Functional Consequences of the SCN5A-p.Y1977N Mutation within the PY Ubiquitylation Motif: Discrepancy between HEK293 Cells and Transgenic Mice

 , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Case Report
2.2. The SCN5A-p.Y1977N Mutation Abolishes the Interaction between Nav1.5 and Nedd4-2 and Nedd4-2-Dependent Ubiquitylation of Nav1.5 in HEK293 Cells
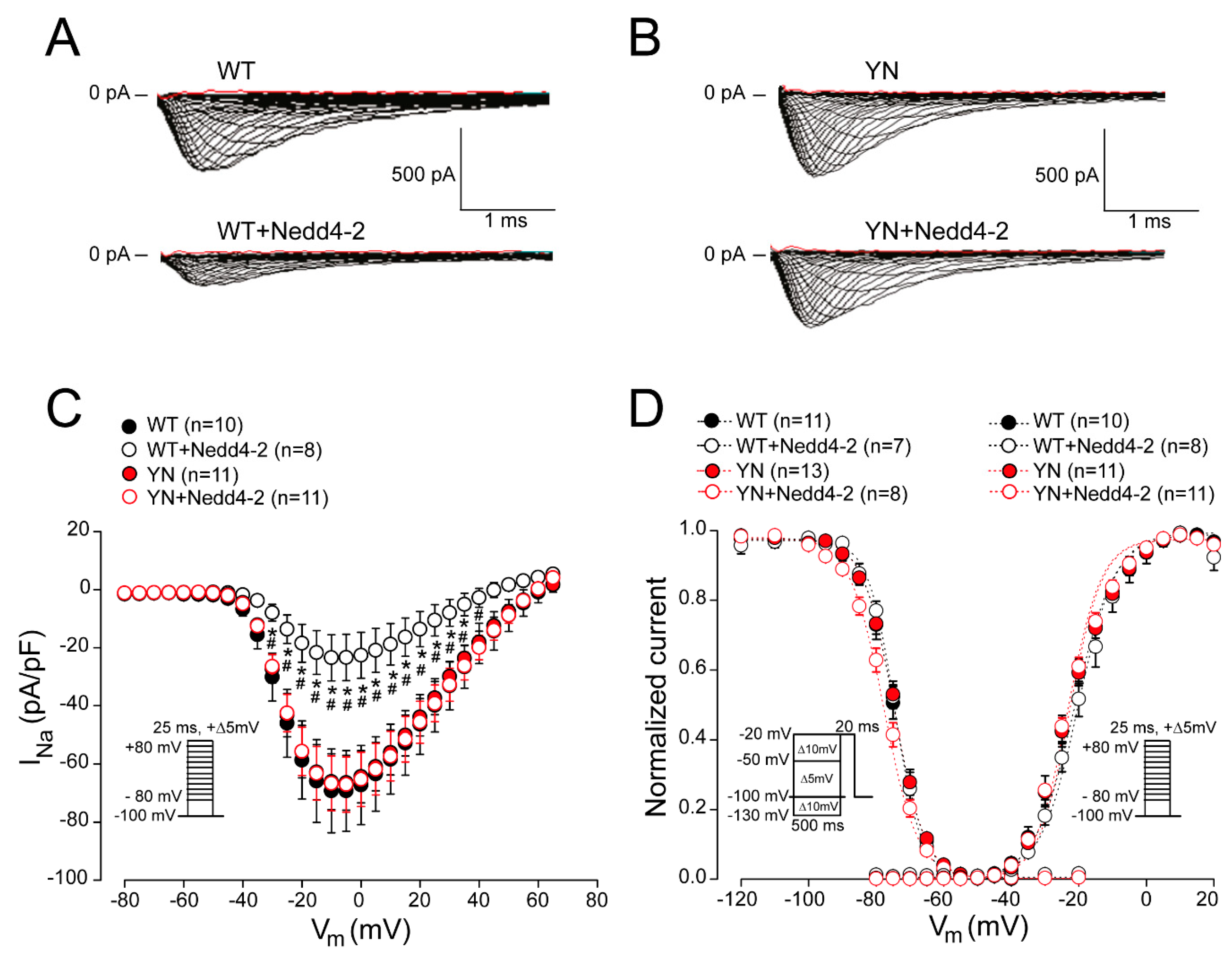
2.3. Nedd4-2-Dependent Regulation of Sodium Current is Abrogated by the SCN5A-p.Y1977N Mutation in HEK293 Cells
2.4. Cell Surface Expression of Nav1.5 in HEK293 Cells is Altered by the SCN5A-p.Y1977N Mutation
2.5. Scn5a-p.Y1981N Mice Show Unaltered Cardiac Electrical Properties In Vivo and Ex Vivo
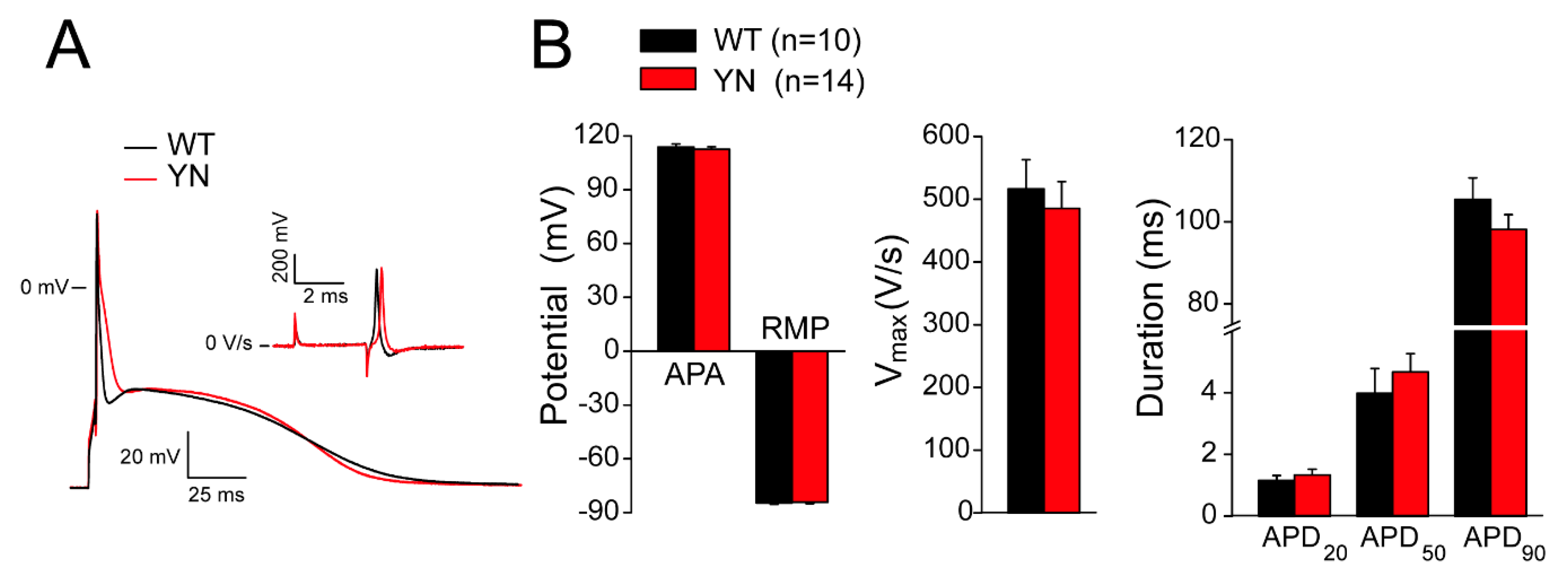
2.6. Action Potential and (Late) Sodium Current Characteristics Are Unaltered in Isolated Scn5a-p.Y1981N Cardiomyocytes
3. Discussion
3.1. PY-motif-Dependent Ubiquitylation of Nav1.5: In Vitro Evidence
3.2. In Vivo Relevance of PY-motif-Dependent Ubiquitylation
3.3. Lack of In Vivo Phenotype in Scn5a-p.Y1981N Mice: Potential Underlying Mechanisms
4. Materials and Methods
4.1. Ethical Statements
4.2. Cell Culture and Molecular Analyses
4.2.1. Cell Culture
4.2.2. Quantitative RT-PCR
4.2.3. Western Blots
4.2.4. Pull-Down Assays.
4.2.5. Cell Surface Biotinylation Assay
4.2.6. Antibodies
4.3. Generation of Homozygous Scn5a-p.Y1981N (YN) Mice
4.4. Surface ECG Analysis
4.5. Epicardial Mapping in Isolated Hearts
4.6. Patch-Clamp Measurements
4.6.1. Isolation of Left Ventricular Cardiomyocytes
4.6.2. Data Acquisition and Analysis
4.6.3. Sodium Current Measurements
4.6.4. Action Potential Measurements
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AP | Action potential |
| APD | Action potential duration |
| APD20 | Action potential duration at 20% repolarization |
| APD50 | Action potential duration at 50% repolarization |
| APD80 | Action potential duration at 80% repolarization |
| APD90 | Action potential duration at 90% repolarization |
| APA | Action potential amplitude |
| Vmax | Maximal upstroke velocity |
| RMP | Resting membrane potential |
