immunology 2019, 157, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Haas, W.; Pereira, P.; Tonegawa, S. Gamma/delta cells. Annu. Rev. Immunol. 1993, 11, 637–685. [Google Scholar] [CrossRef]
- Li, Y.; Toraldo, G.; Li, A.; Yang, X.; Zhang, H.; Qian, W.P.; Weitzmann, M.N. B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood 2007, 109, 3839–3848. [Google Scholar] [CrossRef]
- John, V.; Hock, J.M.; Short, L.L.; Glasebrook, A.L.; Galvin, R.J. A role for CD8+ T lymphocytes in osteoclast differentiation in vitro. Endocrinology 1996, 137, 2457–2463. [Google Scholar] [CrossRef]
- Grcevic, D.; Lee, S.K.; Marusic, A.; Lorenzo, J.A. Depletion of CD4 and CD8 T lymphocytes in mice in vivo enhances 1,25-dihydroxyvitamin D3-stimulated osteoclast-like cell formation in vitro by a mechanism that is dependent on prostaglandin synthesis. J. Immunol. 2000, 165, 4231–4238. [Google Scholar] [CrossRef]
- Hao, L.; Zhu, G.; Lu, Y.; Wang, M.; Jules, J.; Zhou, X.; Chen, W. Deficiency of cathepsin K prevents inflammation and bone erosion in rheumatoid arthritis and periodontitis and reveals its shared osteoimmune role. Febs. Lett. 2015, 589, 1331–1339. [Google Scholar] [CrossRef]
- Bromley, M.; Woolley, D.E. Chondroclasts and osteoclasts at subchondral sites of erosion in the rheumatoid joint. Arthritis Rheum. 1984, 27, 968–975. [Google Scholar] [CrossRef]
- Colucci, S.; Brunetti, G.; Cantatore, F.P.; Oranger, A.; Mori, G.; Quarta, L.; Cirulli, N.; Mancini, L.; Corrado, A.; Grassi, F.R.; et al. Lymphocytes and synovial fluid fibroblasts support osteoclastogenesis through RANKL, TNFalpha, and IL-7 in an in vitro model derived from human psoriatic arthritis. J. Pathol. 2007, 212, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Takayanagi, H. Osteoclasts, rheumatoid arthritis, and osteoimmunology. Curr. Opin. Rheumatol. 2006, 18, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Manoury-Schwartz, B.; Chiocchia, G.; Bessis, N.; Abehsira-Amar, O.; Batteux, F.; Muller, S.; Huang, S.; Boissier, M.C.; Fournier, C. High susceptibility to collagen-induced arthritis in mice lacking IFN-gamma receptors. J. Immunol. 1997, 158, 5501–5506. [Google Scholar] [PubMed]
- Takayanagi, H.; Ogasawara, K.; Hida, S.; Chiba, T.; Murata, S.; Sato, K.; Takaoka, A.; Yokochi, T.; Oda, H.; Tanaka, K.; et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature 2000, 408, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Suematsu, A.; Okamoto, K.; Yamaguchi, A.; Morishita, Y.; Kadono, Y.; Tanaka, S.; Kodama, T.; Akira, S.; Iwakura, Y.; et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J. Exp. Med. 2006, 203, 2673–2682. [Google Scholar] [CrossRef]
- Prabhakara, R.; Harro, J.M.; Leid, J.G.; Harris, M.; Shirtliff, M.E. Murine immune response to a chronic Staphylococcus aureus biofilm infection. Infect. Immun. 2011, 79, 1789–1796. [Google Scholar] [CrossRef]
- Luo, C.Y.; Wang, L.; Sun, C.; Li, D.J. Estrogen enhances the functions of CD4(+).CD25(+).Foxp3(+) regulatory T cells that suppress osteoclast differentiation and bone resorption in vitro. Cell. Mol. Immunol. 2011, 8, 50–58. [Google Scholar] [CrossRef]
