Effects of Membrane and Biological Target on the Structural and Allosteric Properties of Recoverin: A Computational Approach

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
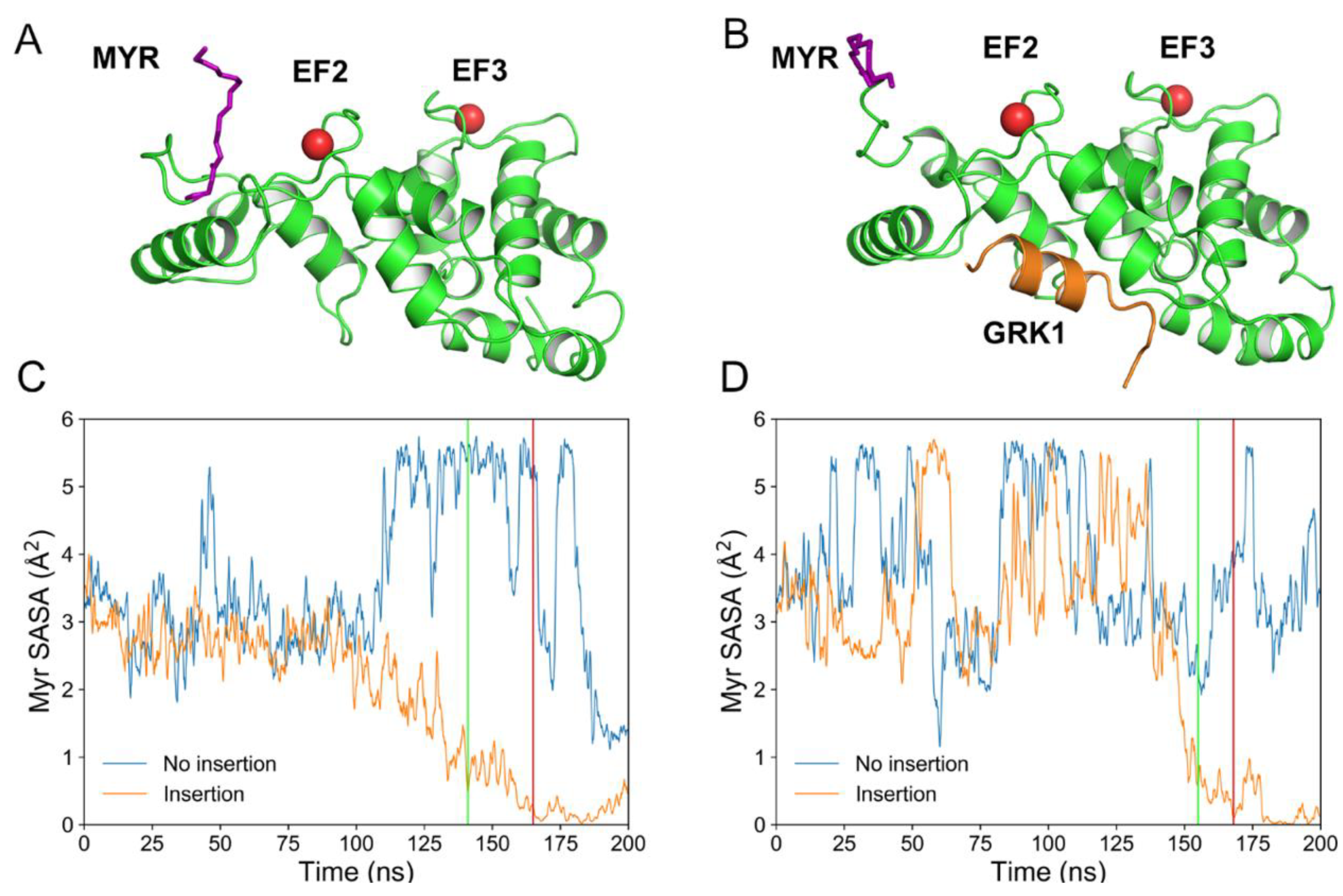
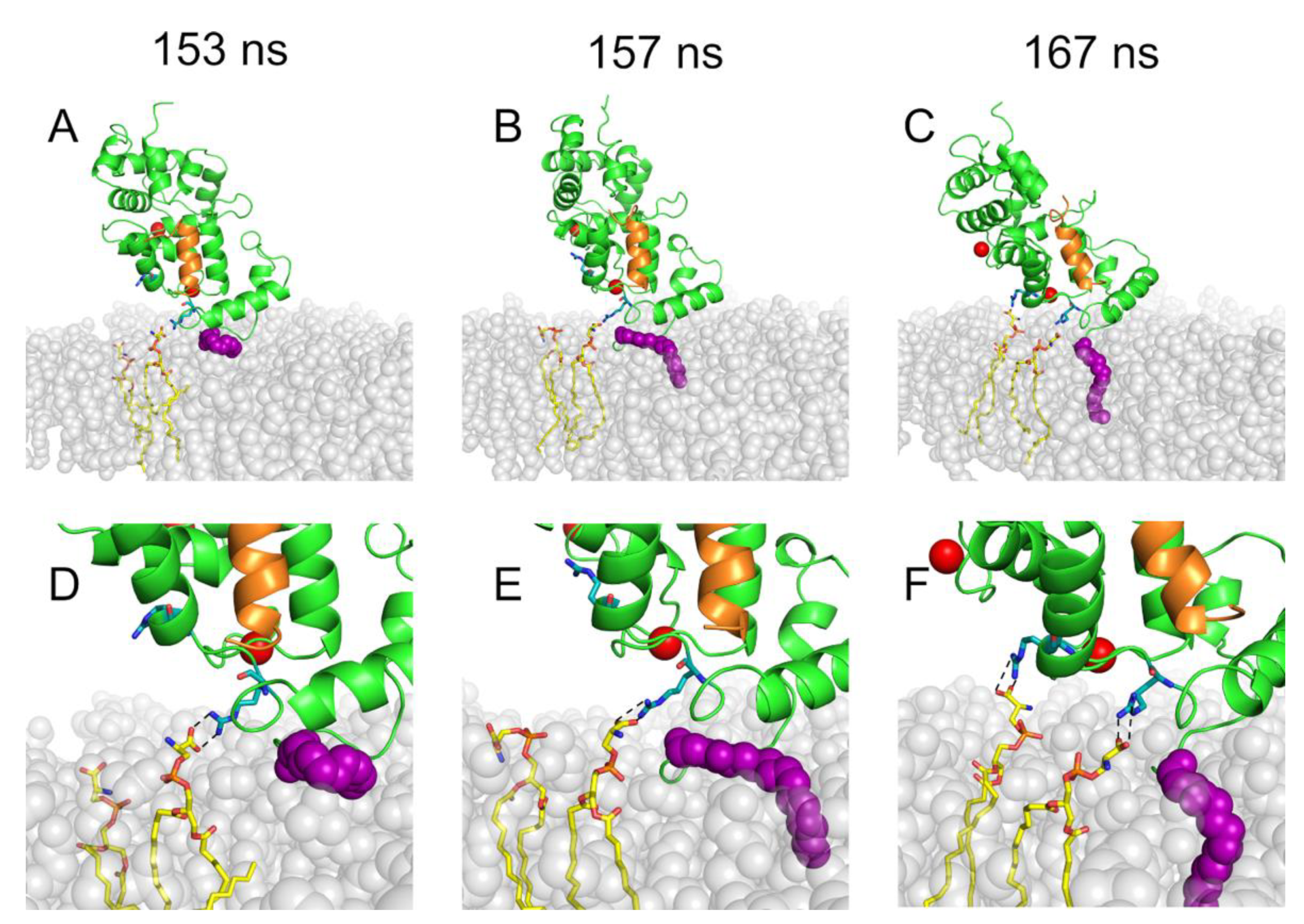
2.1. Spontaneous Insertion of the Myristoyl Moiety of Rec and Rec-GRK1 into the Membrane
2.2. Binding of Rec to the Membrane is a Multi-Step Process Involving Specific Electrostatic Interactions
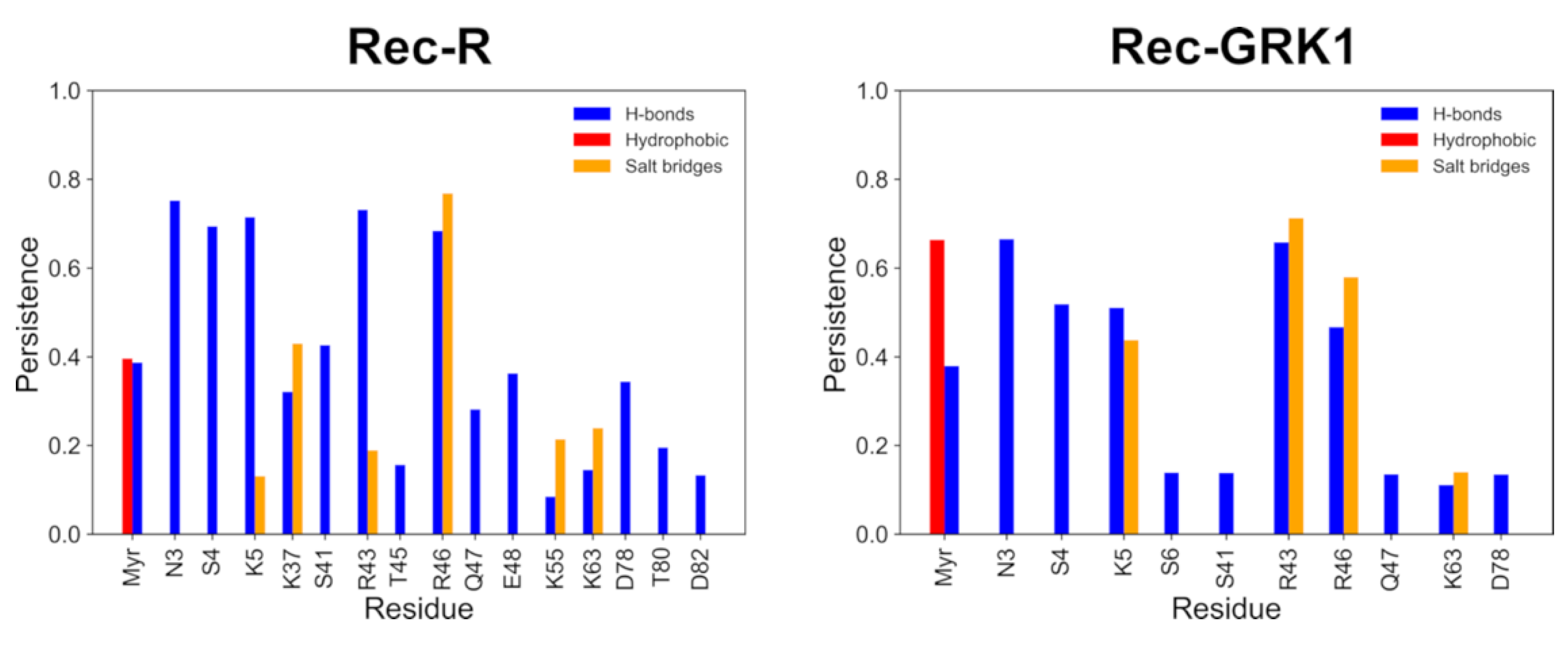
2.3. A Specific Network of Electrostatic Interactions Stabilizes the Membrane-Bound State of Rec-R and Rec-GRK1
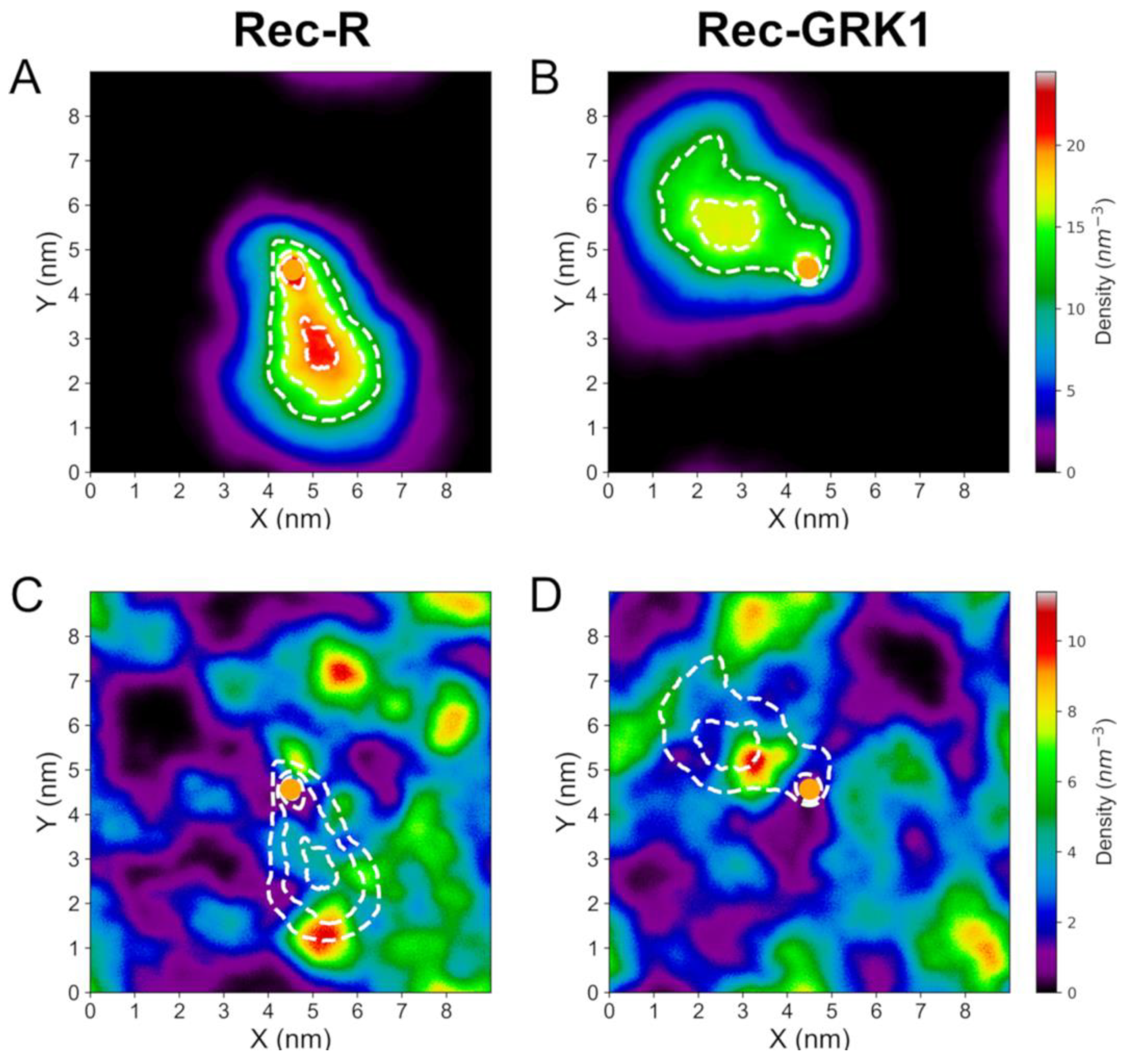
2.4. Dynamics of Rec-Bound Biological Membrane
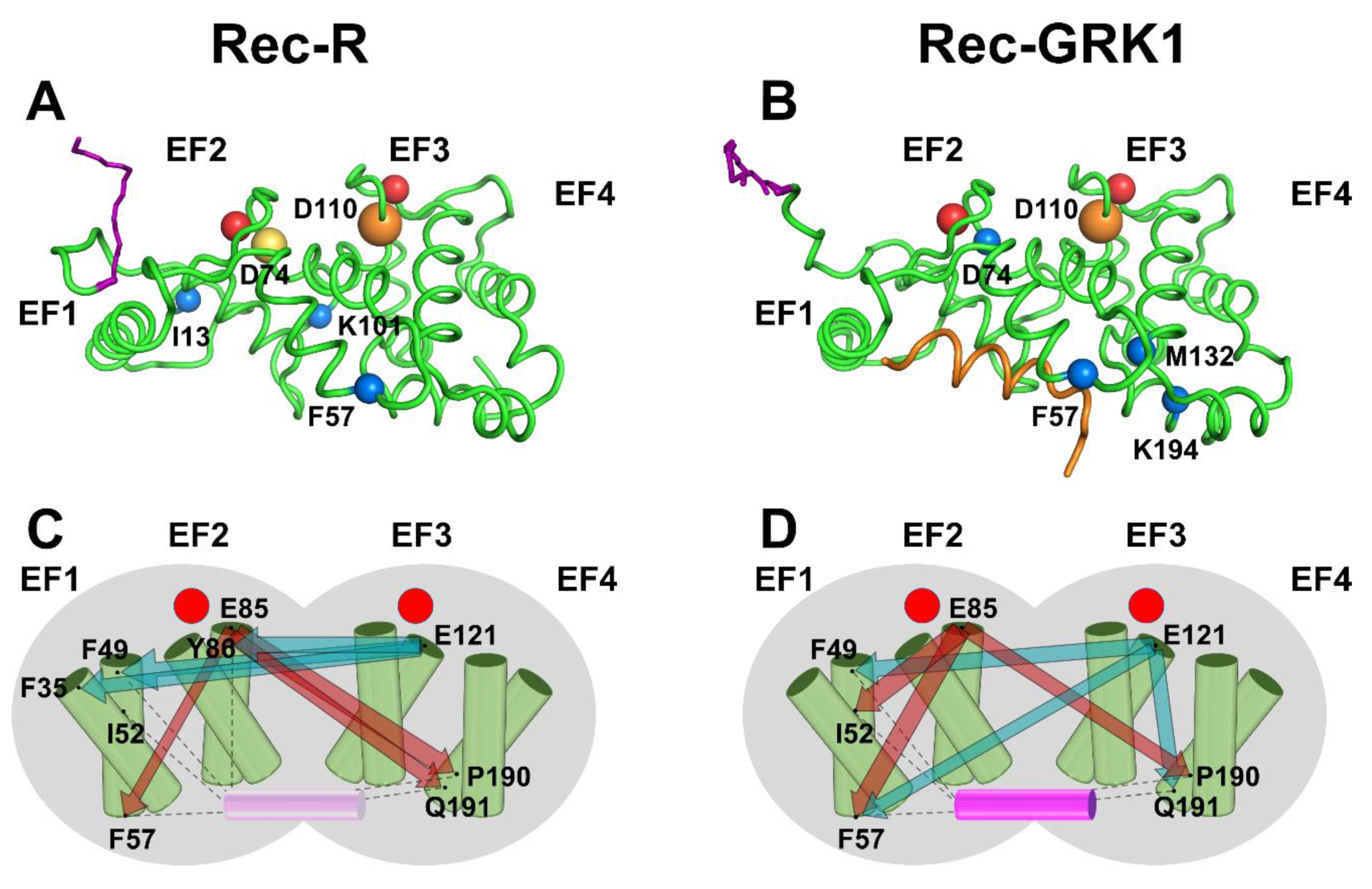
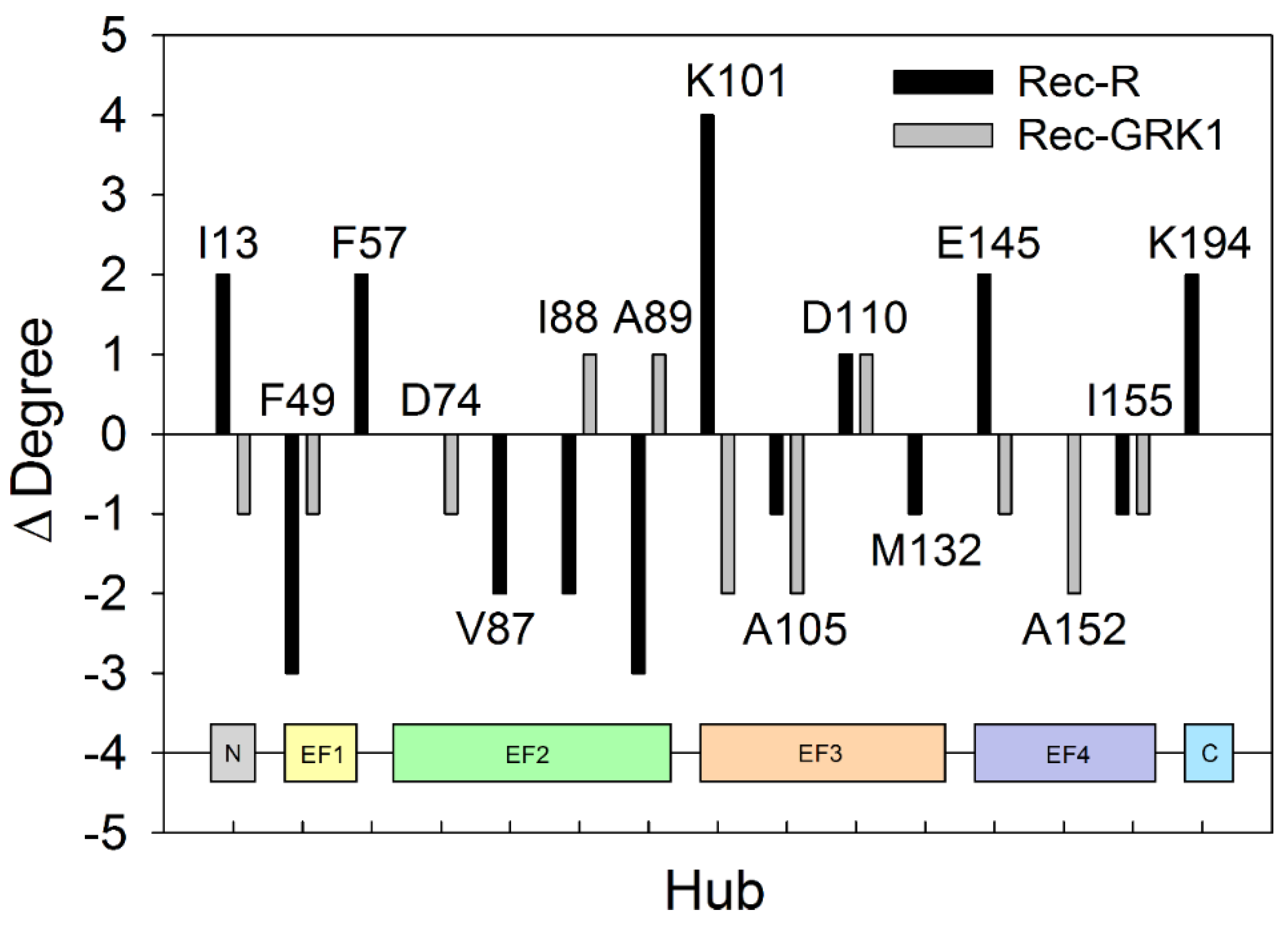
2.5. PSN Analysis of Rec-R and Rec-GRK1 Suggests State-Specific Allosteric Mechanisms
3. Material and Methods
3.1. Membrane Building and Equilibration
3.2. Rec Structures
3.3. System Assembly and MD Simulations
3.4. Sampling Homogeneity Checks
3.5. Lateral Mean Square Displacements
3.6. Analysis of PSN
3.7. Analysis of Protein-Lipid Interactions Persistence
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Rec | Recoverin |
| GRK1 | Rhodopsin Kinase |
| NCS | Neuronal Calcium Sensor |
| Rec-T | Recoverin “Tense” (apo) |
| Rec-R | Recoverin “Relaxed” (Ca2+-loaded) |
| Rec-GRK1 | Ca2+-loaded Recoverin-Rhodopsin Kinase complex |
| MD | Molecular Dynamics |
| PC | Phosphatidylcholine |
| PG | Phosphatidylglycerol |
| ROS | Rod Outer Segment |
| PS | Phosphatidylserine |