| LQT3 | Long QT syndrome type 3 |
| INaL | Late sodium current |
| INa | Sodium current |
| BrS | Brugada syndrome |
| CCD | Cardiac conduction disease |
| SCD | Sudden cardiac death |
| ICD | Implantable cardioverter defibrillator |
| WT | Wild-type |
| TTX | Tetrodotoxin |
| hiPSC-CMs | Human induced pluripotent stem cell-derived cardiomyocytes |
| V1/2 | Half-maximal voltage of (in)activation |
| CVs | Conduction velocities |
| τf | Fast time constant of recovery from inactivation |
| τs | Slow time constant of recovery from inactivation |
References
- Remme, C.A.; Bezzina, C.R. Sodium channel (dys)function and cardiac arrhythmias. Cardiovasc. Ther. 2010, 28, 287–294. [Google Scholar] [CrossRef]
- Remme, C.A. Cardiac sodium channelopathy associated with SCN5A mutations: Electrophysiological, molecular and genetic aspects. J. Physiol. 2013, 591, 4099–4116. [Google Scholar] [CrossRef]
- Bezzina, C.R.; Remme, C.A. Dilated cardiomyopathy due to sodium channel dysfunction: What is the connection? Circ. Arrhythm. Electrophysiol. 2008, 1, 80–82. [Google Scholar] [CrossRef]
- Remme, C.A.; Wilde, A.A. Late sodium current inhibition in acquired and inherited ventricular (dys)function and arrhythmias. Cardiovasc. Drugs Ther. 2013, 27, 91–101. [Google Scholar] [CrossRef]
- Portero, V.; Casini, S.; Hoekstra, M.; Verkerk, A.O.; Mengarelli, I.; Belardinelli, L.; Rajamani, S.; Wilde, A.A.M.; Bezzina, C.R.; Veldkamp, M.W.; et al. Anti-arrhythmic potential of the late sodium current inhibitor GS-458967 in murine Scn5a-1798insD+/- and human SCN5A-1795insD+/- iPSC-derived cardiomyocytes. Cardiovasc. Res. 2017, 113, 829–838. [Google Scholar] [CrossRef]
- Rougier, J.S.; Albesa, M.; Abriel, H. Ubiquitylation and SUMOylation of cardiac ion channels. J. Cardiovasc. Pharmacol. 2010, 56, 22–28. [Google Scholar] [CrossRef]
- Ohtake, F.; Tsuchiya, H. The emerging complexity of ubiquitin architecture. J. Biochem. 2017, 161, 125–133. [Google Scholar] [CrossRef]