- Wing, K.; Yamaguchi, T.; Sakaguchi, S. Cell-autonomous and -non-autonomous roles of CTLA-4 in immune regulation. Trends Immunol. 2011, 32, 428–433. [Google Scholar] [CrossRef]
- Zaiss, M.M.; Axmann, R.; Zwerina, J.; Polzer, K.; Guckel, E.; Skapenko, A.; Schulze-Koops, H.; Horwood, N.; Cope, A.; Schett, G. Treg cells suppress osteoclast formation: A new link between the immune system and bone. Arthritis Rheum. 2007, 56, 4104–4112. [Google Scholar] [CrossRef]
- Volpe, E.; Servant, N.; Zollinger, R.; Bogiatzi, S.I.; Hupe, P.; Barillot, E.; Soumelis, V. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat. Immunol. 2008, 9, 650–657. [Google Scholar] [CrossRef]
- Adamopoulos, I.E.; Bowman, E.P. Immune regulation of bone loss by Th17 cells. Arthritis Res. Ther. 2008, 10, 225. [Google Scholar] [CrossRef] [PubMed]
- LeBien, T.W.; Tedder, T.F. B lymphocytes: How they develop and function. Blood 2008, 112, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Ghia, P.; ten Boekel, E.; Rolink, A.G.; Melchers, F. B-cell development: A comparison between mouse and man. Immunol. Today. 1998, 19, 480–485. [Google Scholar] [CrossRef]
- Koni, P.A.; Joshi, S.K.; Temann, U.A.; Olson, D.; Burkly, L.; Flavell, R.A. Conditional vascular cell adhesion molecule 1 deletion in mice: Impaired lymphocyte migration to bone marrow. J. Exp. Med. 2001, 193, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Yun, T.J.; Tallquist, M.D.; Aicher, A.; Rafferty, K.L.; Marshall, A.J.; Moon, J.J.; Ewings, M.E.; Mohaupt, M.; Herring, S.W.; Clark, E.A. Osteoprotegerin, a crucial regulator of bone metabolism, also regulates B cell development and function. J. Immunol. 2001, 166, 1482–1491. [Google Scholar] [CrossRef]
- Manabe, N.; Kawaguchi, H.; Chikuda, H.; Miyaura, C.; Inada, M.; Nagai, R.; Nabeshima, Y.; Nakamura, K.; Sinclair, A.M.; Scheuermann, R.H.; et al. Connection between B lymphocyte and osteoclast differentiation pathways. J. Immunol. 2001, 167, 2625–2631. [Google Scholar] [CrossRef]
- Coat, J.; Demoersman, J.; Beuzit, S.; Cornec, D.; Devauchelle-Pensec, V.; Saraux, A.; Pers, J.O. Anti-B lymphocyte immunotherapy is associated with improvement of periodontal status in subjects with rheumatoid arthritis. J. Clin. Periodontol. 2015, 42, 817–823. [Google Scholar] [CrossRef]
- Oliver-Bell, J.; Butcher, J.P.; Malcolm, J.; MacLeod, M.K.; Adrados Planell, A.; Campbell, L.; Nibbs, R.J.; Garside, P.; McInnes, I.B.; Culshaw, S. Periodontitis in the absence of B cells and specific anti-bacterial antibody. Mol. Oral Microbiol. 2015, 30, 160–169. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, X.; Lin, J.; Hu, Y.; Zhao, Q.; Kawai, T.; Taubman, M.A.; Han, X. B10 Cells Alleviate Periodontal Bone Loss in Experimental Periodontitis. Infect. Immun. 2017, 85. [Google Scholar] [CrossRef]
- Demoersman, J.; Pochard, P.; Framery, C.; Simon, Q.; Boisrame, S.; Soueidan, A.; Pers, J.O. B cell subset distribution is altered in patients with severe periodontitis. PLoS ONE 2018, 13, e0192986. [Google Scholar] [CrossRef]
