Elevation of Cleaved p18 Bax Levels Associated with the Kinetics of Neuronal Cell Death during Japanese Encephalitis Virus Infection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
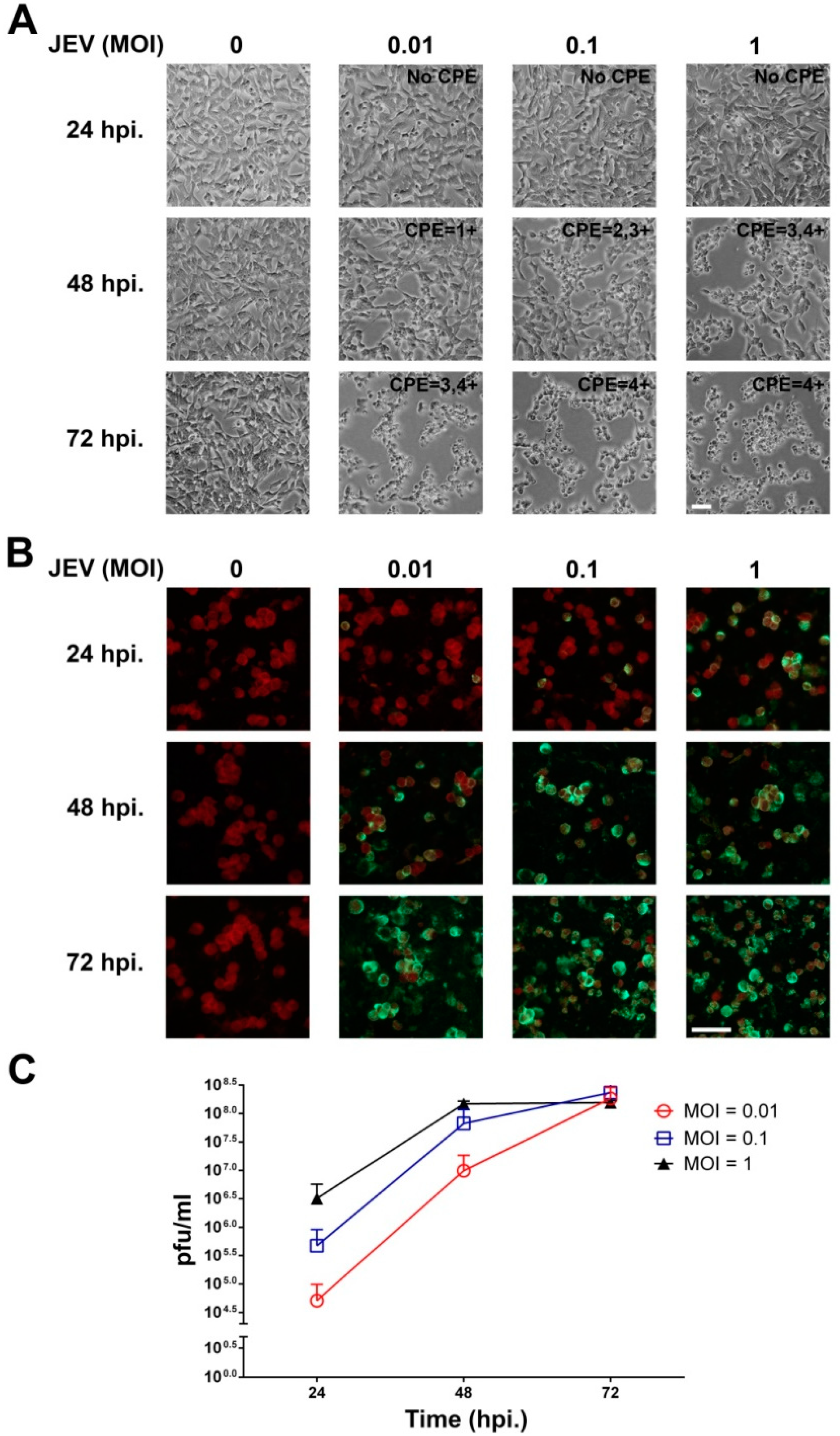
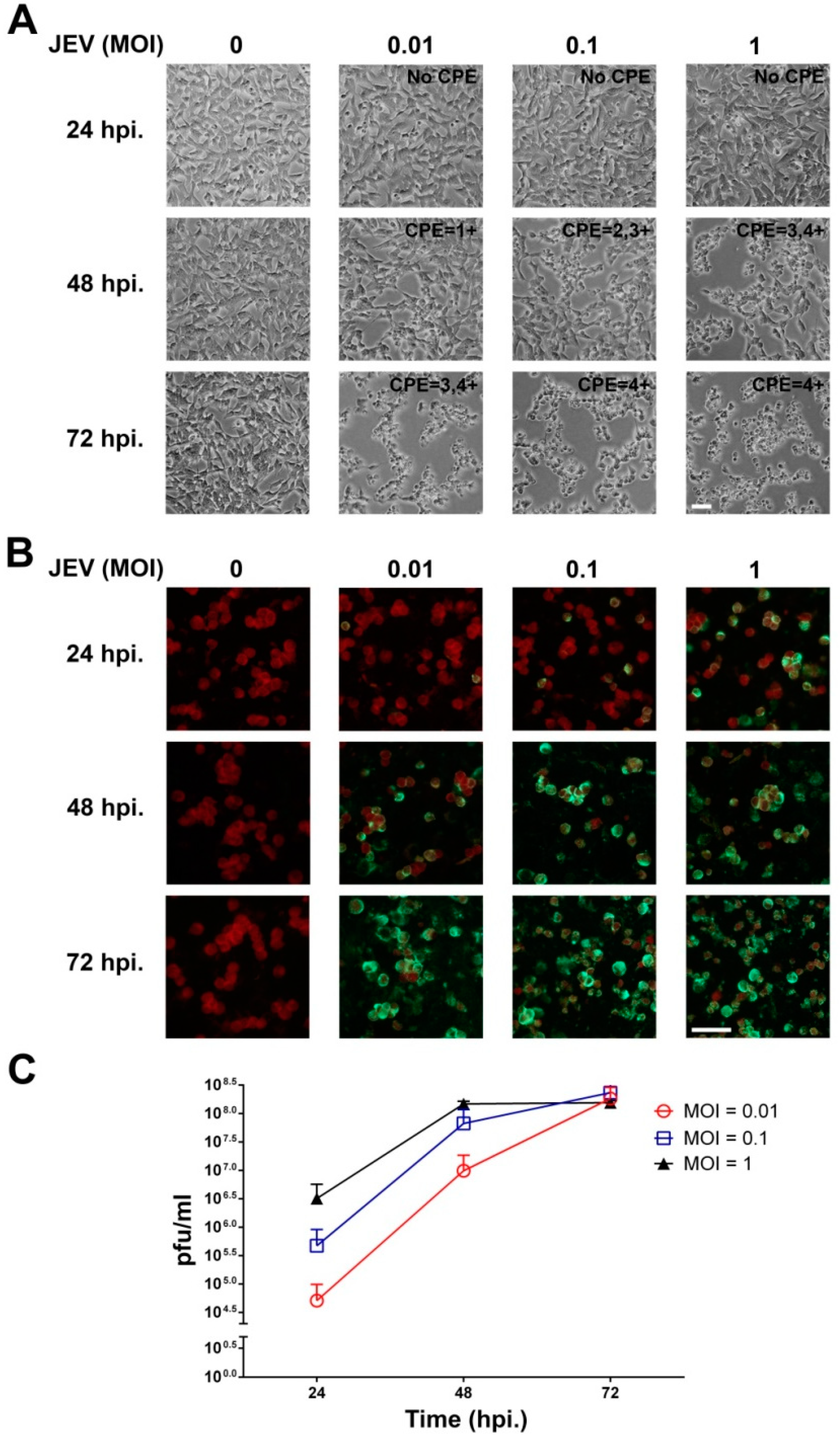
2.1. JEV Infection of SH-SY5Y Cells
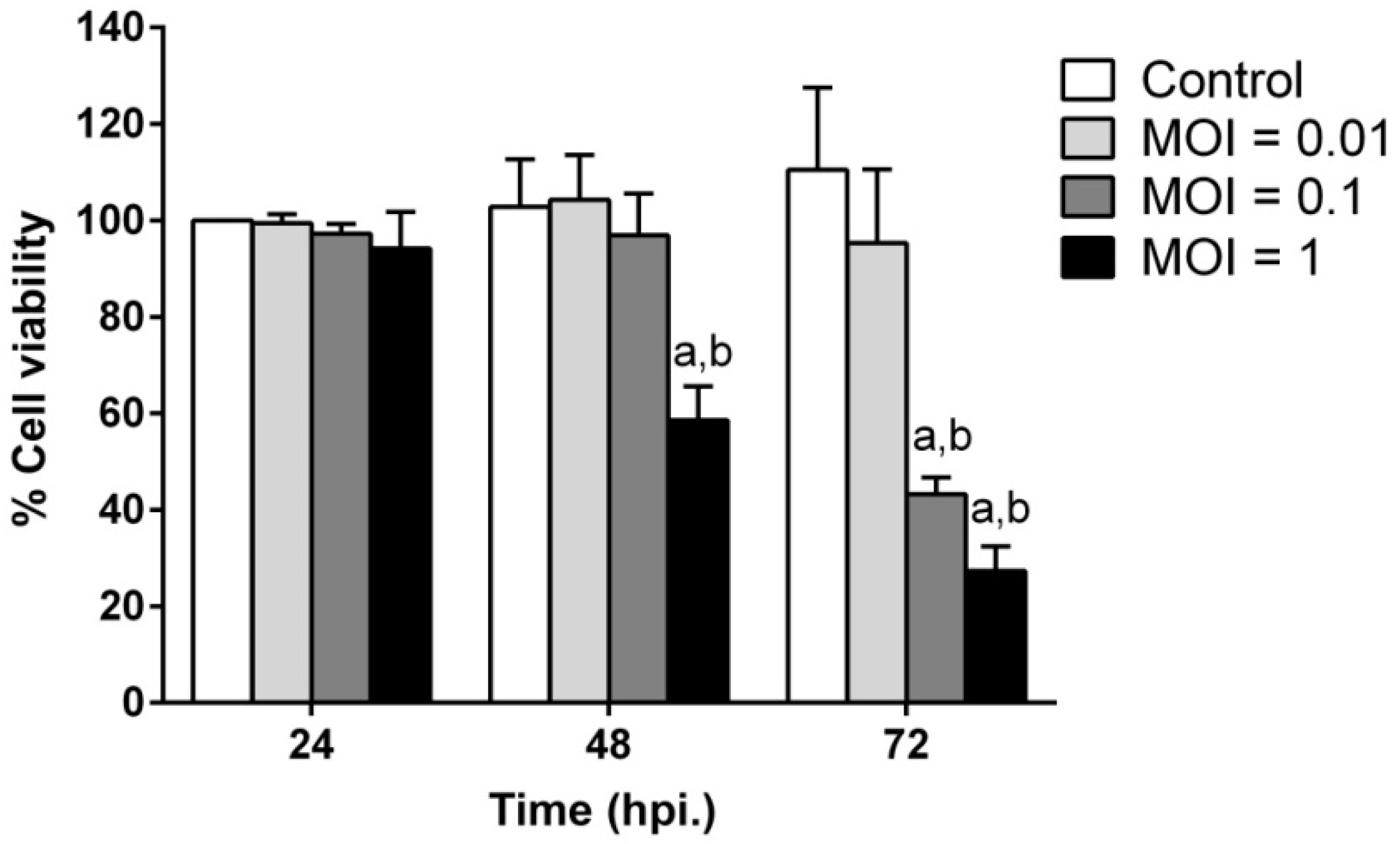
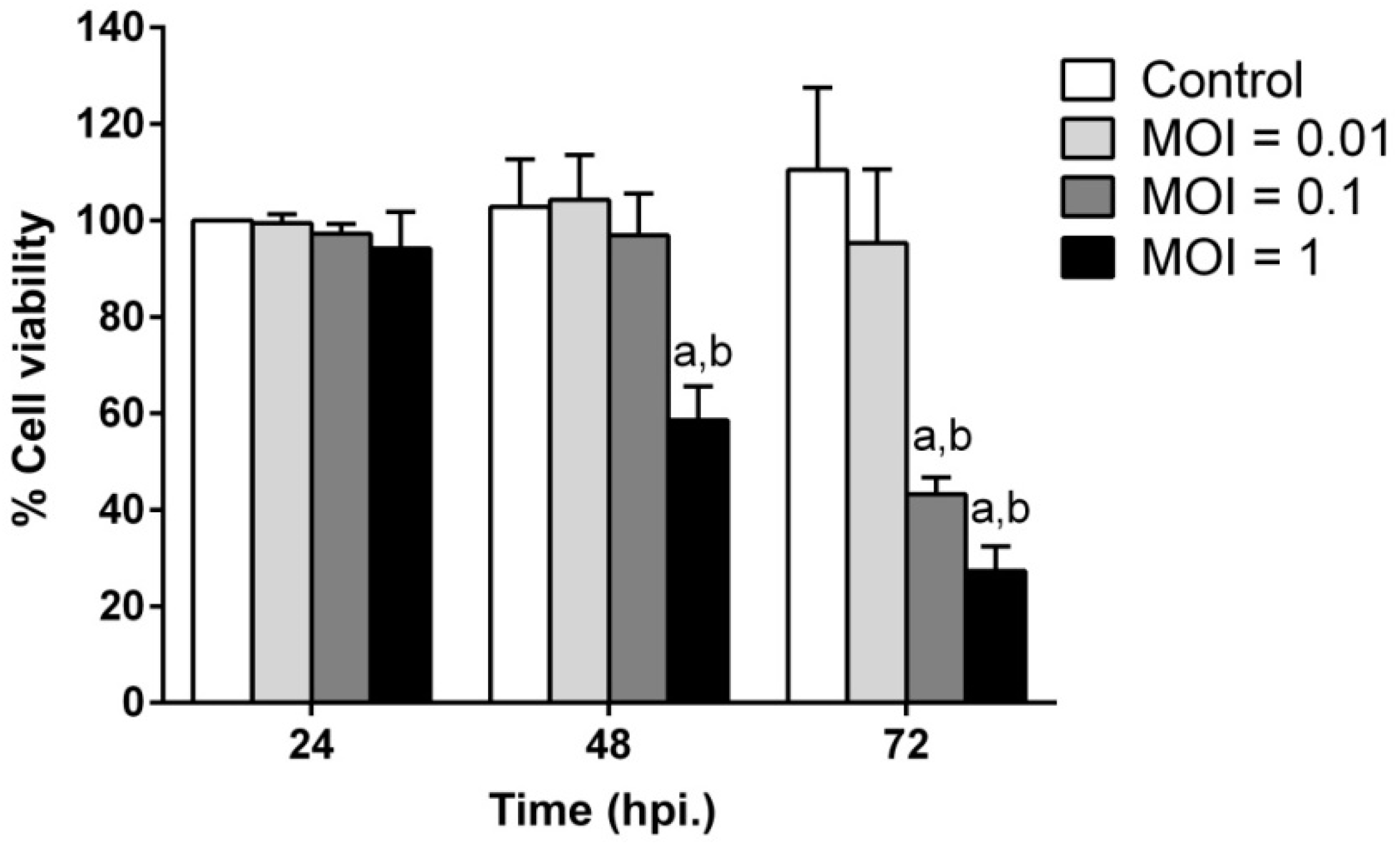
2.2. JEV Infection Induces Cell Death in SH-SY5Y Cells
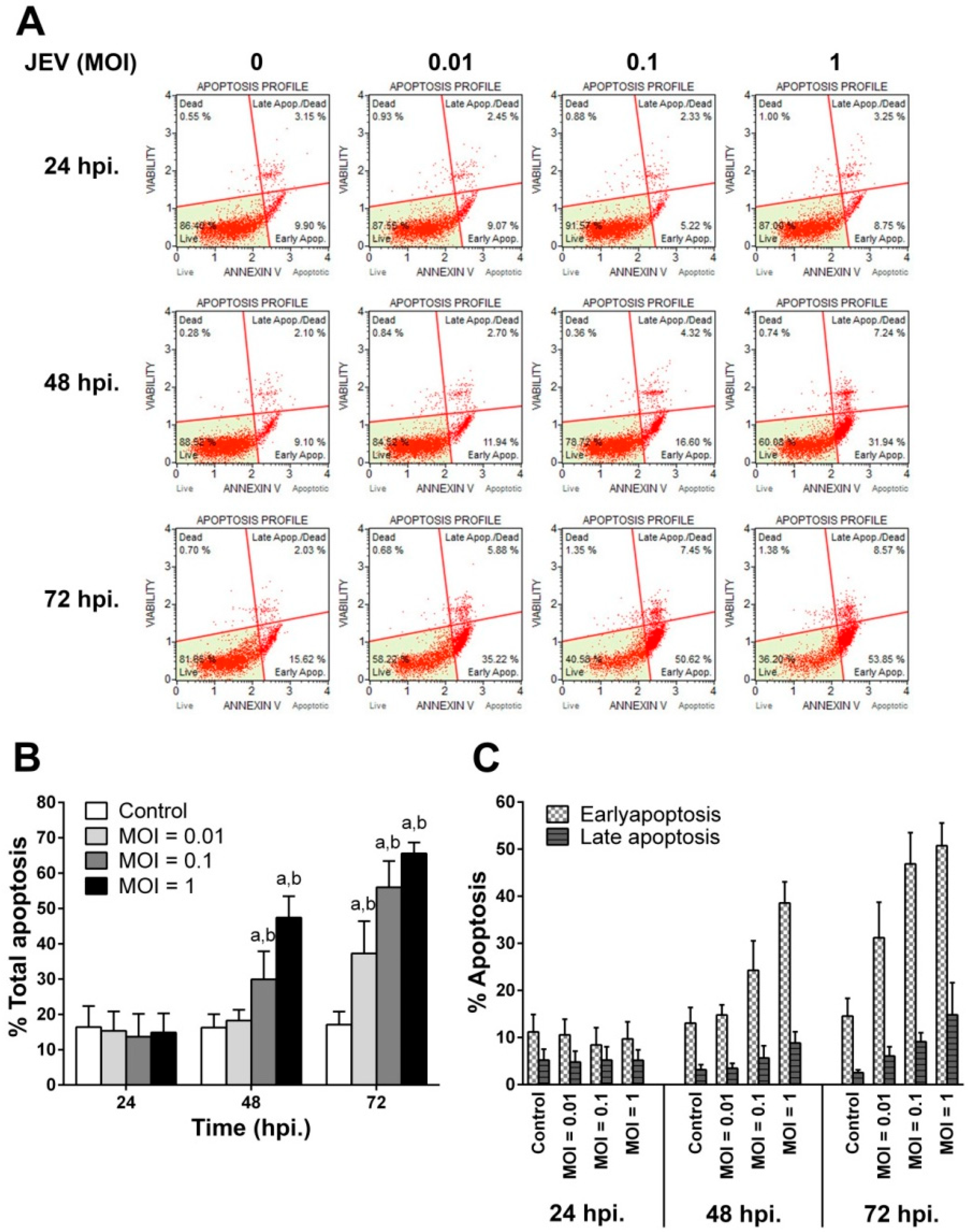
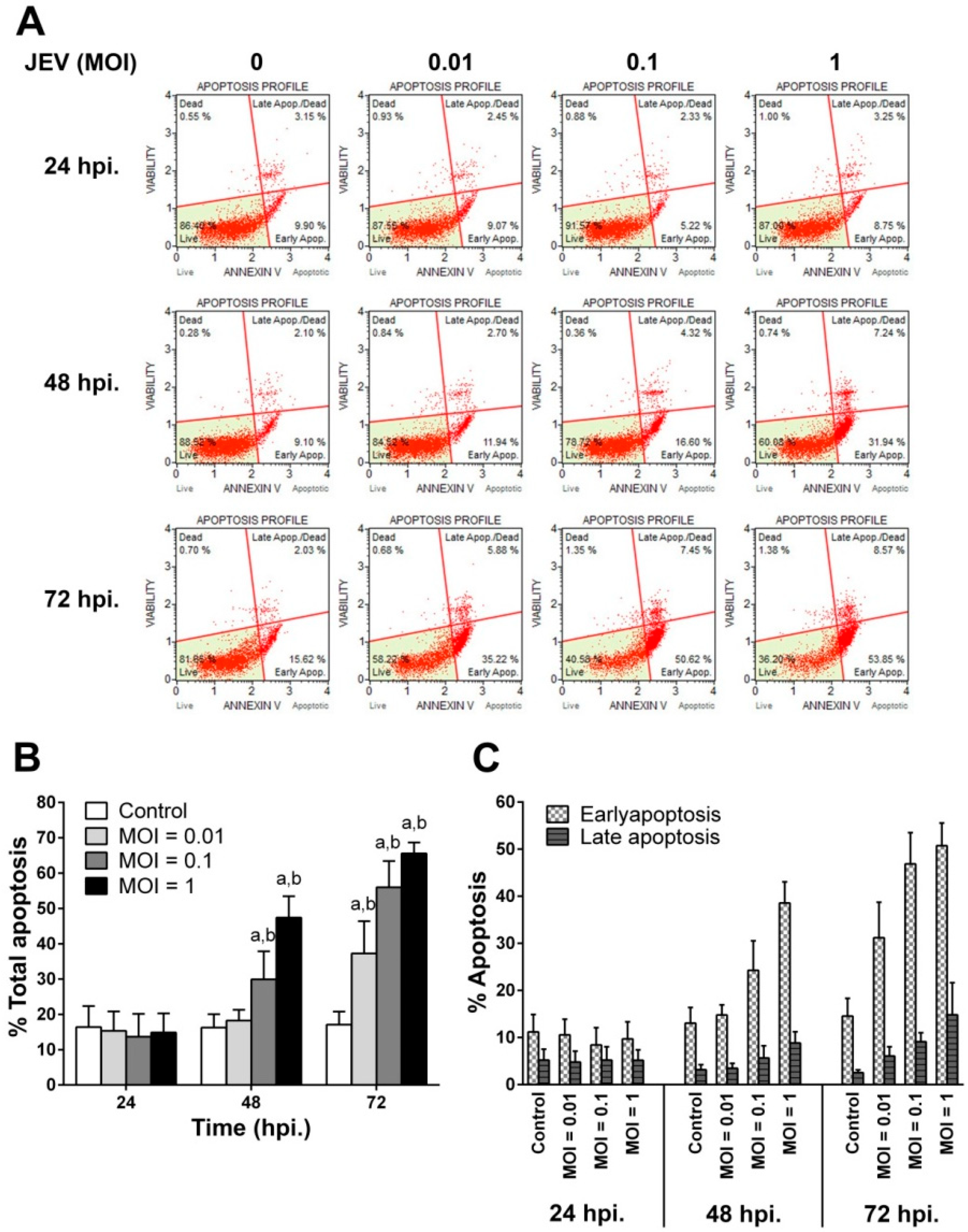
2.3. JEV Infection Induces Apoptosis in SH-SY5Y Cells
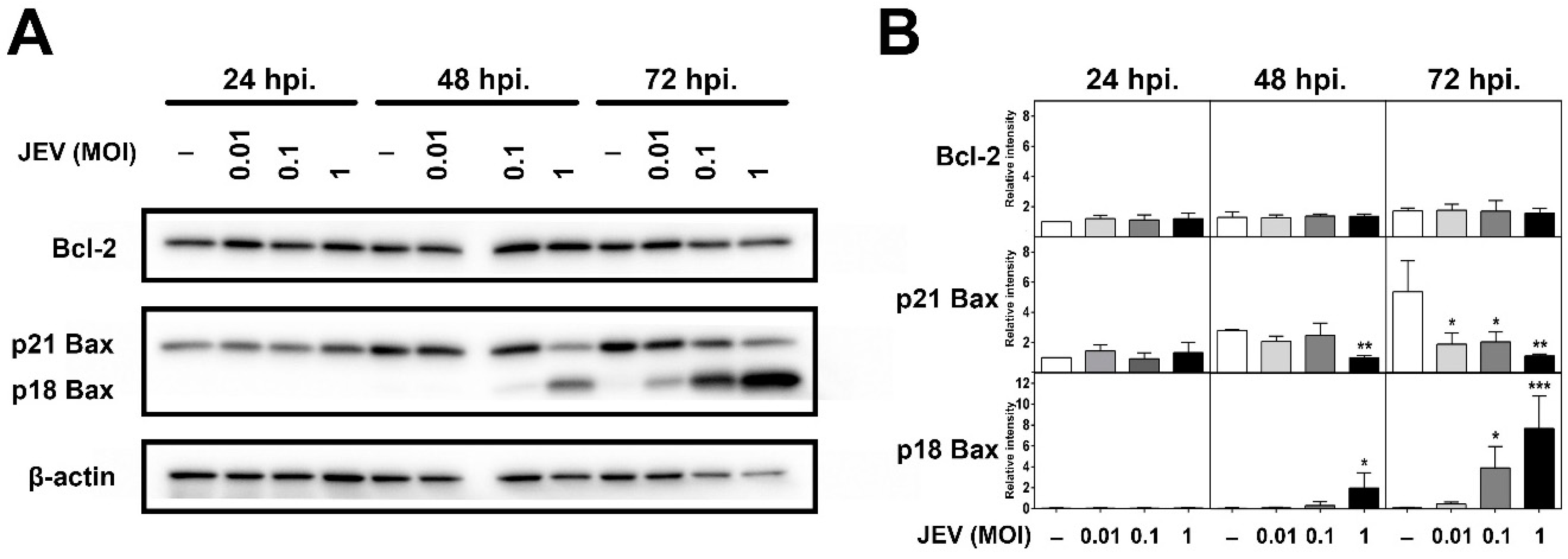
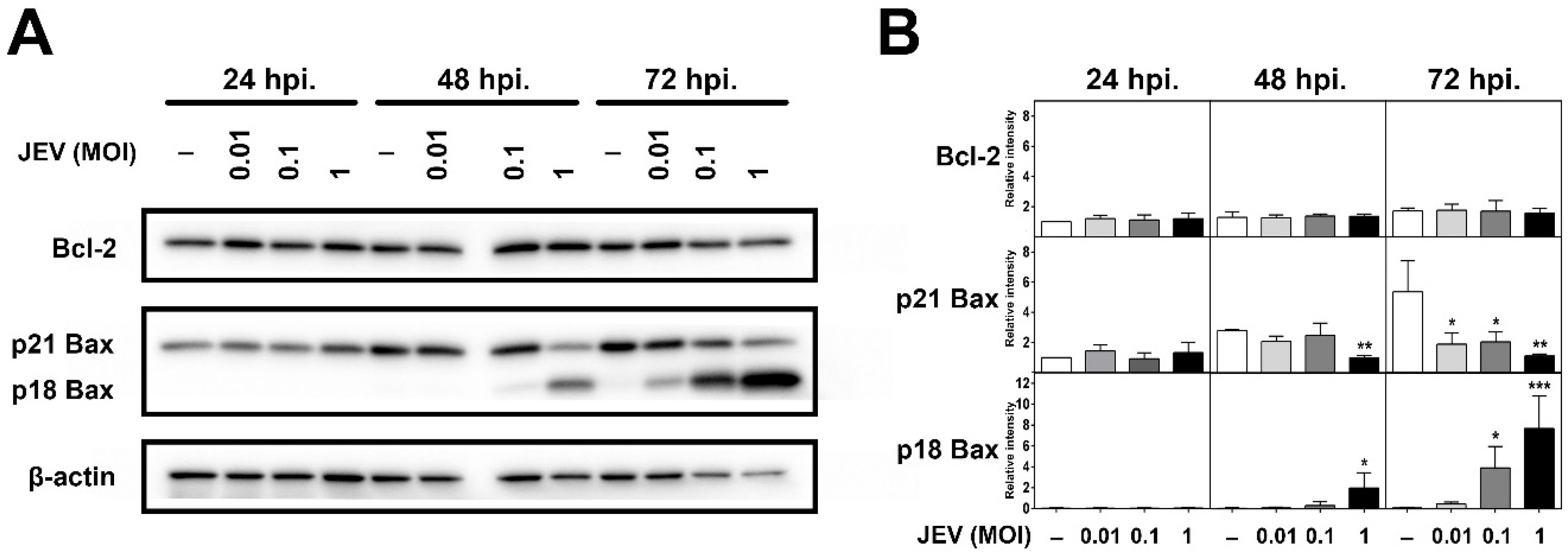
2.4. Expression of Bcl-2 Family Members in JEV-Infected SH-SY5Y Cells
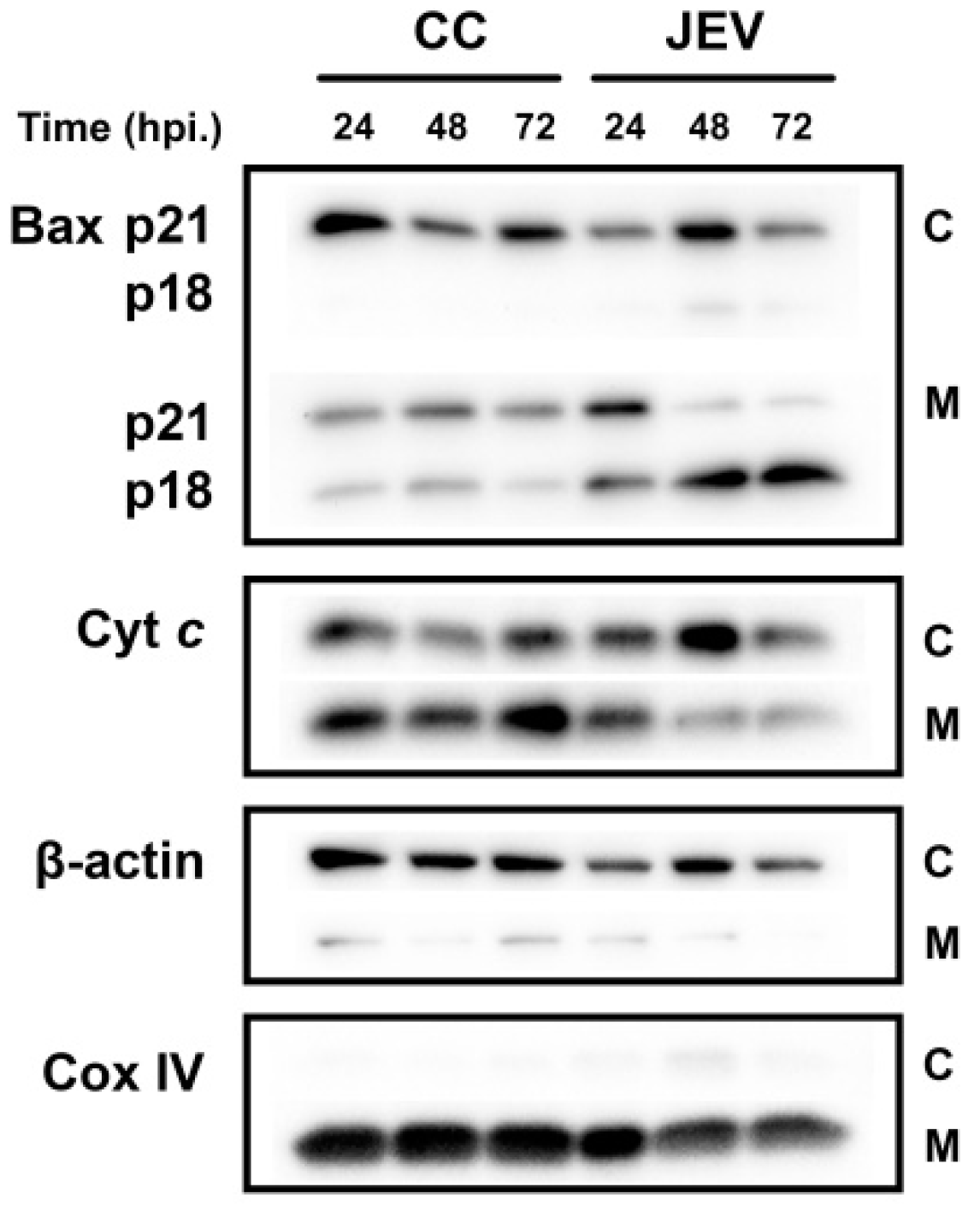
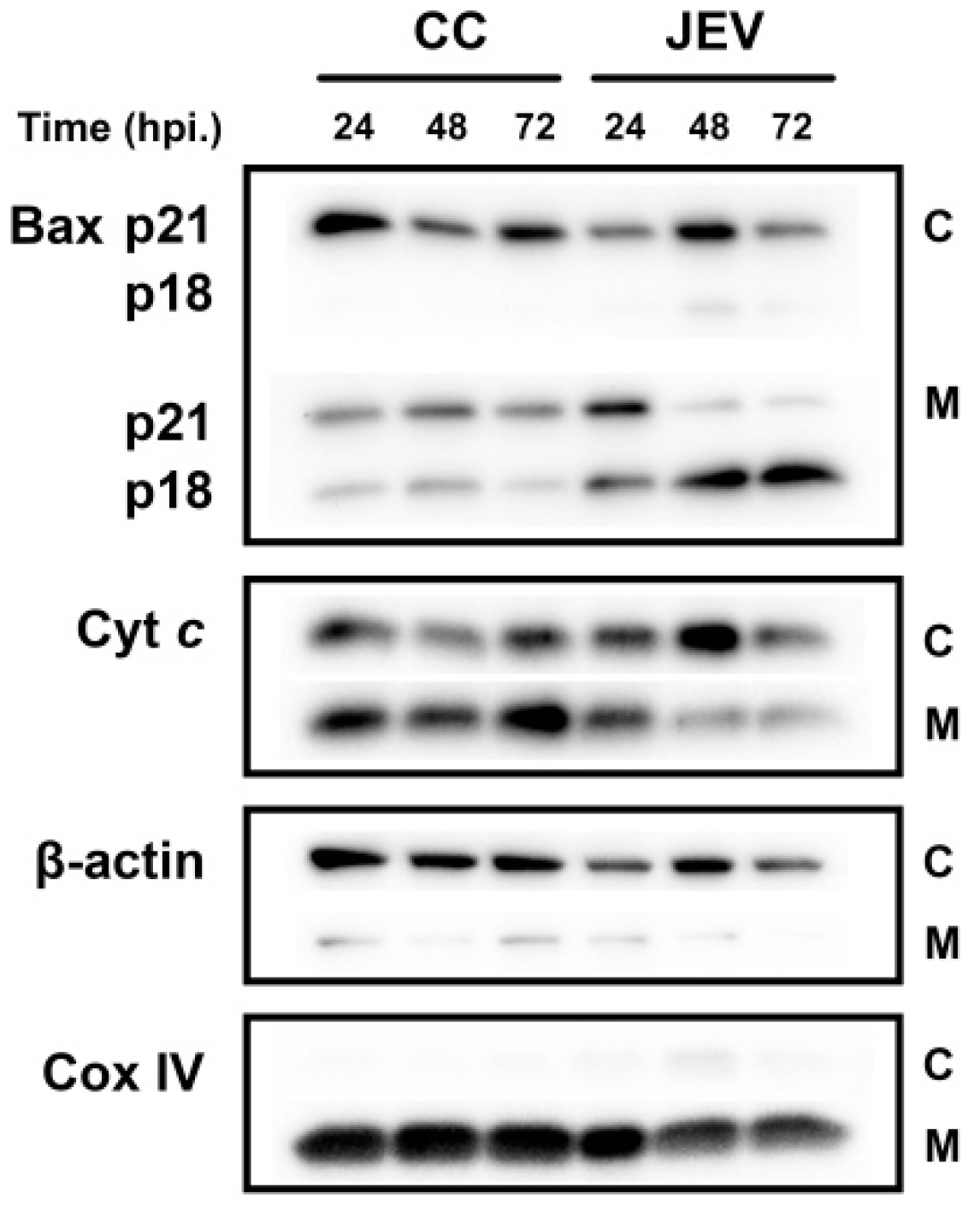
2.5. JEV Infection Increases p18 Bax Accumulation in Mitochondria and Induced Cyt c Release into the Cytosol
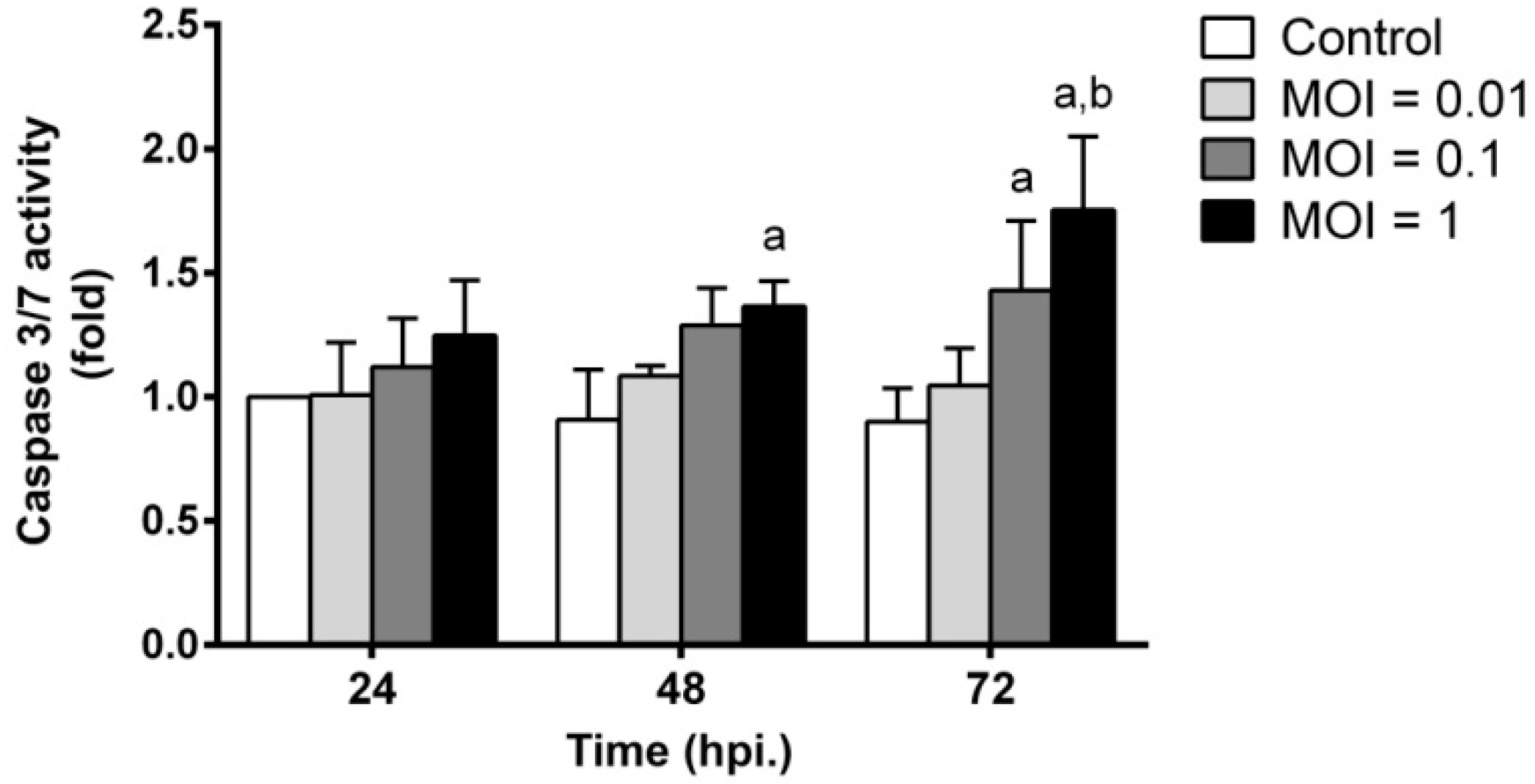
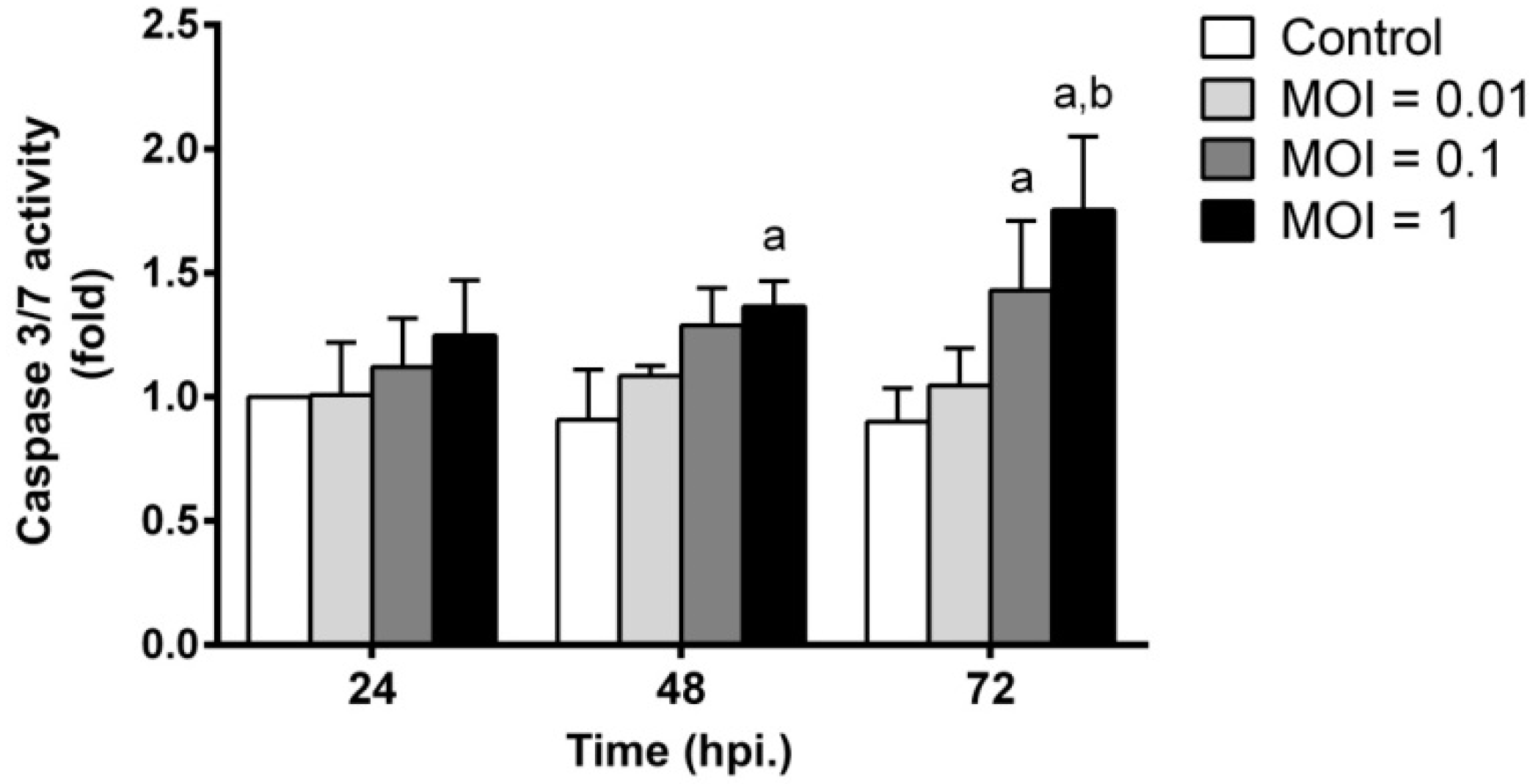
2.6. JEV Infection Triggers Apoptosis by Inducing Caspase-3/7 Activity
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. JEV Propagation in Vero Cells
4.3. Virus Titration Assay
4.4. JEV Infection in Neuronal SH-SY5Y Cells
4.5. Cell Viability Assay
4.6. Western Blot Analysis
4.7. Apoptotic Cell Death Analysis
4.8. Cytochrom c Releasing Assay
4.9. Caspase Activity Assay
4.10. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yun, S.I.; Lee, Y.M. Japanese encephalitis: The virus and vaccines. Hum. Vaccin. Immunother. 2014, 10, 263–279. [Google Scholar] [CrossRef]