- Staub, O.; Abriel, H.; Plant, P.; Ishikawa, T.; Kanelis, V.; Saleki, R.; Horisberger, J.D.; Schild, L.; Rotin, D. Regulation of the epithelial Na+ channel by Nedd4 and ubiquitination. Kidney Int. 2000, 57, 809–815. [Google Scholar] [CrossRef]
- Van Bemmelen, M.X.; Rougier, J.S.; Gavillet, B.; Apotheloz, F.; Daidie, D.; Tateyama, M.; Rivolta, I.; Thomas, M.A.; Kass, R.S.; Staub, O.; et al. Cardiac voltage-gated sodium channel Nav1.5 is regulated by Nedd4-2 mediated ubiquitination. Circ. Res. 2004, 95, 284–291. [Google Scholar] [CrossRef]
- Rougier, J.S.; van Bemmelen, M.X.; Bruce, M.C.; Jespersen, T.; Gavillet, B.; Apotheloz, F.; Cordonier, S.; Staub, O.; Rotin, D.; Abriel, H. Molecular determinants of voltage-gated sodium channel regulation by the Nedd4/Nedd4-like proteins. Am. J. Physiol. Cell Physiol. 2005, 288, C692–C701. [Google Scholar] [CrossRef]
- Casini, S.; Verkerk, A.O.; Remme, C.A. Human iPSC-Derived Cardiomyocytes for Investigation of Disease Mechanisms and Therapeutic Strategies in Inherited Arrhythmia Syndromes: Strengths and Limitations. Cardiovasc. Drugs Ther. 2017, 31, 325–344. [Google Scholar] [CrossRef]
- Deveraux, Q.; Ustrell, V.; Pickart, C.; Rechsteiner, M. A 26 S protease subunit that binds ubiquitin conjugates. J. Biol. Chem. 1994, 269, 7059–7061. [Google Scholar]
- Kapplinger, J.D.; Tester, D.J.; Salisbury, B.A.; Carr, J.L.; Harris-Kerr, C.; Pollevick, G.D.; Wilde, A.A.; Ackerman, M.J. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm. 2009, 6, 1297–1303. [Google Scholar] [CrossRef]
- Laedermann, C.J.; Decosterd, I.; Abriel, H. Ubiquitylation of voltage-gated sodium channels. Handb. Exp. Pharmacol. 2014, 221, 231–250. [Google Scholar]
- Jespersen, T.; Membrez, M.; Nicolas, C.S.; Pitard, B.; Staub, O.; Olesen, S.P.; Baro, I.; Abriel, H. The KCNQ1 potassium channel is down-regulated by ubiquitylating enzymes of the Nedd4/Nedd4-like family. Cardiovasc. Res. 2007, 74, 64–74. [Google Scholar] [CrossRef]
- Fotia, A.B.; Ekberg, J.; Adams, D.J.; Cook, D.I.; Poronnik, P.; Kumar, S. Regulation of neuronal voltage-gated sodium channels by the ubiquitin-protein ligases Nedd4 and Nedd4-2. J. Biol. Chem. 2004, 279, 28930–28935. [Google Scholar] [CrossRef]