| PE | Phosphatidylethanolamine |
| PSN | Protein Structure Network |
| SASA | Solvent Accessible Surface Area |
| NMR | Nuclear Magnetic Resonance |
| MSD | Mean Square Displacement |
| PCA | Principal Component Analysis |
| RMSIP | Root-Mean Square Inner Product |
| ES | Essential Subspace |
| CR | Communication Robustness |
References
- Koch, K.W.; Dell’Orco, D. Protein and Signaling Networks in Vertebrate Photoreceptor Cells. Front. Mol. Neurosci. 2015, 8, 67. [Google Scholar] [CrossRef] [PubMed]
- Zang, J.; Neuhauss, S.C.F. The Binding Properties and Physiological Functions of Recoverin. Front. Mol. Neurosci. 2018, 11, 473. [Google Scholar] [CrossRef] [PubMed]
- Koch, K.W.; Dell’orco, D. A calcium-relay mechanism in vertebrate phototransduction. ACS Chem. Neurosci. 2013, 4, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Dizhoor, A.M.; Ericsson, L.H.; Johnson, R.S.; Kumar, S.; Olshevskaya, E.; Zozulya, S.; Neubert, T.A.; Stryer, L.; Hurley, J.B.; Walsh, K.A. The NH2 terminus of retinal recoverin is acylated by a small family of fatty acids. J. Biol. Chem. 1992, 267, 16033–16036. [Google Scholar] [PubMed]
- Ames, J.B.; Ishima, R.; Tanaka, T.; Gordon, J.I.; Stryer, L.; Ikura, M. Molecular mechanics of calcium-myristoyl switches. Nature 1997, 389, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Ames, J.B.; Harvey, T.S.; Stryer, L.; Ikura, M. Sequestration of the membrane-targeting myristoyl group of recoverin in the calcium-free state. Nature 1995, 376, 444–447. [Google Scholar] [CrossRef]