- Turtle, L.; Solomon, T. Japanese encephalitis—The prospects for new treatments. Nat. Rev. Neurol. 2018, 14, 298–313. [Google Scholar] [CrossRef]
- Unni, S.K.; Ruzek, D.; Chhatbar, C.; Mishra, R.; Johri, M.K.; Singh, S.K. Japanese encephalitis virus: From genome to infectome. Microbes Infect. 2011, 13, 312–321. [Google Scholar] [CrossRef]
- Van den Hurk, A.F.; Ritchie, S.A.; Mackenzie, J.S. Ecology and geographical expansion of Japanese encephalitis virus. Annu. Rev. Entomol. 2009, 54, 17–35. [Google Scholar] [CrossRef]
- Kumar, S.; Kalita, J.; Saxena, V.; Khan, M.Y.; Khanna, V.K.; Sharma, S.; Dhole, T.N.; Misra, U.K. Some observations on the tropism of Japanese encephalitis virus in rat brain. Brain Res. 2009, 1268, 135–141. [Google Scholar] [CrossRef]
- Yang, K.D.; Yeh, W.T.; Chen, R.F.; Chuon, H.L.; Tsai, H.P.; Yao, C.W.; Shaio, M.F. A model to study neurotropism and persistency of Japanese encephalitis virus infection in human neuroblastoma cells and leukocytes. J. Gen. Virol. 2004, 85, 635–642. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Z.; Chen, H.; Chen, Z.; Tian, Y. Antioxidants: Potential antiviral agents for Japanese encephalitis virus infection. Int. J. Infect. Dis. 2014, 24, 30–36. [Google Scholar] [CrossRef]
- Lannes, N.; Summerfield, A.; Filgueira, L. Regulation of inflammation in Japanese encephalitis. J. Neuroinflamm. 2017, 14, 158. [Google Scholar] [CrossRef]
- Srivastava, R.; Kalita, J.; Khan, M.Y.; Misra, U.K. Free radical generation by neurons in rat model of Japanese encephalitis. Neurochem. Res. 2009, 34, 2141–2146. [Google Scholar] [CrossRef]