- Hajishengallis, E.; Hajishengallis, G. Neutrophil homeostasis and periodontal health in children and adults. J. Dent. Res. 2014, 93, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Prichard, J.F.; Ferguson, D.M.; Windmiller, J.; Hurt, W.C. Prepubertal periodontitis affecting the deciduous and permanent dentition in a patient with cyclic neutropenia. A case report and discussion. J. Periodontol. 1984, 55, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, D.; Kagari, T.; Doi, H.; Shimozato, T. Essential role of neutrophils in anti-type II collagen antibody and lipopolysaccharide-induced arthritis. Immunology 2006, 119, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Kraan, M.C.; de Koster, B.M.; Elferink, J.G.; Post, W.J.; Breedveld, F.C.; Tak, P.P. Inhibition of neutrophil migration soon after initiation of treatment with leflunomide or methotrexate in patients with rheumatoid arthritis: Findings in a prospective, randomized, double-blind clinical trial in fifteen patients. Arthritis Rheum. 2000, 43, 1488–1495. [Google Scholar] [CrossRef]
- Chakravarti, A.; Raquil, M.A.; Tessier, P.; Poubelle, P.E. Surface RANKL of Toll-like receptor 4-stimulated human neutrophils activates osteoclastic bone resorption. Blood 2009, 114, 1633–1644. [Google Scholar] [CrossRef]
- Navegantes, K.C.; de Souza Gomes, R.; Pereira, P.A.T.; Czaikoski, P.G.; Azevedo, C.H.M.; Monteiro, M.C. Immune modulation of some autoimmune diseases: The critical role of macrophages and neutrophils in the innate and adaptive immunity. J. Transl. Med. 2017, 15, 36. [Google Scholar] [CrossRef]
- Tak, P.P.; Smeets, T.J.; Daha, M.R.; Kluin, P.M.; Meijers, K.A.; Brand, R.; Meinders, A.E.; Breedveld, F.C. Analysis of the synovial cell infiltrate in early rheumatoid synovial tissue in relation to local disease activity. Arthritis Rheum. 1997, 40, 217–225. [Google Scholar] [CrossRef]
- Bastian, O.W.; Croes, M.; Alblas, J.; Koenderman, L.; Leenen, L.P.H.; Blokhuis, T.J. Neutrophils Inhibit Synthesis of Mineralized Extracellular Matrix by Human Bone Marrow-Derived Stromal Cells In Vitro. Front. Immunol. 2018, 9, 945. [Google Scholar] [CrossRef]
- Rivollier, A.; Mazzorana, M.; Tebib, J.; Piperno, M.; Aitsiselmi, T.; Rabourdin-Combe, C.; Jurdic, P.; Servet-Delprat, C. Immature dendritic cell transdifferentiation into osteoclasts: A novel pathway sustained by the rheumatoid arthritis microenvironment. Blood 2004, 104, 4029–4037. [Google Scholar] [CrossRef]
- Alnaeeli, M.; Park, J.; Mahamed, D.; Penninger, J.M.; Teng, Y.T. Dendritic cells at the osteo-immune interface: Implications for inflammation-induced bone loss. J. Bone Miner. Res. 2007, 22, 775–780. [Google Scholar] [CrossRef]
- Leung, B.P.; Conacher, M.; Hunter, D.; McInnes, I.B.; Liew, F.Y.; Brewer, J.M. A novel dendritic cell-induced model of erosive inflammatory arthritis: Distinct roles for dendritic cells in T cell activation and induction of local inflammation. J. Immunol. 2002, 169, 7071–7077. [Google Scholar] [CrossRef] [PubMed]
- Alnaeeli, M.; Penninger, J.M.; Teng, Y.T. Immune interactions with CD4+ T cells promote the development of functional osteoclasts from murine CD11c+ dendritic cells. J. Immunol. 2006, 177, 3314–3326. [Google Scholar] [CrossRef] [PubMed]
- Arizon, M.; Nudel, I.; Segev, H.; Mizraji, G.; Elnekave, M.; Furmanov, K.; Eli-Berchoer, L.; Clausen, B.E.; Shapira, L.; Wilensky, A.; et al. Langerhans cells down-regulate inflammation-driven alveolar bone loss. Proc. Natl. Acad. Sci. USA 2012, 109, 7043–7048. [Google Scholar] [CrossRef] [PubMed]