- Lange, C.; Koch, K.W. Calcium-dependent binding of recoverin to membranes monitored by surface plasmon resonance spectroscopy in real time. Biochemistry 1997, 36, 12019–12026. [Google Scholar] [CrossRef]
- Senin, I.I.; Koch, K.W.; Akhtar, M.; Philippov, P.P. Ca2+-dependent control of rhodopsin phosphorylation: Recoverin and rhodopsin kinase. Adv. Exp. Med. Biol. 2002, 514, 69–99. [Google Scholar]
- Zozulya, S.; Stryer, L. Calcium-myristoyl protein switch. Proc. Natl. Acad. Sci. USA 1992, 89, 11569–11573. [Google Scholar] [CrossRef]
- Lim, S.; Strahl, T.; Thorner, J.; Ames, J.B. Structure of a Ca2+-myristoyl switch protein that controls activation of a phosphatidylinositol 4-kinase in fission yeast. J. Biol. Chem. 2011, 286, 12565–12577. [Google Scholar] [CrossRef]
- Li, C.; Lim, S.; Braunewell, K.H.; Ames, J.B. Structure and Calcium Binding Properties of a Neuronal Calcium-Myristoyl Switch Protein, Visinin-Like Protein 3. PLoS ONE 2016, 11, e0165921. [Google Scholar] [CrossRef] [PubMed]
- Dell’Orco, D.; Muller, M.; Koch, K.W. Quantitative detection of conformational transitions in a calcium sensor protein by surface plasmon resonance. Chem. Commun. 2010, 46, 7316–7318. [Google Scholar] [CrossRef] [PubMed]
- Senin, I.I.; Fischer, T.; Komolov, K.E.; Zinchenko, D.V.; Philippov, P.P.; Koch, K.W. Ca2+-myristoyl switch in the neuronal calcium sensor recoverin requires different functions of Ca2+-binding sites. J. Biol. Chem. 2002, 277, 50365–50372. [Google Scholar] [CrossRef] [PubMed]