- Kumar, S.; Misra, U.K.; Kalita, J.; Khanna, V.K.; Khan, M.Y. Imbalance in oxidant/antioxidant system in different brain regions of rat after the infection of Japanese encephalitis virus. Neurochem. Int. 2009, 55, 648–654. [Google Scholar] [CrossRef]
- Yang, T.C.; Shiu, S.L.; Chuang, P.H.; Lin, Y.J.; Wan, L.; Lan, Y.C.; Lin, C.W. Japanese encephalitis virus NS2B-NS3 protease induces caspase 3 activation and mitochondria-mediated apoptosis in human medulloblastoma cells. Virus Res. 2009, 143, 77–85. [Google Scholar] [CrossRef]
- Guo, F.; Yu, X.; Xu, A.; Xu, J.; Wang, Q.; Guo, Y.; Wu, X.; Tang, Y.; Ding, Z.; Zhang, Y.; et al. Japanese encephalitis virus induces apoptosis by inhibiting Foxo signaling pathway. Vet. Microbiol. 2018, 220, 73–82. [Google Scholar] [CrossRef]
- Al-Obaidi, M.M.J.; Bahadoran, A.; Har, L.S.; Mui, W.S.; Rajarajeswaran, J.; Zandi, K.; Manikam, R.; Sekaran, S.D. Japanese encephalitis virus disrupts blood-brain barrier and modulates apoptosis proteins in THBMEC cells. Virus Res. 2017, 233, 17–28. [Google Scholar] [CrossRef]
- Hardwick, J.M.; Soane, L. Multiple functions of BCL-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- Kaminskyy, V.; Zhivotovsky, B. To kill or be killed: How viruses interact with the cell death machinery. J. Intern. Med. 2010, 267, 473–482. [Google Scholar] [CrossRef]
- Gao, G.; Dou, Q.P. N-terminal cleavage of bax by calpain generates a potent proapoptotic 18-kDa fragment that promotes bcl-2-independent cytochrome C release and apoptotic cell death. J. Cell. Biochem. 2000, 80, 53–72. [Google Scholar] [CrossRef]