- Bitar, D.; Parvizi, J. Biological response to prosthetic debris. World J. Orthop. 2015, 6, 172–189. [Google Scholar] [CrossRef] [PubMed]
- Pajarinen, J.; Kouri, V.P.; Jamsen, E.; Li, T.F.; Mandelin, J.; Konttinen, Y.T. The response of macrophages to titanium particles is determined by macrophage polarization. Acta. Biomater. 2013, 9, 9229–9240. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.S. Management of bone and joint infections due to Staphylococcus aureus. Intern. Med. J. 2005, 35 (Suppl. 2), S79–S96. [Google Scholar] [CrossRef] [PubMed]
- Conterno, L.O.; Turchi, M.D. Antibiotics for treating chronic osteomyelitis in adults. Cochrane Database Syst. Rev. 2013, CD004439. [Google Scholar] [CrossRef]
- Finzel, S.; Rech, J.; Schmidt, S.; Engelke, K.; Englbrecht, M.; Schett, G. Interleukin-6 receptor blockade induces limited repair of bone erosions in rheumatoid arthritis: A micro CT study. Ann. Rheum. Dis. 2013, 72, 396–400. [Google Scholar] [CrossRef]
- Finzel, S.; Rech, J.; Schmidt, S.; Engelke, K.; Englbrecht, M.; Stach, C.; Schett, G. Repair of bone erosions in rheumatoid arthritis treated with tumour necrosis factor inhibitors is based on bone apposition at the base of the erosion. Ann. Rheum. Dis. 2011, 70, 1587–1593. [Google Scholar] [CrossRef]
- Redlich, K.; Gortz, B.; Hayer, S.; Zwerina, J.; Doerr, N.; Kostenuik, P.; Bergmeister, H.; Kollias, G.; Steiner, G.; Smolen, J.S.; et al. Repair of local bone erosions and reversal of systemic bone loss upon therapy with anti-tumor necrosis factor in combination with osteoprotegerin or parathyroid hormone in tumor necrosis factor-mediated arthritis. Am. J. Pathol. 2004, 164, 543–555. [Google Scholar] [CrossRef]


 Stimulation;
Stimulation;  Inhibition.
Stimulation; Inhibition.
Inhibition.
Stimulation; Inhibition.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells | Cytokines and Mediators | Effect on Osteoclastogenesis |
|---|---|---|
| Th1 | INF-γ | Inhibits |
| TNF-α | Supports | |
| Th2 | IL-4 | Inhibits |
| Th17 | RANKL | Supports |
| IL-17 | Supports | |
| Tregs | IL-10 | Inhibits |
| CTLA-4 | Inhibits | |
| B cells | OPG | Inhibits |
| Osteoblasts | RANKL | Supports |
| M-CSF | Supports | |
| OPG | Inhibits |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, G.; Roger, P.-M. From Crosstalk between Immune and Bone Cells to Bone Erosion in Infection. Int. J. Mol. Sci. 2019, 20, 5154. https://doi.org/10.3390/ijms20205154
Kumar G, Roger P-M. From Crosstalk between Immune and Bone Cells to Bone Erosion in Infection. International Journal of Molecular Sciences. 2019; 20(20):5154. https://doi.org/10.3390/ijms20205154
Chicago/Turabian StyleKumar, Gaurav, and Pierre-Marie Roger. 2019. "From Crosstalk between Immune and Bone Cells to Bone Erosion in Infection" International Journal of Molecular Sciences 20, no. 20: 5154. https://doi.org/10.3390/ijms20205154
APA StyleKumar, G., & Roger, P.-M. (2019). From Crosstalk between Immune and Bone Cells to Bone Erosion in Infection. International Journal of Molecular Sciences, 20(20), 5154. https://doi.org/10.3390/ijms20205154