- Timr, S.; Pleskot, R.; Kadlec, J.; Kohagen, M.; Magarkar, A.; Jungwirth, P. Membrane Binding of Recoverin: From Mechanistic Understanding to Biological Functionality. ACS Cent. Sci. 2017, 3, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Boesze-Battaglia, K.; Albert, A.D. Phospholipid distribution among bovine rod outer segment plasma membrane and disk membranes. Exp. Eye Res. 1992, 54, 821–823. [Google Scholar] [CrossRef]
- Fliesler, S.J.; Anderson, R.E. Chemistry and metabolism of lipids in the vertebrate retina. Prog. Lipid Res. 1983, 22, 79–131. [Google Scholar] [CrossRef]
- Boesze-Battaglia, K.; Hennessey, T.; Albert, A.D. Cholesterol heterogeneity in bovine rod outer segment disk membranes. J. Biol. Chem. 1989, 264, 8151–8155. [Google Scholar] [PubMed]
- Boesze-Battaglia, K.; Fliesler, S.J.; Albert, A.D. Relationship of cholesterol content to spatial distribution and age of disc membranes in retinal rod outer segments. J. Biol. Chem. 1990, 265, 18867–18870. [Google Scholar] [PubMed]
- Albert, A.; Alexander, D.; Boesze-Battaglia, K. Cholesterol in the rod outer segment: A complex role in a “simple” system. Chem. Phys. Lipids 2016, 199, 94–105. [Google Scholar] [CrossRef]
- Senin, I.I.; Hoppner-Heitmann, D.; Polkovnikova, O.O.; Churumova, V.A.; Tikhomirova, N.K.; Philippov, P.P.; Koch, K.W. Recoverin and rhodopsin kinase activity in detergent-resistant membrane rafts from rod outer segments. J. Biol. Chem. 2004, 279, 48647–48653. [Google Scholar] [CrossRef]