- Okamoto, T.; Suzuki, T.; Kusakabe, S.; Tokunaga, M.; Hirano, J.; Miyata, Y.; Matsuura, Y. Regulation of apoptosis during flavivirus infection. Viruses 2017, 9, 243. [Google Scholar] [CrossRef]
- Kim, J.A.; Kim, J.C.; Min, J.S.; Kang, I.; Oh, J.; Ahn, J.K. HSV-1 ICP27 induces apoptosis by promoting Bax translocation to mitochondria through interacting with 14-3-3θ. BMB Reports 2017, 50, 257–262. [Google Scholar] [CrossRef]
- Choi, W.S.; Lee, E.H.; Chung, C.W.; Jung, Y.K.; Jin, B.K.; Kim, S.U.; Oh, T.H.; Saido, T.C.; Oh, Y.J. Cleavage of Bax is mediated by caspase-dependent or -independent calpain activation in dopaminergic neuronal cells: Protective role of Bcl-2. J. Neurochem. 2001, 77, 1531–1541. [Google Scholar] [CrossRef]
- Toyota, H.; Yanase, N.; Yoshimoto, T.; Moriyama, M.; Sudo, T.; Mizuguchi, J. Calpain-induced Bax-cleavage product is a more potent inducer of apoptotic cell death than wild-type Bax. Cancer Lett. 2003, 189, 221–230. [Google Scholar] [CrossRef]
- Wood, D.E.; Newcomb, E.W. Cleavage of Bax enhances its cell death function. Exp. Cell Res. 2000, 256, 375–382. [Google Scholar] [CrossRef]
- Yiang, G.T.; Chen, Y.H.; Chou, P.L.; Chang, W.J.; Wei, C.W.; Yu, Y.L. The NS3 protease and helicase domains of Japanese encephalitis virus trigger cell death via caspase dependent and independent pathways. Mol. Med. Rep. 2013, 7, 826–830. [Google Scholar] [CrossRef]
- Jurgensmeier, J.M.; Xie, Z.; Deveraux, Q.; Ellerby, L.; Bredesen, D.; Reed, J.C. Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl. Acad. Sci. USA 1998, 95, 4997–5002. [Google Scholar] [CrossRef]