- Laedermann, C.J.; Cachemaille, M.; Kirschmann, G.; Pertin, M.; Gosselin, R.D.; Chang, I.; Albesa, M.; Towne, C.; Schneider, B.L.; Kellenberger, S.; et al. Dysregulation of voltage-gated sodium channels by ubiquitin ligase NEDD4-2 in neuropathic pain. J. Clin. Invest. 2013, 123, 3002–3013. [Google Scholar] [CrossRef]
- Lu, C.; Pribanic, S.; Debonneville, A.; Jiang, C.; Rotin, D. The PY-motif of ENaC, mutated in Liddle syndrome, regulates channel internalization, sorting and mobilization from subapical pool. Traffic 2007, 8, 1246–1264. [Google Scholar] [CrossRef]
- Butterworth, M.B. Regulation of the epithelial sodium channel (ENaC) by membrane trafficking. Biochim. Biophys. Acta 2010, 1802, 1166–1177. [Google Scholar] [CrossRef]
- Abriel, H.; Loffing, J.; Rebhun, J.F.; Pratt, J.H.; Schild, L.; Horisberger, J.D.; Rotin, D.; Staub, O. Defective regulation of the epithelial Na+ channel by Nedd4 in Liddle’s syndrome. J. Clin. Invest. 1999, 103, 667–673. [Google Scholar] [CrossRef]
- Staub, O.; Gautschi, I.; Ishikawa, T.; Breitschopf, K.; Ciechanover, A.; Schild, L.; Rotin, D. Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 1997, 16, 6325–6336. [Google Scholar] [CrossRef]
- Shi, P.P.; Cao, X.R.; Sweezer, E.M.; Kinney, T.S.; Williams, N.R.; Husted, R.F.; Nair, R.; Weiss, R.M.; Williamson, R.A.; Sigmund, C.D.; et al. Salt-sensitive hypertension and cardiac hypertrophy in mice deficient in the ubiquitin ligase Nedd4-2. Am. J. Physiol. Renal Physiol. 2008, 295, F462–F470. [Google Scholar] [CrossRef]
- Luo, L.; Ning, F.; Du, Y.; Song, B.; Yang, D.; Salvage, S.C.; Wang, Y.; Fraser, J.A.; Zhang, S.; Ma, A.; et al. Calcium-dependent Nedd4-2 upregulation mediates degradation of the cardiac sodium channel Nav1.5: Implications for heart failure. Acta. Physiol. 2017, 221, 44–58. [Google Scholar] [CrossRef]
- Minegishi, S.; Ishigami, T.; Kawamura, H.; Kino, T.; Chen, L.; Nakashima-Sasaki, R.; Doi, H.; Azushima, K.; Wakui, H.; Chiba, Y.; et al. An Isoform of Nedd4-2 Plays a Pivotal Role in Electrophysiological Cardiac Abnormalities. Int. J. Mol. Sci. 2017, 18, 1268. [Google Scholar] [CrossRef]