- Vishveshwara, S.; Ghosh, A.; Hansia, P. Intra and inter-molecular communications through protein structure network. Curr. Protein Pept. Sci. 2009, 10, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Papaleo, E. Integrating atomistic molecular dynamics simulations, experiments, and network analysis to study protein dynamics: Strength in unity. Front. Mol. Biosci. 2015, 2, 28. [Google Scholar] [CrossRef] [PubMed]
- Vendruscolo, M.; Dokholyan, N.V.; Paci, E.; Karplus, M. Small-world view of the amino acids that play a key role in protein folding. Phys. Rev. E Stat. Nonlin Soft Matter Phys. 2002, 65, 061910. [Google Scholar] [CrossRef] [PubMed]
- Marino, V.; Dell’Orco, D. Allosteric communication pathways routed by Ca2+/Mg2+ exchange in GCAP1 selectively switch target regulation modes. Sci. Rep. 2016, 6, 34277. [Google Scholar] [CrossRef] [PubMed]
- Marino, V.; Dell’Orco, D. Evolutionary-Conserved Allosteric Properties of Three Neuronal Calcium Sensor Proteins. Front. Mol. Neurosci. 2019, 12, 50. [Google Scholar] [CrossRef] [PubMed]
- Galassi, V.V.; Villarreal, M.A.; Montich, G.G. Relevance of the protein macrodipole in the membrane-binding process. Interactions of fatty-acid binding proteins with cationic lipid membranes. PLoS ONE 2018, 13, e0194154. [Google Scholar] [CrossRef] [PubMed]
- Brand, I.; Matyszewska, D.; Koch, K.W. Binding of a Myristoylated Protein to the Lipid Membrane Influenced by Interactions with the Polar Head Group Region. Langmuir 2018, 34, 14022–14032. [Google Scholar] [CrossRef] [PubMed]