- Shi, Y. Caspase activation, inhibition, and reactivation: A mechanistic view. Protein Sci. 2004, 13, 1979–1987. [Google Scholar] [CrossRef] [Green Version]
- Tsao, C.H.; Su, H.L.; Lin, Y.L.; Yu, H.P.; Kuo, S.M.; Shen, C.I.; Chen, C.W.; Liao, C.L. Japanese encephalitis virus infection activates caspase-8 and -9 in a FADD-independent and mitochondrion-dependent manner. J. Gen. Virol. 2008, 89, 1930–1941. [Google Scholar] [CrossRef]
- Gupta, N.; Bhaskar, A.S.; Lakshmana Rao, P.V. Transcriptional regulation and activation of the mitogen-activated protein kinase pathway after Japanese encephalitis virus infection in neuroblastoma cells. FEMS Immunol. Med. Microbiol. 2011, 62, 110–121. [Google Scholar] [CrossRef]
- Hongmei, Z. Extrinsic and intrinsic apoptosis signal pathway review. In Apoptosis and Medicine; Ntuli, T., Ed.; IntechOpen: London, UK, 2012. [Google Scholar]
- Huang, M.; Xu, A.; Wu, X.; Zhang, Y.; Guo, Y.; Guo, F.; Pan, Z.; Kong, L. Japanese encephalitis virus induces apoptosis by the IRE1/JNK pathway of ER stress response in BHK-21 cells. Arch. Virol. 2016, 161, 699–703. [Google Scholar] [CrossRef]
- Yang, T.C.; Lai, C.C.; Shiu, S.L.; Chuang, P.H.; Tzou, B.C.; Lin, Y.Y.; Tsai, F.J.; Lin, C.W. Japanese encephalitis virus down-regulates thioredoxin and induces ROS-mediated ASK1-ERK/p38 MAPK activation in human promonocyte cells. Microbes Infect. 2010, 12, 643–651. [Google Scholar] [CrossRef]