- Lu, P.J.; Zhou, X.Z.; Shen, M.H.; Lu, K.P. Function of WW domains as phosphoserine- or phosphothreonine-binding modules. Science 1999, 283, 1325–1328. [Google Scholar] [CrossRef]
- Bedford, M.T.; Reed, R.; Leder, P. WW domain-mediated interactions reveal a spliceosome-associated protein that binds a third class of proline-rich motif: The proline glycine and methionine-rich motif. Proc. Natl. Acad. Sci. USA 1998, 95, 10602–10607. [Google Scholar] [CrossRef]
- Bedford, M.T.; Chan, D.C.; Leder, P. FBP WW domains and the Abl SH3 domain bind to a specific class of proline-rich ligands. EMBO J. 1997, 16, 2376–2383. [Google Scholar] [CrossRef]
- Ekberg, J.; Schuetz, F.; Boase, N.A.; Conroy, S.J.; Manning, J.; Kumar, S.; Poronnik, P.; Adams, D.J. Regulation of the voltage-gated K+ channels KCNQ2/3 and KCNQ3/5 by ubiquitination. Novel role for Nedd4-2. J. Biol. Chem. 2007, 282, 12135–12142. [Google Scholar] [CrossRef]
- Henke, G.; Maier, G.; Wallisch, S.; Boehmer, C.; Lang, F. Regulation of the voltage gated K+ channel Kv1.3 by the ubiquitin ligase Nedd4-2 and the serum and glucocorticoid inducible kinase SGK1. J. Cell Physiol. 2004, 199, 194–199. [Google Scholar] [CrossRef]
- Kuryshev, Y.A.; Wible, B.A.; Gudz, T.I.; Ramirez, A.N.; Brown, A.M. KChAP/Kvbeta1.2 interactions and their effects on cardiac Kv channel expression. Am. J. Physiol. Cell Physiol. 2001, 281, C290–C299. [Google Scholar] [CrossRef]
- Krueger, B.; Yang, L.; Korbmacher, C.; Rauh, R. The phosphorylation site T613 in the beta-subunit of rat epithelial Na+ channel (ENaC) modulates channel inhibition by Nedd4-2. Pflugers Arch. 2018, 470, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Goel, P.; Manning, J.A.; Kumar, S. NEDD4-2 (NEDD4L): The ubiquitin ligase for multiple membrane proteins. Gene 2015, 557, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, M.; Vermij, S.H.; Wyler, K.; Gillet, L.; Keller, I.; Abriel, H. Transcriptomic analyses of murine ventricular cardiomyocytes. Sci. Data 2018, 5, 180170. [Google Scholar] [CrossRef] [PubMed]