- Valentine, K.G.; Mesleh, M.F.; Opella, S.J.; Ikura, M.; Ames, J.B. Structure, topology, and dynamics of myristoylated recoverin bound to phospholipid bilayers. Biochemistry 2003, 42, 6333–6340. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Jo, S.; Lim, J.B.; Klauda, J.B.; Im, W. CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009, 97, 50–58. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; Vangunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular-Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Crystal Structure and Pair Potentials: A Molecular-Dynamics Study. Phys. Rev. Lett. 1980, 45, 1196–1198. [Google Scholar] [CrossRef]
- Zernii, E.Y.; Komolov, K.E.; Permyakov, S.E.; Kolpakova, T.; Dell’orco, D.; Poetzsch, A.; Knyazeva, E.L.; Grigoriev, I.I.; Permyakov, E.A.; Senin, I.I.; et al. Involvement of the recoverin C-terminal segment in recognition of the target enzyme rhodopsin kinase. Biochem. J. 2011, 435, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Marino, V.; Sulmann, S.; Koch, K.W.; Dell’Orco, D. Structural effects of Mg2+ on the regulatory states of three neuronal calcium sensors operating in vertebrate phototransduction. Biochim. Biophys. Acta 2015, 1853, 2055–2065. [Google Scholar] [CrossRef] [PubMed]
- Ames, J.B.; Levay, K.; Wingard, J.N.; Lusin, J.D.; Slepak, V.Z. Structural basis for calcium-induced inhibition of rhodopsin kinase by recoverin. J. Biol. Chem. 2006, 281, 37237–37245. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Amadei, A.; Linssen, A.B.; Berendsen, H.J. Essential dynamics of proteins. Proteins 1993, 17, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Tiberti, M.; Invernizzi, G.; Lambrughi, M.; Inbar, Y.; Schreiber, G.; Papaleo, E. PyInteraph: A framework for the analysis of interaction networks in structural ensembles of proteins. J. Chem. Inf. Model. 2014, 54, 1537–1551. [Google Scholar] [CrossRef] [PubMed]