- Lixia, H.; Jun, C.; Song, H.; FaHu, Y.; Jinwen, T. Neuroprotective effect of (-)-tetrahydropalmatine in Japanese encephalitis virus strain GP-78 infected mouse model. Microb. Pathog. 2018, 114, 197–203. [Google Scholar] [CrossRef]
- Swarup, V.; Das, S.; Ghosh, S.; Basu, A. Tumor necrosis factor receptor-1-induced neuronal death by TRADD contributes to the pathogenesis of Japanese encephalitis. J. Neurochem. 2007, 103, 771–783. [Google Scholar] [CrossRef]
- Cao, X.; Deng, X.; May, W.S. Cleavage of Bax to p18 Bax accelerates stress-induced apoptosis, and a cathepsin-like protease may rapidly degrade p18 Bax. Blood 2003, 102, 2605–2614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, D.E.; Thomas, A.; Devi, L.A.; Berman, Y.; Beavis, R.C.; Reed, J.C.; Newcomb, E.W. Bax cleavage is mediated by calpain during drug-induced apoptosis. Oncogene 1998, 17, 1069–1078. [Google Scholar] [CrossRef] [Green Version]
- Tamura, H.; Ohtsuru, A.; Kamohara, Y.; Fujioka, H.; Yanaga, K.; Kanematsu, T.; Yamashita, S. Bax cleavage implicates caspase-dependent H2O2-induced apoptosis of hepatocytes. Int. J. Mol. Med. 2003, 11, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.Y.; Paik, S.; Park, S.H.; Kim, H.S.; Lee, I.S.; Kim, S.P.; Baek, W.K.; Suh, M.H.; Kwon, T.K.; Park, J.W.; et al. N-phenethyl-2-phenylacetamide isolated from Xenorhabdus nematophilus induces apoptosis through caspase activation and calpain-mediated Bax cleavage in U937 cells. Int. J. Oncol. 2003, 22, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Yanase, N.; Takada, E.; Yoshihama, I.; Ikegami, H.; Mizuguchi, J. Participation of Bax-α in IFN-α-mediated apoptosis in daudi B lymphoma cells. J. Interf. Cytok. Res. 1998, 18, 855–861. [Google Scholar] [CrossRef]