- Remme, C.A.; Verkerk, A.O.; Nuyens, D.; van Ginneken, A.C.; van Brunschot, S.; Belterman, C.N.; Wilders, R.; van Roon, M.A.; Tan, H.L.; Wilde, A.A.; et al. Overlap syndrome of cardiac sodium channel disease in mice carrying the equivalent mutation of human SCN5A-1795insD. Circulation 2006, 114, 2584–2594. [Google Scholar] [CrossRef]
- Mitchell, G.F.; Jeron, A.; Koren, G. Measurement of heart rate and Q-T interval in the conscious mouse. Am. J. Physiol. 1998, 274, H747–H751. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casini, S.; Albesa, M.; Wang, Z.; Portero, V.; Ross-Kaschitza, D.; Rougier, J.-S.; Marchal, G.A.; Chung, W.K.; Bezzina, C.R.; Abriel, H.; et al. Functional Consequences of the SCN5A-p.Y1977N Mutation within the PY Ubiquitylation Motif: Discrepancy between HEK293 Cells and Transgenic Mice. Int. J. Mol. Sci. 2019, 20, 5033. https://doi.org/10.3390/ijms20205033
Casini S, Albesa M, Wang Z, Portero V, Ross-Kaschitza D, Rougier J-S, Marchal GA, Chung WK, Bezzina CR, Abriel H, et al. Functional Consequences of the SCN5A-p.Y1977N Mutation within the PY Ubiquitylation Motif: Discrepancy between HEK293 Cells and Transgenic Mice. International Journal of Molecular Sciences. 2019; 20(20):5033. https://doi.org/10.3390/ijms20205033
Chicago/Turabian StyleCasini, Simona, Maxime Albesa, Zizun Wang, Vincent Portero, Daniela Ross-Kaschitza, Jean-Sébastien Rougier, Gerard A. Marchal, Wendy K. Chung, Connie R. Bezzina, Hugues Abriel, and et al. 2019. "Functional Consequences of the SCN5A-p.Y1977N Mutation within the PY Ubiquitylation Motif: Discrepancy between HEK293 Cells and Transgenic Mice" International Journal of Molecular Sciences 20, no. 20: 5033. https://doi.org/10.3390/ijms20205033
APA StyleCasini, S., Albesa, M., Wang, Z., Portero, V., Ross-Kaschitza, D., Rougier, J.-S., Marchal, G. A., Chung, W. K., Bezzina, C. R., Abriel, H., & Remme, C. A. (2019). Functional Consequences of the SCN5A-p.Y1977N Mutation within the PY Ubiquitylation Motif: Discrepancy between HEK293 Cells and Transgenic Mice. International Journal of Molecular Sciences, 20(20), 5033. https://doi.org/10.3390/ijms20205033