- Michaud-Agrawal, D.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [PubMed]
- Gowers, R.; Linke, M.; Barnoud, J.; Reddy, T.J.E.; Melo, M.N.; Seyler, S.L.; Domanski, J.; Dotson, D.L.; Buchoux, S.; Kenney, I.M.; et al. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations. In Proceedings of the 15th Python in Science Conference (SCIPY 2016), Austin, TX, USA, 11–17 July 2016; pp. 1–8. [Google Scholar]
- Ames, J.B.; Porumb, T.; Tanaka, T.; Ikura, M.; Stryer, L. Amino-terminal myristoylation induces cooperative calcium binding to recoverin. J. Biol. Chem. 1995, 270, 4526–4533. [Google Scholar] [CrossRef] [PubMed]
- Dell’Orco, D.; Sulmann, S.; Linse, S.; Koch, K.W. Dynamics of conformational Ca2+-switches in signaling networks detected by a planar plasmonic device. Anal. Chem. 2012, 84, 2982–2989. [Google Scholar] [CrossRef]
- Komolov, K.E.; Senin, I.I.; Kovaleva, N.A.; Christoph, M.P.; Churumova, V.A.; Grigoriev, I.I.; Akhtar, M.; Philippov, P.P.; Koch, K.W. Mechanism of rhodopsin kinase regulation by recoverin. J. Neurochem. 2009, 110, 72–79. [Google Scholar] [CrossRef]
- Marino, V.; Astegno, A.; Pedroni, M.; Piccinelli, F.; Dell’Orco, D. Nanodevice-induced conformational and functional changes in a prototypical calcium sensor protein. Nanoscale 2014, 6, 412–423. [Google Scholar] [CrossRef]