- Oh, S.H.; Lee, B.H.; Lim, S.C. Cadmium induces apoptotic cell death in WI 38 cells via caspase-dependent Bid cleavage and calpain-mediated mitochondrial Bax cleavage by Bcl-2-independent pathway. Biochem. Pharmacol. 2004, 68, 1845–1855. [Google Scholar] [CrossRef]
- Megyeri, K.; Orosz, L.; Kemeny, L. Vesicular stomatitis virus infection triggers apoptosis associated with decreased DeltaNp63alpha and increased Bax levels in the immortalized HaCaT keratinocyte cell line. Biomed. Pharmacother. 2007, 61, 254–260. [Google Scholar] [CrossRef]
- Grandgirard, D.; Studer, E.; Monney, L.; Belser, T.; Fellay, I.; Borner, C.; Michel, M.R. Alphaviruses induce apoptosis in Bcl-2-overexpressing cells: Evidence for a caspase-mediated, proteolytic inactivation of Bcl-2. EMBO J. 1998, 17, 1268–1278. [Google Scholar] [CrossRef]
- Cartron, P.F.; Oliver, L.; Juin, P.; Meflah, K.; Vallette, F.M. The p18 truncated form of Bax behaves like a Bcl-2 homology domain 3-only protein. J. Biol. Chem. 2004, 279, 11503–11512. [Google Scholar] [CrossRef]
- Liao, C.L.; Lin, Y.L.; Shen, S.C.; Shen, J.Y.; Su, H.L.; Huang, Y.L.; Ma, S.H.; Sun, Y.C.; Chen, K.P.; Chen, L.K. Antiapoptotic but not antiviral function of human bcl-2 assists establishment of japanese encephalitis virus persistence in cultured cells. J. Virol. 1998, 72, 9844. [Google Scholar]