- Sulmann, S.; Dell’Orco, D.; Marino, V.; Behnen, P.; Koch, K.W. Conformational changes in calcium-sensor proteins under molecular crowding conditions. Chemistry 2014, 20, 6756–6762. [Google Scholar] [CrossRef]
- Orban, T.; Jastrzebska, B.; Palczewski, K. Structural approaches to understanding retinal proteins needed for vision. Curr. Opin. Cell Biol. 2014, 27, 32–43. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borsatto, A.; Marino, V.; Abrusci, G.; Lattanzi, G.; Dell’Orco, D. Effects of Membrane and Biological Target on the Structural and Allosteric Properties of Recoverin: A Computational Approach. Int. J. Mol. Sci. 2019, 20, 5009. https://doi.org/10.3390/ijms20205009
Borsatto A, Marino V, Abrusci G, Lattanzi G, Dell’Orco D. Effects of Membrane and Biological Target on the Structural and Allosteric Properties of Recoverin: A Computational Approach. International Journal of Molecular Sciences. 2019; 20(20):5009. https://doi.org/10.3390/ijms20205009
Chicago/Turabian StyleBorsatto, Alberto, Valerio Marino, Gianfranco Abrusci, Gianluca Lattanzi, and Daniele Dell’Orco. 2019. "Effects of Membrane and Biological Target on the Structural and Allosteric Properties of Recoverin: A Computational Approach" International Journal of Molecular Sciences 20, no. 20: 5009. https://doi.org/10.3390/ijms20205009
APA StyleBorsatto, A., Marino, V., Abrusci, G., Lattanzi, G., & Dell’Orco, D. (2019). Effects of Membrane and Biological Target on the Structural and Allosteric Properties of Recoverin: A Computational Approach. International Journal of Molecular Sciences, 20(20), 5009. https://doi.org/10.3390/ijms20205009