- Lee, C.J.; Liao, C.L.; Lin, Y.L. Flavivirus activates phosphatidylinositol 3-kinase signaling to block caspase-dependent apoptotic cell death at the early stage of virus infection. J. Virol. 2005, 79, 8388–8399. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.L.; Lin, Y.L.; Wang, J.J.; Huang, Y.L.; Yeh, C.T.; Ma, S.H.; Chen, L.K. Effect of enforced expression of human bcl-2 on Japanese encephalitis virus-induced apoptosis in cultured cells. J. Virol. 1997, 71, C5963–C5971. [Google Scholar]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wongchitrat, P.; Samutpong, A.; Lerdsamran, H.; Prasertsopon, J.; Yasawong, M.; Govitrapong, P.; Puthavathana, P.; Kitidee, K. Elevation of Cleaved p18 Bax Levels Associated with the Kinetics of Neuronal Cell Death during Japanese Encephalitis Virus Infection. Int. J. Mol. Sci. 2019, 20, 5016. https://doi.org/10.3390/ijms20205016
Wongchitrat P, Samutpong A, Lerdsamran H, Prasertsopon J, Yasawong M, Govitrapong P, Puthavathana P, Kitidee K. Elevation of Cleaved p18 Bax Levels Associated with the Kinetics of Neuronal Cell Death during Japanese Encephalitis Virus Infection. International Journal of Molecular Sciences. 2019; 20(20):5016. https://doi.org/10.3390/ijms20205016
Chicago/Turabian StyleWongchitrat, Prapimpun, Arisara Samutpong, Hatairat Lerdsamran, Jarunee Prasertsopon, Montri Yasawong, Piyarat Govitrapong, Pilaipan Puthavathana, and Kuntida Kitidee. 2019. "Elevation of Cleaved p18 Bax Levels Associated with the Kinetics of Neuronal Cell Death during Japanese Encephalitis Virus Infection" International Journal of Molecular Sciences 20, no. 20: 5016. https://doi.org/10.3390/ijms20205016